

Abstract
Myasthenia gravis (MG) is an autoimmune disorder caused by target-specific pathogenic antibodies directed toward postsynaptic neuromuscular junction (NMJ) proteins, most commonly the skeletal muscle nicotinic acetylcholine receptor (AChR). In MG, high-affinity anti-AChR Abs binding to the NMJ lead to loss of functional AChRs, culminating in neuromuscular transmission failure and myasthenic symptoms. Intravenous immune globulin (IVIg) has broad therapeutic application in the treatment of a range of autoimmune diseases, including MG, although its mechanism of action is not clear. Recently, the anti-inflammatory and anti-autoimmune activities of IVIg have been attributed to the IgG Fc domains. Soluble immune aggregates bearing intact Fc fragments have been shown to be effective treatment for a number of autoimmune disorders in mice, and fully recombinant multimeric Fc molecules have been shown to be effective in treating collagen-induced arthritis, murine immune thrombocytopenic purpura, and experimental inflammatory neuritis. In this study, a murine model of MG (EAMG) was used to study the effectiveness of this novel recombinant polyvalent IgG2a Fc (M045) in treating established myasthenia, with a direct comparison to treatment with IVIg. M045 treatment had profound effects on the clinical course of EAMG, accompanied by down-modulation of pathogenic antibody responses. These effects were associated with reduced B cell activation and T cell proliferative responses to AChR, an expansion in the population of FoxP3+ regulatory T cells, and enhanced production of suppressive cytokines, such as IL-10. Treatment was at least as effective as IVIg in suppressing EAMG, even at doses 25–30 fold lower. Multimeric Fc molecules offer the advantages of being recombinant, homogenous, available in unlimited quantity, free of risk from infection and effective at significantly reduced protein loads, and may represent a viable therapeutic alternative to polyclonal IVIg.
Keywords: IgG, Fc, IVIg, multimers, EAMG, T cells, Regulatory T cells, B cells, Dendritic cells
1. Introduction
Myasthenia gravis (MG) is an autoimmune disorder characterized in most cases by T cell and antibody (Ab) responses to the skeletal muscle nicotinic acetylcholine receptor (AChR). High-affinity, anti-AChR Abs bind to the muscle endplate leading to AChR dysfunction or loss via activation of complement, cross-linking of AChR receptors, or direct blockade of acetylcholine binding sites [1,2]. MG is typically managed with acetylcholinesterase inhibitors and immunosuppressive medications, but acute exacerbations are treated using either therapeutic plasma exchange or intravenous immune globulin (IVIg). The effectiveness of IVIg in MG has been demonstrated in a randomized clinical trial [3], and it is often preferred due to its ease of administration, although it has definite limitations due to its expense, potential side effects, and the high volume load of a therapeutic dose [4].
Although the mode of action of IVIg in MG is still not clear, several possibilities have been proposed, including actions related to the Fc portion of IgG. In fact, recent studies suggest that the anti-inflammatory and anti-autoimmune effects of IVIg reside primarily in the Fc fragment [5–7]. While the exact mechanisms of Fc-mediated immune tolerance are controversial, it is likely that Fc interactions with Fc gamma receptors (FcγRs) are critically involved. FcγRs play an essential role in antibody-mediated effector functions, and blocking of activating FcγRs results in the abrogation of antibody activity in autoimmune models [7]. It is also well-known that the majority of FcγRs are low-affinity receptors, binding Fc bearing immune aggregates more efficiently than homodimeric Fc fragments that comprise normal IVIg [7]. Along these lines, aggregated IgG fragments have been shown to be required for suppression of inflammation in immune thrombocytopenic purpura (ITP) and inflammatory arthritis animal models [8–11].
Fc-based fusion protein therapeutics have recently emerged as a significant class of highly efficient pharmaceuticals, in which the Fc region of an antibody of the IgG isotype is joined to a different protein [12,13]. Moreover, their effectiveness is commonly believed to be due to their interaction with specific effector proteins, such as the neonatal Fc receptor (FcRn), which increases IgG serum half-life and prolongs therapeutic activity [14,15]. Fc fragments have also been tested along with adjuvants for the stimulation of protective immunity or induction of tolerance against specific antigens due to their ability to activate specific FCγRs[16]. However, current approaches that use Fc fragments to deliver Ag to immune cells have a major disadvantage in that the stalk from their monomeric structure are unable to cross-link multiple FcγRs required for enhanced cell signaling [17]. Thus, it has been a long-sought goal to develop a strategy to couple homodimeric IgG Fc-fusion proteins efficiently into polymeric immune complexes.
Murine IgG2a is the homologue of human IgG1, and both molecules have a high affinity for FcγRI [18,19], share the ability to fix complement and bind to protein antigens [20,21]. The IgG1 is the most abundant human immunoglobulin and thus the major component of IVIG [22–24]. Therefore, to develop a platform for clinical translation, fully recombinant Fc molecules consisting of multimerized murine IgG2a Fc (termed M045) were developed and shown to bind with high affinity to canonical FcγRs, and to effectively ameliorate collagen-induced arthritis and murine immune thrombocytopenic purpura [25]. In the present study, we sought to compare the therapeutic efficacy of M045 with that of IVIg in a murine model of autoimmune myasthenia gravis (EAMG). Our findings show that M045 effectively down-modulated ongoing EAMG, and these clinical effects were accompanied by lowered serum autoantibody levels, reduced splenic B cell number and T cell proliferative responses, an expansion in the population of splenic FoxP3+ regulatory T cells and a reduction in the expression of co-stimulatory molecules on B cells and dendritic cells.
2. Materials and Methods
2.1 Antibodies and cell culture reagents
APC-conjugated anti-mouse CD11c, CD19 and FOXP3, FITC-conjugated anti-mouse CD80, CD25 and BAFF-R, PE-conjugated anti-mouse CD40, eFluor 450-conjugated anti-mouse CD4 and CD16/CD32 (for FCγRIIB), and respective isotype controls were purchased from eBioscience, CA, USA. RPMI 1640 medium supplemented with 1% sodium pyruvate, 1% non-essential amino acids, 2mM L-glutamine, 20mM HEPES, 50 U/ml penicillin and 50 μg/ml streptomycin (GIBCO, CA), 50μM 2-ME, 10% heat inactivated FBS (Invitrogen, CA) was used as culture medium. Anti-mouse CD3 (clone OKT3) and carboxyfluorescein succinimidyl ester (CFSE) were purchased from eBioscience and Invitrogen, respectively. Complete Freund’s adjuvant (CFA) and incomplete Freund’s adjuvant (IFA) were purchased from Sigma, Saint Louis.
2.2 Mice
Eight-week-old female C57BL/6J mice were purchased from The Jackson Laboratory. Mice were housed in the Biologic Resources Laboratory facilities at the University of Illinois at Chicago and provided food and water ad libitum. All mice were cared for in accordance with the guidelines set forth by the University of Illinois Animal Care and Use committee.
2.3 Purification of Torpedo AChR (tAChR) and induction of EAMG
tAChR was purified from the electric organs of Torpedo californica by affinity chromatography using a conjugate of neurotoxin coupled to agarose as described previously [26,27]. Purity of the isolated product was tested by SDS-PAGE. The purified tAChR was used to induce EAMG and as Ag for in vitro testing of immune responses. To induce EAMG, mice were immunized with 40 μg of tAChR emulsified in CFA in a total volume of 200 μl s.c. along the back and at the base of the tail on day -1. Mice were boosted with 20 μg of tAChR emulsified in IFA in 200 μl of volume injected in the flanks and tail base on day 26 after first immunization.
2.4 Clinical scoring of EAMG
For clinical examination, mice were observed on a flat platform for a total of 2 min. They were then exercised by gently dragging them suspended by the base of the tail across a cage top grid repeatedly (20–30 times) as they attempted to grip the grid. They were then placed on a flat platform for 2 min and again observed for signs of EAMG. Clinical muscle weakness was graded as follows: grade 0, mouse with normal posture, muscle strength, and mobility at baseline and after exercise; grade 1, normal at rest but with muscle weakness characteristically shown by a hunchback posture, restricted mobility, and difficulty in raising the head after exercise; grade 2, grade 1 symptoms without exercise during observation period; grade 3, dehydrated and moribund with grade 2 weakness; and grade 4, dead.
2.5 Generation of recombinant IgG2a Fc multimers (M045)
M045 and human IVIg were kindly provided by Gliknik, Baltimore, MD, USA. To test the efficacy of polyvalent FcR-binding fragments in the treatment of EAMG, fully recombinant forms of polyvalent murine IgG2a Fc were constructed by linking the hinge-CH2-CH3 domain of murine IgG2a Fc to a multimerization domain at the carboxy terminus (M045) as described previously [25]. These proteins were manufactured in a shake flask system using transient transfection of an HEK cell line and purified on a GE AktaXpress system using GE mAb Select Proetin A affinity columns [15]. Enhanced formation of highly ordered IgG2a Fc multimers was confirmed by SDS-PAGE. Upon purification, M045 exists as homodimers and highly ordered multimers of the homodimer, as defined by both SDS-PAGE and analytical ultracentrifugation.
2.6 Purification of mouse AChR
To purify AChR, mouse muscle was used to prepare extracts containing mouse AChR, according to the method published by Wu et al [28]. Briefly, mouse muscle was homogenized in buffer A containing 0.1M NaCl; 10mM NaN3; 0.01M EDTA; 0.01M EGTA; 0.01M iodacetamide; 1mM PMSF; 1mM sodium phosphate buffer; pH 7.5). The resulting homogenate was clarified at 17,000×g for 30 min at 4 °C. The resultant pellet was resuspended in buffer A containing 0.1% Triton X-100, agitated at low speed 3 to 4h at 4°C and centrifuged at 17,000×g for 30min at 4°C. The resultant supernatant was centrifuged at 50,000×g for 30 min at 4 °C to yield supernatants containing crude AChR. This crude preparation was then applied to a neurotoxin-3 affinity column [29] to prepare AChR. Fractions rich in AChR (as determined by protein quantitation assay) were collected, pooled, and further fractionated by hydroxylapatite column chromatography. Highly pure AChR was then aliquotted and stored at −70°C for use as antigen in the ELISA assays.
2.7 Treatment with recombinant IgG2a Fc multimers (M045) and IVIG
Mice were immunized with tAChR and then divided into five groups (n=15 per group). Group - I: Control mice received PBS. Group - II: Mice received M045 at a dose of 20mg/kg body weight. Group - III: Mice received M045 at a dose of 40mg/kg body weight. Group - IV: Mice received M045 at a dose of 60mg/kg body weight. Group - V: Mice received IVIG at a dose of 1g/kg body weight. The experiment was blinded with respect to all treatment groups except IVIG. M045 was administered via tail vein in a single dose on day +1. IVIG was administered via intraperitoneally on two consecutive days. PBS was administered on day 2. Treatments were repeated on day 14 and day 28. Mice received AChR immunizations on day -1 and boost on day 26 and were scored every other day after initiation of treatment using a standard EAMG clinical severity grading scale. Mice were bled on days 0, and 45 to obtain sera and then sacrificed on day 45. Spleens were collected for use in various immunological assays. Separate groups of mice were immunized (day 1) and boosted (day 2) and scored for 10 days. We then divided these animals into three groups (n=12) with an equal balance of the various disease severities in each group. Group - I: Control mice received PBS. Group - II: Mice received M045 at a dose of 400mg/kg body weight for 5 consecutive days. Group - III: Mice received IVIG at a dose of 0.8g/kg body weight for 5 consecutive days. Clinical muscle weakness was scored every other day after initiation of treatment.
2.8 T cell proliferation assay
Single cell suspension of splenocytes prepared in PBS (0.1% BSA) was used at 2×106 cells/ml and incubated with CFSE (final concentration 2 μM) for 10 min at 37°C. Cells were washed and re-suspended in culture medium for 15 min to stabilize the CFSE staining. After a final wash, cells were re-suspended in culture medium and seeded in triplicate into 96-well, round-bottom plates at 5×104 cells per well in 200 μl of medium with/without the addition of anti-CD3 (2 μg/ml) or tAChR (5 μg/ml). After 5 days, cells were harvested, stained for CD4 and the gated lymphocytes were analyzed by flow cytometry (CyAn ADP, DakoCytomation) to determine CFSE dilution. Supernatant was collected for cytokine measurement.
2.9 Phenotypic analysis
Single-cell suspension of splenocyte was prepared, washed once and re-suspended in flow cytometry (FACS) buffer (PBS + 0.5% BSA + 2 mM EDTA). After addition of 0.25μg anti-mouse CD4, CD25, CD11c, CD19, CD40, CD80, BAFF-R and CD16/CD32, the cell suspension was mixed gently and incubated at 4°C for 30 min. Cells were washed, pelleted and re-suspended in 100 μl FACS buffer. Intracellular staining to determine FOXP3 expression was performed as per the manufacturer’s recommendations (eBioscience, CA, USA). Briefly, CD4 and CD25 stained cells were washed once with FACS buffer and fixed overnight. After washing again with FACS buffer, cell pellet was re-suspended in 100μl FACS buffer. Anti-mouse FOXP3 was added to the cell suspension and incubated at room temperature for 30 min. After incubation, FACS buffer was added to each sample and cells were pelleted, resuspended in 200 μl buffer and subjected to FACS analyses. Isotype controls were used to determine the gating parameters.
2.10 Reverse Transcription-Polymerase Chain Reaction
Splenocytes (1×106) were washed once and re-suspended in 500μl FACS buffer in a 5ml polystyrene round bottom tube. After addition of 0.5μg APC-anti-mouse CD19, the cell suspension was mixed gently and incubated at 4°C for 30 min. Cells were then washed once in cold FACS buffer. Labeled CD19+ B cells were sorted using a MoFlo (Becton Dickinson). The gates for the sorted B cell subsets were set to include only those events exhibiting the CD19-specific fluorescence. Isotype controls and unstained cells were used to determine the gating parameters. The resulting B cell preparation was >85 pure. Total RNA was isolated from the sorted B cells using RNase Micro kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The purity of RNA obtained was >1.75. For cDNA synthesis, a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used according to manufacturer’s instructions. Briefly, 2 μg of total RNA was reverse transcribed using MultiScribe™ Reverse Transcriptase (50 U/μl) in the presence of 2 μl Random primers, 0.8 μl 100 mM dNTP Mix, 1 μl of RNase Inhibitor and 10xRT Buffer in a final volume of 20 μl. The reaction was carried out in an iCycler (Biorad, Germany) thermocycler at 25°C for 10 min, 37°C for 120 min and 85°C for 5 min. The following specific oligonucleotide primers were used in the multiplex PCR: i) Mice FcγRIIB (PubMed Nucleotide Accession No. NM_010187.2) sense primer 5′-GGATTGCTGTCGCAGCCATTGTTA-3′, and the antisense primer 5′-TGCTCTGTTTCTTCATCCAGGGCT-3′. The predicted size of the amplified fragment by multiplex PCR is 172 base pairs (bp). ii) Mouse β-actin (PubMed Nucleotide Accession No. M12481.1) sense primer 5′-TCTTGGGTATGGAATCCTGTGGCA-3′, and the antisense primer 5′-ACTCCTGCTTGCTGATCCACATCT-3′. The predicted size of the amplified fragment by multiplex PCR is 288 bp. Reaction was performed in triplicate in 50 μL using 200 nM of specific primers, 1μl cDNA, and 2x QIAGEN Multiplex PCR Master Mix (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Thirty-five cycles of amplification reaction was carried out as follows: initial PCR activation at 95 °C for 15 min, denaturation for 30 sec at 94 °C, annealing for 1.5 min at 58 °C, extension for 1.5 min at 72 °C, with a final extens ion at 72 °C for 10 min. Finally, the reaction mixture containing PCR products were separated by electrophoresis using a 2% agarose gel along with 100 bp marker DNA. Gels were densitometrically (Bio-Rad) scanned and the cDNAs were normalized to that of the house keeping gene or internal control (β-actin), which was co-amplified along with the cDNA of interest.
2.11 Cytokine Assay
Culture supernatants were collected and frozen at −20°C until quantification. Cytokine levels were measured by mouse Th1/Th2 enzyme-linked immunosorbent assay (ELISA) Ready SET Go! Kit (eBioscience, San Diego, CA). We measured interleukin (IL)-2 and interferon (IFN)-γ as Th1-type and IL-4 and IL-10 as Th2-type cytokines. The sensitivity of the assay for each cytokine was as follows: 2 pg/ml for IL-2, 15 pg/ml for IFN-γ, 4 pg/ml for IL-4 and 30 pg/ml for IL-10.
2.12 ELISA for anti-mouse AChR antibody isotypes
Affinity-purified mouse AChR (0.5 μg/ml) was used to coat 96-well microtiter plates (Corning Costar) with 0.1 M carbonate-bicarbonate buffer (pH 9.6) overnight at 4°C. The plates were blocked with 10% FBS in PBS at room temperature for 30 min. Serum samples, obtained from mice bled on day 0 prior to the treatment and day 45 after the treatment, were diluted 1:2000 to 1:10,000 in blocking buffer were added and incubated at 37°C for 90 min. After four washes, horseradish peroxidase (HRPO)-conjugated goat anti-mouse IgG, IgG2b (Caltag Laboratories, Burlingame) diluted 1/2000 in blocking buffer was added and incubated at 37°C for 90 min. Subsequently, tetramethylbenzidine substrate solution (eBioscience) was added and color was allowed to develop at room temperature in the dark for 15 min. The reaction was stopped by adding 2M H2SO4 and absorbance values were measured at a wavelength of 450 nm using a Bio-Rad microplate reader. Serially diluted anti-tAChR sera (from tAChR immunized mice) were used as a positive control, and normal mouse serum was used for background determination. The results were expressed as OD values.
2.13 Statistical analysis
The data were subjected to statistical analysis using Student’s t test and Wilcoxon signed test to assess the significance of individual variations between the control and treatment groups using a computer based software (SPSS 7.5 for windows student version). Non-parametric Wilcoxon signed test was used for statistical analysis of clinical severity; Student’s t test was used for ELISA, proliferation assays, and flowcytometry. A value of p<0.05 was considered significant.
3. Results
3.1 Suppression of clinical EAMG by M045 is comparable to IVIg and is associated with reduced serum anti-AChR antibody levels
To study the therapeutic efficacy of M045 in EAMG, mice were immunized with tAChR and treated with different doses of M045, or IVIG or PBS as described under Materials and Methods. Mice were scored on every 2nd or 3rd day. Mice treated with M045 and IVIG developed significantly less frequent and severe disease, compared with control animals, in which continuous worsening and pronounced ongoing disease were observed(Fig. 1a). Mice treated with either 40 or 60mg of M045 showed significant decreases in disease severity when compared to those treated with 20mg of M045. The disease suppression in mice treated with 40mg of M045 was comparable to the disease suppression seen in mice treated with IVIg 1g/kg (Fig. 1b).
Figure 1. Recombinant IgG2a Fc multimers (M045) improve disease severity in EAMG.

Clinical score was evaluated in M045, IVIG and PBS treated mice as described in materials and methods. (a) The average clinical scores during the 45 day observation period are shown for the four groups. (b) Disease severity was significantly lower in M045 and IVIG-treated mice compared with the PBS treated tAChR-immunized controls. (c) M045 and IVIG treatment significantly suppresses the disease severity in mice with established EAMG. The values in the bar diagram represent the mean ± SEM of three separate experiments. Significance at p<0.05. *, compared to PBS.
To evaluate the antibody response to AChR in treated versus untreated mice, circulating anti-mouse AChR antibody levels were determined by ELISA. By the end of the observation period, at which time the animals had received three doses of treatment, circulating anti-AChR total IgG (Fig. 2a) and IgG2b (Fig. 2b) antibody levels were significantly reduced in animals treated with either 40 or 60 mg of M045 or IVIg compared to those treated with 20mg of M045 and PBS. These data, demonstrate the therapeutic effectiveness of M045 in a classic antibody-mediated autoimmune disease, with effects comparable to that of IVIg even at 25–30 fold lower doses. Since a dose of 40mg of M045 was most beneficial, we used this dose in further mechanistic studies.
Figure 2. Effect of M045 on serum anti-AChR Ab levels in EAMG.

Anti-AChR Abs and isotypes were analyzed by ELISA at baseline and at the end of treatment in the four experimental groups. (a) total serum anti-AChR IgG; (b) anti-AChR IgG2b serum levels. The values in the bar diagram represent the mean ± SEM of duplicate values of three separate experiments. Significance at p<0.05. *, compared to PBS.
3.1.1 Effect of M045 on ongoing EAMG
To scrutinize whether M045 is also potentially suitable for treatment of myasthenia gravis, we tested its efficacy on mice suffering from the disease. For that, administration of M045 and IVIG were initiated 10 days after induction of EAMG and was continued for 5 consecutive days. Mice treated with M045 and IVIG developed significantly less frequent and severe disease, compared with PBS treated animals, in which continuous worsening and pronounced ongoing disease were observed (Fig. 1c). The suppressive effect of M045 and IVIG on EAMG lasted at least up to 2 weeks after cessation of the treatment.
3.2 M045-mediated amelioration of EAMG is associated with a decrease in splenic B cell numbers, down-regulation of CD40 and BAFF-R expression, and up-regulation of inhibitory FcγRIIb expression
M045-induced suppression of anti-AChR antibody levels in the serum could result from either inhibition of B cell activation or a reduction in their numbers. Therefore, we determined the B cell frequency as well as the expression levels of activation markers CD40 and B cell activating factor receptor (BAFF-R). As expected, compared to mice treated with PBS (63%), mice treated with either M045 (53%) or IVIg (52%) showed significant reductions in B cell numbers (Fig. 3a) as well as in the percentage of cells expressing activation markers CD40 (Fig. 3b) and BAFF-R (Fig. 3c). Interestingly, the percentage of cells expressing CD40 and BAFF-R was lower in M045-treated mice (24% vs 43% in PBS treated mice) than in IVIG treated mice (35%). Since B cell activation can be prevented by a negative feedback upon engagement of an inhibitory FcγRIIb, we measured the levels of inhibitory FcγRIIB expression in B cells. We observed a significant increase in surface expression of FcγRIIB protein (Fig. 3d) and its mRNA levels in B cells from M045 and IVIg treated mice (Fig. 3e) relative to B cells from PBS treated mice, although this increase was more pronounced in B cells from IVIg treated mice.
Figure 3. Recombinant IgG2a Fc multimers (M045) downregulate B cell activation markers in EAMG.

Splenocytes were isolated and stained with APC-labeled anti-mouse CD19, FITC-labeled anti-mouse BAFF-R, eFluor 450-labeled anti-mouse CD16/CD32 (for FCγRIIB) and PE-labeled CD40. Representative plots and bar diagrams showing the percentage of CD19+ B cells (a), CD19+CD40+ B cells (b), CD19+BAFF-R+ B cells (c) and the intensity of cell surface inhibitory FcγRIIb protein expression (d) are shown. Inhibitory FcγRIIb mRNA expression was determined by Multiplex PCR. Total RNA, extracted from B cells, was reverse transcribed and a 172bp fragment corresponding to FcγRIIb was amplified and separated on a 2% agarose gel. Gels were densitometrically scanned and cDNAs were normalized to that of &beta:-actin, which was co-amplified along with the cDNA of interest. (e) Representative gel and bar diagram showing relative FcγRIIb mRNA expression in B cells from recombinant IgG2a Fc multimers (M045) and IVIG treated compared to PBS treated controls. The values in the bar diagram represents the mean ± SEM of triplicate values of three separate experiments. Significance at p<0.05. *, compared to PBS; ♠, compared to IVIG.
3.3 Suppression of AChR-specific T cell proliferation in M045-treated mice is coupled with expansion of Foxp3+ T cells
To evaluate the effects of M045 on T cells, we analyzed tAChR stimulated T cell proliferation. Both anti-CD3-induced polyclonal and auto-antigen (tAChR) specific T cell proliferation were significantly decreased in M045 (21% and 12% respectively) and IVIg (32% and 15% respectively) treated mice when compared to PBS (57% and 32% respectively) treated animals (Fig. 4a & b). Interestingly, M045 treatment suppressed both anti-CD3-induced and antigen-specific T cell proliferation more effectively than did IVIG treatment. Foxp3+ regulatory T cells (Tregs) are a specialized subpopulation of T cells that actively suppress T cell responses and help maintain self-tolerance. Therefore, we analyzed for CD4+CD25+ as well as Foxp3+ T cells. The percentages of both CD4+CD25+ (Fig. 5a) as well as CD4+CD25+Foxp3+ (Fig. 5b) T cells were significantly higher in M045 (14.2 and 11.3 respectively) and IVIg (11% and 7.9% respectively) treated mice compared to the PBS group (6.3 and 4.6% respectively). Moreover, M045 and IVIG treatment significantly increased CD4+CD25−Foxp3+ T cells (17% and 12% respectively) compared to PBS treated mice (9%) (Fig. 5b).
Figure 4. Recombinant IgG2a Fc multimer (M045) suppresses polyclonal and antigen-specific T cell proliferation.

CFSE-labeled splenocytes were stimulated with anti-mouse CD3 or tAChR as described in Materials and methods. (a) Representative flow cytometry plots illustrate CFSE dilution profiles of anti-CD3 and tAChR driven T cell proliferation. (b) Bar diagram shows the percent of CD4+ T cell proliferation from splenocyte cultures upon stimulation with anti-CD3 and tAChR. The values in the bar diagram represent the mean ± SEM of duplicate values of three separate experiments. Significance at p<0.05. *, compared to PBS; ♠, compared to IVIG.
Figure 5. Recombinant IgG2a Fc multimer (M045) induces expansion of CD4+CD25+ and FOXP3 expressing T cells.
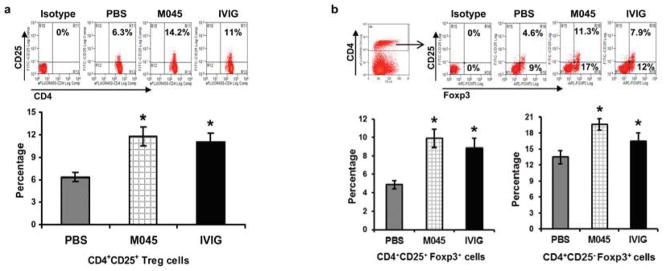
Splenocytes were isolated and stained with CD4, CD25 and Foxp3 antibodies. Representative plots and bar diagrams show the percentages of CD4+CD25+ T cell (a), CD4+CD25+Foxp3+ and CD4+CD25−Foxp3+ T cells (b). The values in the bar diagram represent the mean ± SEM of duplicate values of three separate experiments. Significance at p<0.05. *, compared to PBS.
3.4 Treatment with M045 decreases co-stimulatory molecule expression on dendritic cells
To investigate the effect of M045 on dendritic cells (DC), we assessed the DC phenotype and observed a significant decrease in the percentages of CD11c+CD40+ (Fig. 6a) and CD11c+CD80+ dendritic cells (Fig. 6b) in splenocytes from M045 (4.5 and 6.1% respectively) and IVIg (1.0% and 1.45% respectively) treated mice relative to PBS (7.0% and 8.7% respectively) treated mice. Treatment with IVIg also decreased the percentage of CD11c+CD80+ cells more significantly than did treatment with M045.
Figure 6. Recombinant IgG2a Fc multimers (M045) down-modulate dendritic cell maturation markers in EAMG.
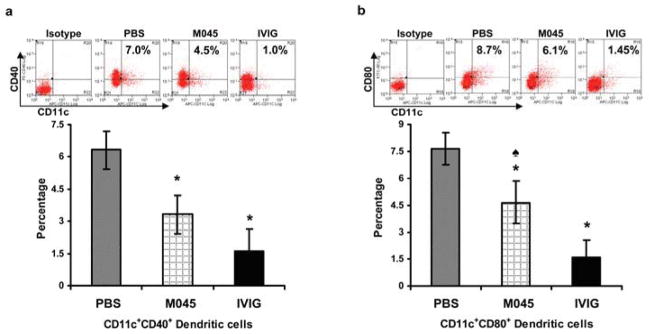
Splenocytes were isolated and stained with CD11c, CD40 and CD80 antibodies. Representative plots and bar diagrams show the percentage of CD11c+CD40+ dendritic cells (a) and CD11c+CD80+ dendritic cells (b). The values in the bar diagram represents the mean ± SEM of duplicate values of three separate experiments. Significance at p<0.05. *, compared to PBS; ♠, compared to IVIG.
3.5 Suppression of T cell proliferation in M045 treated mice is associated with decreased pro-inflammatory cytokines and increased anti-inflammatory cytokines
Levels of IL-2, IFN-γ IL-4 and IL-10 were measured in the supernatants obtained from cultures stimulated with anti-CD3 and tAChR as described above. Compared to the PBS-treated group, splenocytes from M045 and IVIg-treated mice secreted significantly lower amounts of pro-inflammatory cytokines such as IL-2 (Fig. 7a&e) and IFN-γ (Fig. 7b&f), while they produced much higher amounts of anti-inflammatory cytokines such as IL-10 (Fig. 7c&g) and IL-4 (Fig. 7d&h). Interestingly, the increase in the levels of IL-10 was more pronounced in M045-treated group when compared to the IVIg-treated group.
Figure 7. Down modulated T cell proliferation in splenocytes obtained from recombinant IgG2a Fc multimers (M045)-treated mice is associated with decreased levels of pro-inflamatory cytokines and elevated level of IL-4 and IL-10.

The cytokines IL-2 (a&e), INF-γ (b&f), IL-10 (c&g) and IL-4 (d&h) were measured in the supernatants obtained from the proliferation assay cultured in which anti-CD3 and tAChR were used to stimulate T cells. The values in the bar diagram represents the mean ± SEM of duplicate values of two separate experiments. Significance at p<0.05. *, compared to PBS; ♠, compared to IVIG.
4. Discussion
IVIg has been shown to have broad therapeutic application in the treatment of a number of autoimmune diseases, including MG (3,30). As noted, the anti-inflammatory activities of IVIg are now generally attributed to the IgG Fc domain [16,31], and soluble immune aggregates bearing intact Fc fragments have previously been shown to be effective in the treatment of autoimmune disorders in mice [9–11]. We directly compared treatment with IVIg to novel fully recombinant IgG2a Fc multimers (M045) and determined the effects on myasthenic weakness and immune regulatory functions in EAMG.
Our results indicate that IVIg and multimerized murine IgG Fc (M045) (at two of the tested doses) have similar therapeutic efficacy. The presentation of multimerized, polyvalent Fc to FcγRs by M045 is hypothesized to enhance binding to these low-affinity receptors, thereby promoting immune modulation. In support of this hypothesis, the therapeutic potential of IgG containing Fc multimers has been demonstrated in animal models [10,11] and the translational relevance is suggested by clinical studies in ITP, in which the therapeutic effects of IVIg directly correlate with the presence of immune aggregates in the sera [32]. Despite these data, there have been few previous studies investigating the therapeutic potential of recombinant IgG Fc multimers.
In this study, we have demonstrated that multimerized murine IgG Fc is capable of protecting mice against subsequently induced EAMG and suppressing ongoing disease. Administration of M045 to mice with EAMG significantly reduced muscle weakness when compared to mice treated with PBS. The disease suppression in mice receiving 40 mg of M045 was comparable to that noted in the IVIg-treated group. Moreover, the observed improvement in muscle strength in both M045 and IVIg treatment groups was associated with diminished serum anti-AChR-antibody levels. The 20 mg dose exhibited lesser (although non-significant compared to placebo) effects, while the 60 mg dose was somewhat less beneficial compared to the 40 mg dose. These observations indicate that the therapeutic effect of M045 was dose-dependent and was as effective as IVIg, even when used at 25–30-fold lower amounts. Our findings in the IVIg-treated mice are consistent with an earlier study [33] which showed that IVIg can suppress disease in the rat model of EAMG. In addition, these results add to previously published studies in which fully recombinant IgG2a Fc multimers have been shown to alleviate collagen-induced arthritis in a dose-dependent fashion, to prevent idiopathic thrombocytopenic purpura in mice [25], and to ameliorate clinical, electrophysiologic, and histologic disease in experimental autoimmune neuritis (EAN) [34]. Interestingly, we noted a rapid decline in the clinical score shortly after each treatment with M045 and IVIG. Although we do not know the underlying cause for this effect, we suspect that M045 and IVIG directly acted at the neuromuscular junction, by scavenging the active complement components and thus diverting the harmful action of complement on the end plates [35–37]. This may have provided the transient clinical relief noted in the treated animals.
B cells play a key role in the pathogenesis of MG and EAMG by producing autoantibodies [2,38]. While higher levels of BAFF-R and CD40 expression in B cells enhance their survival [39,40], the inhibitory receptors FcγRIIB negatively regulate antigen-specific proliferation and differentiation of B cells [41,42]. Interestingly, both M045 and IVIg treatments decreased B cell numbers as well as the levels of expression of activation markers CD40 and BAFF-R on B cells. In contrast, the level of expression of the inhibitory FcγRIIb mRNA and protein was increased. These results suggest that M045 may suppress B cell activation, and as a consequence AChR antibody production, by up-regulating the levels of expression of inhibitory FcγRIIb in B cells. However, higher levels of FcγRIIb transcripts were found in B cells from IVIg-treated mice relative to M045-treated mice and it correlated somewhat better with a more significant reduction in anti-AChR antibody levels (Fig-2). In contrast, the B cell activation markers were more potently suppressed in M045-treated mice. These somewhat discordant results may be due to differences in the mechanisms of action of M045 and IVIG, or may also be attributable to the fact that M045 is mouse-derived and IVIg is human-derived.
Interaction of B cells with auto-reactive AChR-specific CD4+ T cells is necessary for full activation of the B cells and high affinity autoantibody production in EAMG and MG [2,43]. The interaction of CD40L expressed on activated T cells with CD40 expressed on B cells induces B-cell proliferation and differentiation into antibody-secreting cells [44]. Diminished polyclonal as well as tAChR-specific T cell proliferation noted in splenocytes from M045 and IVIG treated mice imply that loss of optimum T cell help may also have contributed to blunting of B cell activation. It is well known that Foxp3+ Tregs can suppress the activation and proliferation of, and cytokine production by, autoreactive T cells and thus control autoimmunity [45–47]. Interestingly, the decreased T cell proliferation noted in M045-treated mice was associated with an increase in the number of splenic Foxp3+ Tregs, with these cells possibly mediating the suppression of T cell proliferation. Furthermore, we not only observed an expansion of CD4+CD25+FOXP3+ cells, but also CD4+CD25−FOXP3+ cells having a phenotype consistent with type 1 Tregs (Tr1). This may be consistent with the hypothesis that naturally occurring CD4+CD25+ Tregs play a role in the induction and differentiation of Tr1 cells, which are induced upon Ag exposure under certain tolerogenic conditions [48–50]. Future studies will help more clearly define the role of these two Tregs subset in disease suppression noted in both M045 and IVIG treated mice.
The activation of T cells is determined by their interactions with antigen presenting cells (APCs). DCs are the most potent APCs and play an important role in MG by presenting self-antigenic peptides required for priming and/or activation of AChR-specific T cells [51–53]. Furthermore, signaling through CD40 expressed on DCs can up-regulate the expression of CD80 co-stimulatory molecule [54,55]. Blockade of T cell interaction with CD80 on DCs inhibits immune responses including autoimmune responses [56]. In addition, DCs, which are not fully activated have a better ability to induce Tregs than CD40 and CD80 expressing matured DCs [27,46,47,57]. We found that the levels of expression of DC maturation markers CD40 and CD80 was significantly or IVIG. reduced in mice treated with M045 or IVIG. In this respect, it is worth noting that FcγRIIB-deficient DCs, or DCs incubated with a mAb that blocks immune complex binding to FcγRIIB, show a spontaneous maturation [−58–60]. These observations imply that M045 decreased DC maturation, which in turn led to suboptimal T cell activation and increased Treg frequency.
Helper T cells can differentiate into two major subtypes known as Th1 and Th2 cells. The Th1-type cells secrete pro-inflammatory cytokines, mainly IL-2 and IFN-γ; the Th2-type cells produce among others IL-4 and IL-10. While Th1-type cytokines are thought to play a critical role in the pathogenesis of autoimmune diseases [61] including EAMG [62,63], by facilitating the production of complement activating IgG2b isotypes [64,65], while the Th2 cytokines have been shown to be protective in EAMG [66,67] by facilitating the production of non-complement activating IgG1 antibodies [64,65]. Apart from Th2 cells, tolerogenic DCs are also known to express high amounts of IL-10 and TGF-α [68], and induce/expand Tregs in vitro and in vivo [69]. Our results showed that reduced T cell proliferation as well as DC activation in M045 or IVIg treated mice were accompanied by a reduction in the levels of IFN-γ and IL-2 and an increase in IL-4 and IL10. Taken together, this suggests that M045 or IVIG can down regulate pathogenic T cell responses by modulating DC maturation as well as Th1/Th2 cytokine balance. These findings are fully consistent with our earlier studies which showed that GM-CSF mediated reversal of myasthenogenic manifestations in EAMG was accompanied by decreased T cell proliferation, increased Treg numbers and altered levels of Th1 and Th2 cytokines [27]. It is persuasive to speculate that the selective response of T cells and dendritic cells in EAMG mice to M045 treatment may result in Th2-type responses and thus favor IgG1 production, which are non-pathogenic due to their inability to activate complement [65]. Further investigation including examining IgG1 or IgG2a anti AChR titers and complement activation may help clarify the underlying mechanism.
IgG mediates its effector functions by interacting with its cognate cellular Fc-γ receptors (FcγR) through their Fc portion [16]. These receptors can be either activating or inhibitory, and both types of FcγR are expressed on most hematopoietic cells except T cells [70]. While FcγRI and FcγRIII are activating receptors, the FcγRIIb is inhibitory. The immunomodulatory effects of IgG results from the net effect of engaging both types of FcγR. In B cells, FcγRIIb, upon IgG binding and co-ligation of the B cell receptor, transduces an inhibitory signal thereby preventing B cells with low affinity or self-reactive receptors from entering the germinal center and becoming antibody producing plasma cells [16]. FcγRIIb receptors are also important negative regulators of DC activation and function [71]. Immature DCs express high levels of FcγRIIb [66] and their engagement limits phagocytosis and decreases cytokine production by DCs, leading to reduced expansion of memory T lymphocytes [72] and induction of regulatory T cells [68]. The immunomodulatory effects of M045 are believed to be mediated through interactions with the low/intermediate affinity FcRs, the stability of which is directly related to the degree of Fc multimerization [25]. Although the present study did not investigate IgG2aFc-FcRs interaction, it is likely that IgG2a Fc interaction with FcγRs could be involved in mediating the aforementioned M045-induced changes in DCs, T cells and B cells. In accordance with this idea, Mekhaiel et al [73] have also reported that polymeric IgG1-Fc molecules are capable of binding selectively to FcγRs, with significantly increased affinity owing to their increased valency. Further studies are needed to fully elucidate the precise mechanism of action of M045 in the treatment of EAMG along these lines, and to determine whether Fc:FcγR interactions are a primary mode of action for IVIg’s demonstrated clinical effects in MG.
In conclusion, treatment with recombinant IgG2a Fc multimers (M045) produced effective suppression of established murine EAMG. The therapeutic response is dose-dependent and comparable to that of IVIg. M045 is formed and exists as multimers in solution similar to the naturally occurring aggregates found in IVIg, although these aggregates comprise the minority of the IVIg preparation. Perhaps as a consequence, M045 is therapeutically as effective as IVIg, but at much lower concentrations. The immunomodulatory effects of M045 in EAMG include suppression of autoantibody responses, B cell and T cell activation, and DC maturation, as well as expansion of Tregs. Multimerized Fc constructs have been manufactured utilizing recombinant human IgG1 and can be produced in unlimited quantity using recombinant DNA technology, therefore ensuring homogeneity and freedom from risk of infection. Notably, our findings indicate that the therapeutic effect of Fc multimers would be at least as effective as IVIg at significantly reduced protein loads, thus providing an additional advantage as a therapeutic alternative to IVIg in the treatment of MG.
Highlights.
Recombinant Fc multimers effectively ameliorate experimental myasthenia gravis.
Effects are accompanied by a reduction in B cell activation, autoantibody production and enhancement of Tregs
Effects are dose-dependent and comparable to IVIg at 25–30 fold lower dose.
Acknowledgments
Gliknik Inc. provided study material [recombinant IgG2a Fc (M045)] and funding for these studies.
Footnotes
Conflict of Interest Statement
The authors declare no competing financial interests.
Gliknik Inc. provided study material “recombinant IgG2a Fc multimer” (M045) and funding for these studies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–54. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–90. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68:837–41. doi: 10.1212/01.wnl.0000256698.69121.45. [DOI] [PubMed] [Google Scholar]
- 4.Bertorini TE, Nance AM, Horner LH, Greene W, Gelfand MS, Jaster JH. Complications of intravenous gamma globulin in neuromuscular and other diseases. Muscle Nerve. 1996;19:388–91. doi: 10.1002/(SICI)1097-4598(199603)19:3<388::AID-MUS20>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through inhibition of Fc receptor. Science. 2001;291:484–86. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 6.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–33. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 8.Morgan EL, Tempelis CH. The Requirement for the Fc Portion of Antibody in Antigen-Antibody Complex-Mediated Suppression. J Immunol. 1978;120:1669–71. [PubMed] [Google Scholar]
- 9.Siragam V, Brinc D, Crow AR, Song S, Freedman J, Lazarus AH. Can antibodies with specificity for soluble antigens mimic the therapeutic effects of intravenous IgG in the treatment of autoimmune disease? J Clin Invest. 2005;115:155–60. doi: 10.1172/JCI22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bazin R, Lemieux R, Tremblay T, St-Amour I. Tetramolecular immune complexes are more efficient than IVIg to prevent antibody-dependent in vitro and in vivo phagocytosis of blood cells. Br J Haematol. 2004;127:90–96. doi: 10.1111/j.1365-2141.2004.05105.x. [DOI] [PubMed] [Google Scholar]
- 11.Teeling JL, Jansen-Hendriks T, Kuijpers TW, de Haas M, van de Winkel JG, Hack CE, et al. Therapeutic efficacy of intravenous immunoglobulin preparations depends on the immunoglobulin G dimers: studies in experimental immune thrombocytopenia. Blood. 2001;98:1095–99. doi: 10.1182/blood.v98.4.1095. [DOI] [PubMed] [Google Scholar]
- 12.Strohl WR, Knight DM. Discovery and development of biopharmaceuticals: current issues. Curr Opin Biotech. 2009;20:668–72. doi: 10.1016/j.copbio.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 13.Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODYTM technology. Curr Opin Biotech. 2009;20:692–99. doi: 10.1016/j.copbio.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 15.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–714. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 17.Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4:1015–28. doi: 10.1002/emmm.201201379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark MR. IgG effector mechanisms. Chem Immunol. 1997;65:88–110. [PubMed] [Google Scholar]
- 19.Taylor L, Bachler M, Duncan I, Keen S, Fallon R, Mair C, et al. In vitro and in vivo activities of OX40 (CD134)-IgG fusion protein isoforms with different levels of immune-effector functions. J Leukoc Biol. 2002;72:522–29. [PubMed] [Google Scholar]
- 20.Scott MG, Briles DE, Nahm MH. Selective IgG subclass expression: biologic, clinical and functional aspects. In: Shakib F, editor. The human IgG subclasses: molecular analysis of structure and function. Oxford: Pergamon Press; 1990. pp. 161–83. [Google Scholar]
- 21.Hussain R, Dawood G, Abrar N, Toossi Z, Minai A, Dojki M, et al. Selective increases in antibody isotypes and immunoglobulin G subclass responses to secreted antigens in tuberculosis patients and healthy household contacts of the patients. Clin Diagn Lab Immunol. 1995;2:726–32. doi: 10.1128/cdli.2.6.726-732.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negi VS, Elluru S, Sibéril S, Graff-Dubois S, Mouthon L, Kazatchkine MD, et al. Intravenous immunoglobulin: an update on the clinical use and mechanisms of action. J Clin Immunol. 2007;27:233–45. doi: 10.1007/s10875-007-9088-9. [DOI] [PubMed] [Google Scholar]
- 23.Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. 2008;29:608–15. doi: 10.1016/j.it.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Cholette JM, Lerner NB. Use of Blood Products. In: Lucking SE, Maffei FA, Tamburro RF, Thomas NJ, editors. Pediatric Critical Care Study Guide Text and Review. London: Springer-Verlag; 2012. pp. 427–50. [Google Scholar]
- 25.Jain A, Olsen HS, Vyzasatya R, Burch E, Sakoda Y, Mérigeon EY, et al. Fully recombinant IgG2a Fc multimers (stradomers) effectively treat collagen-induced arthritis and prevent idiopathic thrombocytopenic purpura in mice. Arthritis Res Ther. 2012;14:R192. doi: 10.1186/ar4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellone M, Ostlie N, Lei S, Conti-Tronconi BM. Experimental myasthenia gravis in congenic mice: sequence mapping and H-2 restriction of T helper epitopes on the α subunits of Torpedo californica and murine acetylcholine receptors. Eur J Immunol. 1991;21:2303–310. doi: 10.1002/eji.1830211003. [DOI] [PubMed] [Google Scholar]
- 27.Sheng JR, Li L, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of experimental autoimmune myasthenia gravis by granulocyte-macrophage colony-stimulating factor is associated with an expansion of FoxP3+ regulatory T cells. J Immunol. 2006;177:5296–306. doi: 10.4049/jimmunol.177.8.5296. [DOI] [PubMed] [Google Scholar]
- 28.Wu B, Goluszko E, Christadoss P. Experimental autoimmune myasthenia gravis in the mouse. Curr Protoc Immunol. 2001;Chapter 15(Unit 15.8) doi: 10.1002/0471142735.im1508s21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindstrom J, Einarson B, Tzartos S. Production and assay of antibodies to acetylcholine receptors. Methods Enzymol. 1981;74(Pt C):432–460. doi: 10.1016/0076-6879(81)74031-x. [DOI] [PubMed] [Google Scholar]
- 30.Jolles S, Sewell WA, Misbah SA. Clinical uses of intravenous immunoglobulin. Clin Exp Immunol. 2005;142:1–11. doi: 10.1111/j.1365-2249.2005.02834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIg anti-inflammatory activity with recombinant IgG Fc. Science. 2008;320:373–76. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Augener W, Friedman B, Brittinger B. Are aggregates of IgG the effective part of high-dose immunoglobulin therapy in adult idiopathic thrombocytopenic purpura (ITP)? Blut. 1985;50:249–52. doi: 10.1007/BF00320302. [DOI] [PubMed] [Google Scholar]
- 33.Zhu KY, Feferman T, Maiti PK, Souroujon MC, Fuchs S. Intravenous immunoglobulin suppresses experimental myasthenia gravis: immunological mechanisms. J Neuroimmunol. 2006;176:187–97. doi: 10.1016/j.jneuroim.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Niknami M, Wang MX, Nguyen T, Pollard JD. Beneficial effect of a multimerized immunoglobulin Fc in an animal model of inflammatory neuropathy (experimental autoimmune neuritis) J Peripher Nerv Syst. 2013;18:141–52. doi: 10.1111/jns5.12022. [DOI] [PubMed] [Google Scholar]
- 35.Basta M, Fries LF, Frank MM. High doses of intravenous Ig inhibit in vitro uptake of C4 fragments onto sensitized erythrocytes. Blood. 1991;77:376–80. [PubMed] [Google Scholar]
- 36.Lutz HU, Stammler P, Jelezarova E, Nater M, Späth PJ. High doses of immunoglobulin G attenuate immune aggregate-mediated complement activation by enhancing physiologic cleavage of C3b in C3bn-IgG complexes. Blood. 1996;88:184–93. [PubMed] [Google Scholar]
- 37.Boros P, Gondolesi G, Bromberg JS. High dose intravenous immunoglobulin treatment: mechanisms of action. Liver Transpl. 2005;11:1469–480. doi: 10.1002/lt.20594. [DOI] [PubMed] [Google Scholar]
- 38.Berrih-Aknin S, Ragheb S, Le Panse R, Lisak RP. Ectopic germinal centers, BAFF, and anti-B-cell therapy in myasthenia gravis. Autoimmun Rev. 2013;12:885–93. doi: 10.1016/j.autrev.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Lee BO, Moyron-Quiroz J, Rangel-Moreno J, Kusser KL, Hartson L, Sprague F, et al. CD40, but not CD154, expression on B cells is necessary for optimal primary B cell responses. J Immunol. 2003;171:5707–17. doi: 10.4049/jimmunol.171.11.5707. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, Kobata T. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol. 2007;37:110–18. doi: 10.1002/eji.200636623. [DOI] [PubMed] [Google Scholar]
- 41.Venkatesh J, Kawabata D, Kim S, Xu X, Chinnasamy P, Paul E, et al. Selective regulation of autoreactive B cells by FcgammaRIIB. J Autoimmun. 32:149–57. doi: 10.1016/j.jaut.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328–43. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conti-Fine BM, Milani M, Wang W. CD4+ T cells and cytokines in the pathogenesis of acquired myasthenia gravis. Ann NY Acad Sci. 2008;1132:193–209. doi: 10.1196/annals.1405.042. [DOI] [PubMed] [Google Scholar]
- 44.Engel P, Gómez-Puerta JA, Ramos-Casals M, Lozano F, Bosch X. Therapeutic targeting of B cells for rheumatic autoimmune diseases. Pharmacol Rev. 2011;63:127–56. doi: 10.1124/pr.109.002006. [DOI] [PubMed] [Google Scholar]
- 45.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 46.Cheatem D, Ganesh BB, Gangi E, Vasu C, Prabhakar BS. Modulation of dendritic cells using granulocyte-macrophage colony-stimulating factor (GM-CSF) delays type 1 diabetes by enhancing CD4+CD25+ regulatory T cell function. Clin Immunol. 2009;131:260–70. doi: 10.1016/j.clim.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c+CD8a--dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol. 2009;21:269–82. doi: 10.1093/intimm/dxn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dieckmann D, Bruett CH, Ploettner H, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory, contact-dependent T cells induce interleukin 10-producing, contact-independent type 1-like regulatory T cells. J Exp Med. 2002;196:247–53. doi: 10.1084/jem.20020642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 50.Veldman C, Nagel A, Hertl M. Type I regulatory T cells in autoimmunity andinflammatory diseases. Int Arch Allergy Immunol. 2006;140:174–83. doi: 10.1159/000092576. [DOI] [PubMed] [Google Scholar]
- 51.Nagane Y, Utsugisawa K, Obara D, Yamagata M, Tohgi H. Dendritic cells in hyperplastic thymuses from patients with myasthenia gravis. Muscle Nerve. 2003;27:582–89. doi: 10.1002/mus.10362. [DOI] [PubMed] [Google Scholar]
- 52.Xiao BG, Duan RS, Link H, Huang YM. Induction of peripheral tolerance to experimental autoimmune myasthenia gravis by acetylcholine receptor- pulsed dendritic cells. Cell Immunol. 2003;223:63–69. doi: 10.1016/s0008-8749(03)00118-7. [DOI] [PubMed] [Google Scholar]
- 53.Yang H, Kala M, Scott BG, Goluszko E, Chapman HA, Christadoss P. Cathepsin S is required for murine autoimmune myasthenia gravis pathogenesis. J Immunol. 2005;174:1729–37. doi: 10.4049/jimmunol.174.3.1729. [DOI] [PubMed] [Google Scholar]
- 54.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–52. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–78. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 56.Sayegh MH, Turka LA. T cell costimulatory pathways: promising novel targets for immunosuppression and tolerance induction. J Am Soc Nephrol. 1995;6:1143–150. doi: 10.1681/ASN.V641143. [DOI] [PubMed] [Google Scholar]
- 57.Mahnke K, Johnson TS, Ring S, Enk AH. Tolerogenic dendritic cells and regulatory T cells: A two-way relationship. J Dermatol Sci. 2007;46:159–67. doi: 10.1016/j.jdermsci.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–23. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci USA. 2005;102:2910–15. doi: 10.1073/pnas.0500014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Théry C, Rescigno M, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–80. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicholson LB, Kuchroo VK. Manipulation of the Th1/Th2 balance in autoimmune disease. Curr Opin Immunol. 1996;8:837–42. doi: 10.1016/s0952-7915(96)80013-6. [DOI] [PubMed] [Google Scholar]
- 62.Baggi F, Andreetta F, Caspani E, Milani M, Longhi R, Mantegazza R, et al. Oral administration of an immunodominant T-cell epitope downregulates Th1/Th2 cytokines and prevents experimental myasthenia gravis. J Clin Invest. 1999;104:1287–95. doi: 10.1172/JCI7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balasa B, Deng C, Lee J, Bradley LM, Dalton DK, Christadoss P, et al. Interferon gamma (IFN-gamma) is necessary for the genesis of acetylcholine receptor-induced clinical experimental autoimmune myasthenia gravis in mice. J Exp Med. 1997;186:385–91. doi: 10.1084/jem.186.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 65.Gomez AM, Van Den Broeck J, Vrolix K, Janssen SP, Lemmens MA, Van Der Esch E, et al. Antibody effector mechanisms in myasthenia gravis-pathogenesis at the neuromuscular junction. Autoimmunity. 2010;43:353–70. doi: 10.3109/08916930903555943. [DOI] [PubMed] [Google Scholar]
- 66.Zhang GX, Navikas V, Link H. Cytokines and the pathogenesis of myasthenia gravis. Muscle Nerve. 1997;20:543–41. doi: 10.1002/(sici)1097-4598(199705)20:5<543::aid-mus2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 67.Shi FD, Li H, Wang H, Bai X, van der Meide PH, Link H, et al. Mechanisms of nasal tolerance induction in experimental autoimmune myasthenia gravis: identification of regulatory cells. J Immunol. 1999;162:5757–763. [PubMed] [Google Scholar]
- 68.Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–440. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- 69.Belkaid Y, Oldenhove G. Tuning microenvironments: Induction of regulatory T cells by dendritic cells. Immunity. 2008;29:362–71. doi: 10.1016/j.immuni.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karsten CM, Köhl J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology. 2012;217:1067–79. doi: 10.1016/j.imbio.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 71.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–23. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Y, Gao X, Masuda E, Redecha PB, Blank MC, Pricop L. Regulated expression of FcgammaR in human dendritic cells controls cross-presentation of antigen-antibody complexes. J Immunol. 2006;177:8440–47. doi: 10.4049/jimmunol.177.12.8440. [DOI] [PubMed] [Google Scholar]
- 73.Mekhaiel DN, Czajkowsky DM, Andersen JT, Shi J, El-Faham M, Doenhoff M, et al. Polymeric human Fc-fusion proteins with modified effector functions. Sci Rep. 2011;1:124. doi: 10.1038/srep00124. [DOI] [PMC free article] [PubMed] [Google Scholar]