

Abstract
The recent establishment of recombination-based cloning systems has greatly facilitated the analysis of gene function by allowing rapid and high-efficiency generation of plasmid constructs. However, the use of such an approach in zebrafish requires the availability of recombination-compatible plasmids that are appropriate for functional studies in zebrafish embryos. In this work, we describe the construction and validation of Gateway compatible vectors based on commonly used zebrafish plasmids. We have generated pCS-based plasmids that allow rapid generation of both N-terminal and C-terminal fusion proteins, and we demonstrate that mRNA synthesized from these plasmids encodes functional native or fusion proteins in injected zebrafish embryos. In parallel, we have established similar Gateway plasmids containing Tol2 cis elements that promote efficient integration into the zebrafish genome and allow expression of native or fusion proteins in a tissue-specific manner in the zebrafish embryo. Finally, we demonstrate the use of this system to rapidly identify tissue-specific cis elements to aid the establishment of blood vessel-specific transgenic constructs. Taken together, this work provides an important platform for the rapid functional analyses of open reading frames in zebrafish embryos.
Keywords: zebrafish, Gateway cloning
Introduction
The zebrafish has emerged as an ideal vertebrate model organism in which to study a wide variety of biological processes. The transparency of the zebrafish embryo, coupled with its rapid external development, make it an excellent system for visualizing cell and tissue movements in a live organism (Beis and Stainier, 2006). Many researchers have taken further advantage of these aspects of the zebrafish through the development of transgenic lines that express fluorescent proteins in a tissue-specific manner (Amsterdam and Becker, 2005). These lines allow detailed cellular analysis through serial observation and time lapse in a live embryo. Along with these powerful analytical tools, there are several genetic techniques that can be used to manipulate signaling pathways of interest in zebrafish. For example, it is possible to perform targeted knockdown of gene products through the injection of anti-sense morpholino oligonucleotides (Nasevicius and Ekker, 2000). In addition, zebrafish researchers commonly use transient expression of mRNA or stable expression of transgenes to perform gain-of-function studies. Together, these approaches can provide a rapid determination of gene function during development.
Despite these benefits, generation of the appropriate plasmid constructs for functional and transgenic studies is often hampered by the inefficiency of standard ligation-mediated cloning. This finding is especially apparent when generating plasmid constructs for transgenesis. These plasmids often contain large pieces of genomic DNA, and their construction usually requires numerous subcloning steps. The difficulty in generating these plasmids often precludes the straightforward construction of multiple related versions of a particular construct, for example, to test several different epitope tags or mutant forms of an open reading frame (ORF) of interest. The widespread availability to the zebrafish community of a more efficient cloning system would greatly simplify standard day-to-day cloning efforts. Importantly, the accessibility of high-efficiency cloning techniques would facilitate the application of large-scale functional genomics approaches used in other model organisms (Walhout et al., 2000a; Deplancke et al., 2004, 2006) to the analysis of zebrafish genes.
The Gateway cloning system is a recombination-based cloning technique that allows efficient and rapid transfer of DNA fragments between plasmids (Hartley et al., 2000; Walhout et al., 2000b). It comprises two distinct recombination reactions (see Fig. 1) that rely on cis elements and excision/integration enzyme complexes from bacteriophage λ. The “BP” recombination reaction describes recombination between homologous attB and attP sites on two different DNA fragments (Fig. 1A). Typically, this reaction is used to recombine a polymerase chain reaction (PCR) product flanked by attB sites into a vector that contains attP sites (usually referred to as a “DONOR” vector; see Fig. 1A). Combinations of different att sites have been engineered to allow site-specific recombination that facilitates directional as well as multi-fragment cloning (see below). Bacteriophage λ-integrase (Int) and integration host factor (IHF; together referred to as BP clonase) catalyze the BP recombination yielding two plasmids or DNA fragments that now contain corresponding attL and attR sites (Fig. 1A). The attL-containing plasmid is referred to as an “ENTRY” plasmid, while the attR-flanked clone is a byproduct plasmid or fragment. To promote high efficiency recovery of ENTRY plasmids, the Gateway system relies on a counter-selection method. In this case, DONOR vectors contain a ccdB cassette that is toxic to standard bacterial strains used for cloning (e.g., DH10B, DH5α); an adjacent chloramphenicol resistance gene allows maintenance of the cassette in ccdB-tolerant cells. Therefore, un-recombined DONOR vector, or attR-containing by-products will not be propagated after a BP reaction resulting in high efficiency recovery of the desired ENTRY clone. Once an attL-flanked ENTRY clone is generated, it can be used in an “LR” reaction with corresponding Destination vectors that contain homologous attR sites (Fig. 1B). Usually, Destination vectors contain elements for eukaryotic or prokaryotic expression, as well as epitope tags to monitor ORF expression and to allow functional analysis. The LR recombination is catalyzed by Int, IHF, and excisionase (referred to as LR clonase) and results in a plasmid in which the ORF of interest is flanked by attB sites (usually referred to as an Expression clone) along with a by-product plasmid that contains a ccdB/chloramphenicol cassette flanked by attP sites (Fig. 1B). As with the BP reaction, both un-recombined Destination vector and the attP-containing by-product are selected against by the presence of the ccdB gene in an LR reaction. Additionally, Destination vectors contain ampicillin resistance, while DONOR or ENTRY plasmids contain kanamycin resistance cassettes. Thus, introduction of an LR reaction into standard cloning bacteria and selection on ampicillin results in high efficiency recovery of the desired Expression clone. The resulting Expression clone that contains the attB-flanked ORF can then be used for appropriate functional studies.
Fig. 1.

Gateway recombination reactions. A: Depiction of a BP reaction for a polymerase chain reaction amplified open reading frame. B: LR reaction between an ENTRY clone and a Destination vector. Note that att sites are not to scale.
A central benefit of the Gateway system is that it obviates the need for standard ligation-mediated cloning and greatly facilitates the generation of multiple related constructs. The efficiency of the Gateway system has also allowed the generation of large gene sets that serve as an important resource for a variety of model organisms. For example, C. elegans researchers have used Gateway cloning to generate clone collections of both ORFs and promoters that can be used together to rapidly generate constructs for transgenesis (Reboul et al., 2003; Dupuy et al., 2004). Similar projects have generated extensive Gateway compatible gene collections for human and Schizosaccharomyces pombe genes (Matsuyama et al., 2006; Lamesch et al., 2007). An important benefit of the Gateway system is its use to simultaneously clone multiple contiguous DNA fragments within a single vector (Dupuy et al., 2004; Hope et al., 2004; Yahata et al., 2005). This unique use of recombination-based cloning allows the rapid construction of a single plasmid containing two to four fragments (for example, a promoter, ORF, and epitope tag) in a single reaction. However, the ability to take advantage of Gateway cloning requires the establishment of the appropriate Destination vectors and ORF- or promoter-containing Entry clones that are suited for functional studies in a particular model organism. In this study, we describe the construction of Gateway compatible vectors to analyze gene function in the zebrafish. These plasmids are based on vectors commonly used for both transient and transgenic expression studies in zebrafish embryos. We demonstrate that these vectors allow rapid cloning of native, N-terminal and C-terminal epitope tagged ORFs. We have validated these vectors by generating injection constructs and testing their function in zebrafish embryos. Together this collection of Gateway compatible plasmids provides an important foundation for making this technology easily accessible to the general zebrafish community and will serve as a starting point to initiate large-scale functional genomics efforts.
Results and Discussion
Gateway-Compatible pCS-Based Vectors
As a first step to adapt the Gateway system for use in zebrafish, we constructed Gateway Destination vectors based on the standard pCS2 backbone commonly used to generate mRNA for injection into Xenopus laevis and zebrafish embryos (Turner and Weintraub, 1994). We constructed a pCS2 Destination plasmid (pCSDest) by cloning an attR1-ccdB-chloramphenicol resistance-attR2 Gateway cassette into the multicloning site of pCS2 (Fig. 2A). We also constructed parallel vectors in which a Gateway cloning cassette lies immediately downstream of a 6x myc epitope, enhanced green fluorescent protein (egfp; Zhang et al., 1996), or the monomeric red fluorescent protein mCherry (Shaner et al., 2004; Yasuda et al., 2006; Fig. 2A). These plasmids are referred to as pCSMTDest, pCSegfpDest, and pCSmcherryDest, respectively. In each of the pCSDest vectors with N-terminal epitope tags, a Kozak consensus sequence initiates expression of the epitope and each is in the standardized Gateway reading frame to allow creation of in-frame fusion proteins (Hartley et al., 2000).
Fig. 2.

pCS-based destination vectors. A: Schematic drawing of Gateway cassettes transferred into a pCS-backbone plasmid. See Table 1 for a complete list. B: Example of an LR reaction used to generate the pCSegfpnotch3icd construct. In this case, the entry plasmid pENTRnotch3icd was used in an LR reaction with pCSegfpDest resulting in pCSegfpnotch3icd. Note that att sites are not to scale.
To demonstrate the utility of these vectors and confirm their functionality, we generated a Gateway compatible Entry clone containing a constitutively active form of the Notch3 receptor intracellular domain (notch3icd) by BP recombination (pENTRnotch3icd; data not shown). The notch3icd coding sequence was then simultaneously transferred from the Entry vector into pCSMTDest, pCSegfpDest, and pCSmcherryDest in parallel LR reactions (an example of the LR reaction between pENTR-notch3icd and pCSegfpDest is shown in Fig. 2B). In each case, the successful transfer of the notch3icd fragment from the Entry plasmid into each Destination vector was greater than 90%, and usually all analyzed clones were correct (data not shown). The final product of the Gateway LR reaction leaves 25 base pair attB sites flanking the original ORF (Fig. 2B). To confirm that the plasmids generated by Gateway cloning were functional, we used them as templates to synthesize mRNA for injection into wild-type zebrafish embryos. In parallel, we used a pCS3 plasmid encoding a myctagged Notch3ICD that was generated with standard ligation mediated cloning techniques (pCS3myc-notch3icd; data not shown). After injection of mRNA, we observed embryos at 50% epiboly to view fluorescent protein expression. In the case of myc-tagged Notch3ICD, we fixed embryos for immunostaining to detect the myc epitope. To determine whether Notch3ICD fusion proteins were functional, we observed injected embryos at 24 to 26 hours postfertilization (hpf) to assay for loss of eye formation, a defect previously reported to be associated with ectopic activation of Notch signaling early in development (Scheer et al., 2001). In embryos injected with mRNA from the pCS3myc-notch3icd plasmid generated by standard cloning, we could detect Myc-Notch3ICD protein in a punctate pattern consistent with the nuclear localization of intracellular Notch (Fig. 3A). At 26 hpf, sibling embryos injected with pCS3 derived myc-notch3icd mRNA displayed loss of eyes (Fig. 3B; compare with Fig. 3N). We could similarly detect Myc-Notch3ICD encoded by mRNA from the Gateway-generated construct by immunostaining (Fig. 3C). Furthermore, Myc-Notch3ICD from the Gateway construct caused identical phenotypic abnormalities as the pCS3 construct (Fig. 3D). We observed comparable results with Egfp and mCherry-Notch3ICD constructs generated by Gateway cloning. In embryos injected with mRNA encoding mCherry-Notch3ICD, we were able to detect red fluorescence at 50% epiboly (Fig. 3E,F). Embryos injected with mcherry-notch3icd mRNA displayed defects identical to those described above for the Myc-Notch3ICD fusion proteins at 26 hpf (compare Fig. 3B,D,G). Similarly, we observed green fluorescence at 50% epiboly in embryos injected with mRNA encoding Egfp-Notch3ICD (Fig. 3H,I), and these embryos also displayed loss of eyes at 26 hpf (Fig. 3J). For both mCherry and Egfp-tagged Notch3ICD, we observed loss of eyes in nearly all injected embryos at the injected dose (100 pg). As a control, we generated a dual fluorescent protein by Gateway cloning egfp coding sequence into pCSmcherryDest. At 50% epiboly, embryos injected with mcherry-egfp mRNA displayed both red and green fluorescence (Fig. 3K–M) and all embryos displayed normal morphology at 26 hpf (Fig. 3N). These observations indicate that Gateway-modified pCS vectors can be used to rapidly generate constructs for synthesis of mRNA and subsequent injection into zebrafish embryos. In addition, the attB sites that remain after the LR reaction did not have any noticeable effect on the function of the expressed ORF, as previously reported in other model organisms (Walhout et al., 2000a; Walhout and Vidal, 2001).
Fig. 3.
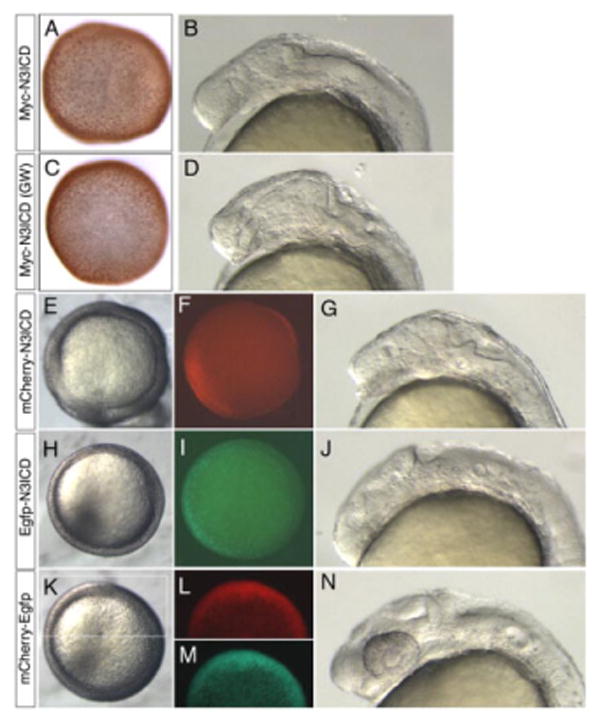
Validation of Gateway compatible N-term pCS vectors. A,B: Embryo injected with myc-notch3icd mRNA from plasmid generated through standard ligation-mediated cloning. C,D: Embryos injected with myc-notch3icd mRNA from plasmid generated through Gateway cloning (GW). A,C: Embryos fixed at 50% epiboly and immunostained to detect the myc epitope; dorsal view. B,D: Live embryos at 26 hpf, lateral view, anterior to the left. E–G: Embryos injected with mRNA encoding mCherry-Notch3ICD. H–J: Embryos injected with mRNA encoding Egfp-Notch3ICD. K–N: Embryos injected with mRNA encoding an mCherry-Egfp fusion. E,H,K: Dorsal views at 50% epiboly, images generated under standard transmitted light illumination of live embryos. F,L: Visualization of red fluorescence at 50% epiboly, dorsal views. I,M: Visualization of green fluorescence, dorsal views at 50% epiboly. G,J,N: Lateral views under transmitted light, dorsal is up, anterior to the left. All embryos injected with 100 pg of indicated mRNA. N3ICD, notch3 intracellular domain.
In some cases, N-terminal epitope tags may interfere with protein function. This is often the case with membrane-spanning proteins or growth factors that have secretion signals located at their N-termini. Therefore, we constructed a pCS-based Destination vector that allows simultaneous Gateway cloning of ORFs together with C-terminal epitopes. In this case, we relied on the ability to perform Gateway reactions involving more than one Entry clone (known as multisite Gateway cloning; Hope et al., 2004). To generate the appropriate Entry clones for multi-site Gateway recombination, we PCR-amplified the egfp coding sequence using forward and reverse primers containing attB1 and attB2 sequences, respectively. We designed the attB2 primer so that it did not contain a stop codon and would be in frame with the C-terminal cassette after a multisite LR reaction. The resulting PCR product was then used in a BP reaction with pDONR221 to generate pENTRegfp3 (Fig. 4A). In parallel, we generated a 3′ Entry clone encoding mCherry as a carboxy epitope tag by PCR with a forward primer containing an attB2 site and a reverse primer containing an attB3 site (Fig. 4B). We then used this PCR product in a BP reaction with the pDONRP2R-P3 plasmid to generate a 3′ Entry clone in which the mcherry ORF is flanked by attR2 and attL3 sites (p3Emcherry; Fig. 4B). The pENTRegfp3 and p3Emcherry ENTRY plasmids were then used in a multisite LR reaction with a pCS Destination plasmid that we modified to contain an attR1-attR3 flanked cassette (referred to as pCSDest2). Because the subsequent LR recombination is restricted to homologous att sites (attL sites can only recombine with corresponding attR sites; Hartley et al., 2000, Hope et al., 2004), this reaction creates a plasmid in which the C-terminal mCherry tag is placed immediately downstream of and in frame with the egfp coding sequence in the pCS-backbone vector (Fig. 4C). To demonstrate that this plasmid encoded a functional dual fluorescent protein, we used it to synthesize mRNA for injection into zebrafish embryos. As shown in Figure 4D–F, an embryo injected with egfp-mcherry mRNA displays both red and green fluorescence. We have also successfully used a 3′ clone containing egfp as an epitope tag as well (p3Eegfp; data not shown). These results indicate that it is possible to rapidly generate C-terminal fusion proteins in a pCS-based vector using a multisite Gateway reaction and the pCSDest2 construct.
Fig. 4.

Multisite Gateway approach to generate C-terminal fusion proteins in a pCS-based vector. A: Generation of the pENTR-egfp3 plasmid by BP recombination between an attB1/attB2 flanked PCR product and pDONR221. The reverse attB2 primer was engineered to eliminate the stop codon in the Egfp coding sequence. B: Generation of a C-terminal mCherry tag (p3Emcherry) by BP cloning. In this case, the forward primer contains an attB2 site and the reverse primer an attB3 site. Recombination of the attB2-attB3 PCR product with pDONRP2r-P3 results in an ENTRY clone in which the mcherry coding sequence is flanked by attR2 and attL3. C: Depiction of the two-way multisite Gateway LR reaction between pENTRegfp3, p3Emcherry and pCSDest2. Recombination can only occur between corresponding attL and attR sites resulting in the proper orientation of egfp and mcherry ORFs in pCSegfp-mcherry. Note that att sites are not to scale. D–F: A 24 hours postfertilization embryo injected with 100 pg of egfp-mcherry mRNA synthesized from the plasmid generated in C; lateral view, dorsal is up, anterior to the left; all pictures are from the same embryo. D: Transmitted light illumination. E: Illumination to visualize green fluorescence. F: Illumination to visualize red fluorescence.
Gateway Compatible Transgenic Constructs
In many cases, injection of mRNA leads to unwanted developmental defects due to the lack of spatial and temporal control over expression. Therefore, it is often desirable to drive expression of an ORF in a particular cell type and/or at a specific time point in development. In zebrafish, this can be accomplished by injecting into embryos a plasmid containing the appropriate promoter elements to drive tissue-specific expression of an ORF. However, construction of these plasmids is often complicated by the presence of multiple restriction sites within promoter elements or in other regions of the plasmid itself. To alleviate this problem and to establish a system that would allow rapid and easy generation of transgenic constructs, we transferred each of the Gateway cassettes from the pCS-based plasmids described above into a plasmid containing cis elements from the Tol2 transposon. Originally isolated from the Japanese Medaka fish (Oryzias latipes), cis sequences from Tol2 have been shown to facilitate high efficiency transgene integration into the zebrafish genome in the presence of expressed Tol2 Transposase (Kawakami and Shima, 1999; Kawakami et al., 2004). To generate Tol2-Destination vectors, we first introduced a unique restriction site (EcoRV) into the original pTol2000 vector (Kawakami et al., 2004) to facilitate standard cloning of promoter elements upstream of the Gateway cassettes. Each of the four cassettes described in Figure 2A was then transferred from the respective pCS-Dest vectors into the modified Tol2 plasmid (Fig. 5A). To test the function of the Tol2-based Destination cassettes, we cloned a fragment of the lmo2 promoter into the EcoRV site in each of the four pTolDest constructs (Fig. 5B). Previous studies have shown that this fragment of the lmo2 promoter can drive transgene expression in both blood and vascular cells in the zebrafish embryo (Zhu et al., 2005). We then performed LR reactions to introduce fluorescent protein coding sequences into pTol-lmo2-Dest vectors. An example depicting an LR reaction with pTol-lmo2-Dest and pENTRegfp2 is shown in Figure 5B. We subsequently co-injected Tol2 constructs with mRNA encoding the Tol2 Transposase into one-cell stage wild-type zebrafish embryos and observed them at approximately 26 hpf for fluorescent protein expression.
Fig. 5.
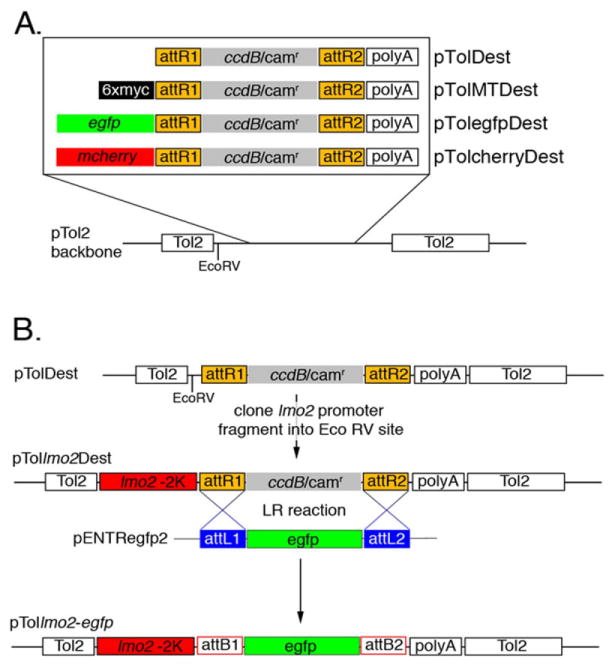
Generation of pTol2-Destintation constructs for tissue-specific gene expression. A: Gateway cassettes from the pCSDest plasmids described in Figure 2A were transferred into a modified pTol2 vector. The presence of an EcoRV site immediately upstream of the cassettes in each plasmid allows subsequent transfer of promoter elements. B: An lmo2 promoter fragment was cloned into the pTolDest plasmid to generate pTollmo2Dest. This plasmid was then used in an LR reaction with pENTRegfp2 to yield the injection construct pTol2lmo2:egfp. Note that att sites are not to scale.
Figure 6A,B shows an embryo injected with a Tol2 construct generated by Gateway cloning in which the lmo2 promoter drives expression of egfp (pTollmo2:egfp). Egfp expression is readily detectable in regions of the embryo known to be associated with blood and vascular development, including developing segmental arteries and the intermediate cell mass (Fig. 6B) consistent with previous studies (Zhu et al., 2005). We also tested N-terminal fusion cassettes as well. Figure 6C–N shows embryos injected with pTollmo2:mcherry-egfp that was generated through an LR reaction between an Egfp entry plasmid (pENTRegfp2) and the pTollmo2:mcherDest plasmid. In this case, the mcherry and egfp coding sequences are in frame to generate a single dual-fluorescent protein. As expected, both green and red fluorescence can be detected at 26 hpf in embryos co-injected with pTollmo2:mcher-egfp and Tol2 transposase mRNA (Fig. 6D,E). Furthermore, both green and red fluorescence appears in locations coincident with blood and vascular development, such as developing segmental arteries (Fig. 6I–K) and blood cells within the caudal vein (Fig. 6G,H,L–N). We obtained similar results with a Tol2-based construct in which the dsRedEx coding sequence was recombined in frame with an N-terminal Egfp tag (data not shown). We did not note significant ectopic expression in injected embryos consistent with previous results that the attB sites do not usually influence transgene expression (Deplancke et al., 2004).
Fig. 6.
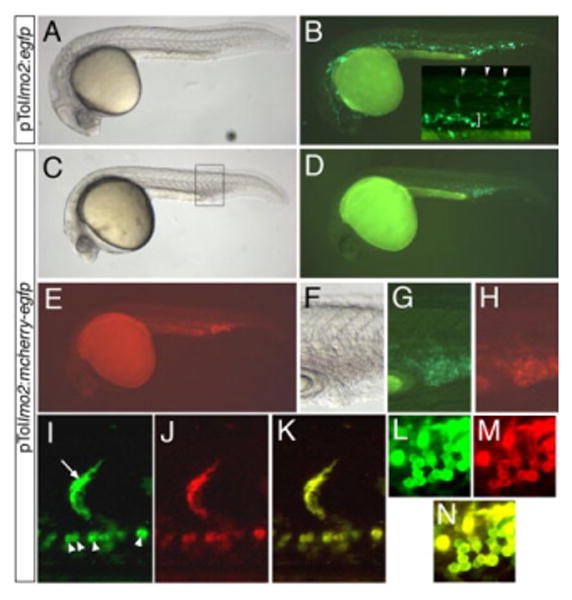
Validation of pTollmo2-Dest vectors. A–N: Injected embryos at 28 hours postfertilization, lateral views, dorsal is up, anterior to the left. A,B: Embryo injected with 25 pg of pTollmo2:egfp and 25 pg of transposase mRNA. A: Transmitted light image. B: Fluorescent image of embryo in A to visualize enhanced green fluorescent protein (Egfp) expression. Inset, higher magnification of trunk region, indicating Egfp expression in numerous segmental arteries (arrowheads) and intermediate cell mass (bracket). C–N: Embryos injected with 25 pg of pTollmo2:mcherry-egfp and 25 pg of transposase mRNA. C,F: Transmitted light image. D,G: Epifluorescence image to visualize Egfp expression. E,H: Epifluorescence image to visualize mCherry expression. F–H: Higher magnification view of caudal vein expression from embryo in C (box). I–N: Confocal micrographs; embryo is different than the one pictured in C. I: Green fluorescence of mCherry-Egfp in a segmental artery (arrow) and red blood cells (arrowheads). J: Red fluorescence of mCherry-Egfp. K: Overlay of green and red fluorescence from images in I and J. L: Green fluorescence of mCherry-Egfp in blood cells. M: Red fluorescence of mCherry-Egfp in blood cells. N: Overlay of green and red fluorescence from images in L and M.
We next tested the use of multisite Gateway cloning to generate Tol2 constructs for transgenic expression of a C-terminally tagged ORF. In this case, we relied on the ability to perform a three-way multisite Gateway reaction (three entry vectors with a single Destination vector) to incorporate a promoter fragment upstream of the ORF and its C-terminal tag. We used a bactin2 promoter entry plasmid (p5Ebactin2; Kwan et al., 2007) and a pTol-based Destination construct that contains an attR4-attR3 cassette (referred to as pDestTol2pA). To generate a Tol2 injection construct in which the bactin2 promoter drives expression of a C-terminally tagged ORF, we performed a three-way multisite LR reaction using p5E-bactin2, pENTRegfp3, and p3Emcherry together with the pDestTol2pA plasmid (Fig. 7A). This created a plasmid in which the bactin2 promoter was placed upstream of an egfp-mcherry fusion coding sequence. We then co-injected this plasmid with transposase mRNA into 1-cell stage zebrafish embryos. Figure 7B–G shows that embryos injected with this construct displayed both green and red fluorescence in identical mosaic patterns. These results indicate that it is possible to generate Tol2-based constructs in which the expression of C-terminally tagged fusion proteins are driven by a tissue-specific promoter in a single cloning step.
Fig. 7.

Multisite Gateway cloning to generate a tissue-specific Tol2 construct for expression of C-terminal fusion proteins. A: Three-way multisite reaction into pDestTol2pA. The egfp and mcherry Entry clones are the same as those used in Figure 4. Note that att sites are not to scale. B–G: Embryos injected with 25 pg transposase mRNA and 25 pg pTolbactin2:egfp-mcherry; lateral view, dorsal is up, anterior to the left. B: Transmitted light illumination. C: Illumination to visualize green fluorescence. D: Illumination to visualize red fluorescence. E–G: Confocal micrographs; embryo is different than the one pictured in B. E: Mosaic green fluorescence of Egfp-mCherry observed in epidermal cells (arrows) and muscle cells (arrowheads). F: Red fluorescence of Egfp-mCherry. G: Overlay of images in E,F.
Construction of an Endothelial Specific Destination Vector
In previous studies, we took advantage of the fli1a promoter to drive expression of transgenes within endothelial cells (Lawson et al., 2001; Lawson and Weinstein, 2002; Roman et al., 2002). However, a drawback to this approach is that the size of the fli1a fragments we have previously used often make plasmid construction challenging. Therefore, we used Gateway cloning to rapidly identify an endothelial enhancer element in fli1a. We subsequently used this enhancer fragment to generate Gateway compatible vectors to drive endothelial gene expression.
To identify a putative endothelial enhancer in the fli1a first intron, we PCR-amplified overlapping 1-kb fragments from the fli1a first intron using forward primers and reverse primers containing attB4 and attB1 sites, respectively (Fig. 8A). The resulting PCR products were then used in a BP recombination reaction with the Gateway donor vector pDONRP4-P1r to generate entry clones. The putative enhancer entry clones were then used in a two-way multisite Gateway LR reaction along with pENTRbas-egfp-pa, which contains a basal promoter/TATA box element in front of egfp, and a Tol2 vector containing an attR4/attR2 Destination cassette (Fig. 8B). We co-injected the resulting pTol-enhancer:reporter constructs individually along with mRNA encoding Tol2 Transposase into one-cell stage zebrafish embryos and assayed for Egfp expression by immunostaining at 24 to 28 hpf. As a control, we injected embryos with a pTol2 construct containing 900 bp upstream and 6 kb downstream of the fli1a exon 1 flanking egfp that is sufficient to drive endothelial-specific expression in both transient assays and stable transgenic lines (Lawson et al., 2001; Roman et al., 2002). Consistent with our previous results, the fli1a −0.9/+5.9 fragment drove robust endothelial cell-specific expression of Egfp (Fig. 8C), whereas three fragments within the fli1a intron were capable of driving expression in zebrafish embryos (Fig. 8D,E, and data not shown). We identified two fragments that drove expression in nonendothelial cell types. For example, pTol fli1a +1.2/+2.2:bas-egfp drove expression in cells throughout the brain and notochord (Fig. 8D), but not in blood vessels. By contrast, we found that pTol fli1+2.2/+3.2:bas-egfp drove expression in blood vessels within the head and trunk of zebrafish embryos at 26 hpf (Fig. 8E).
Fig. 8.
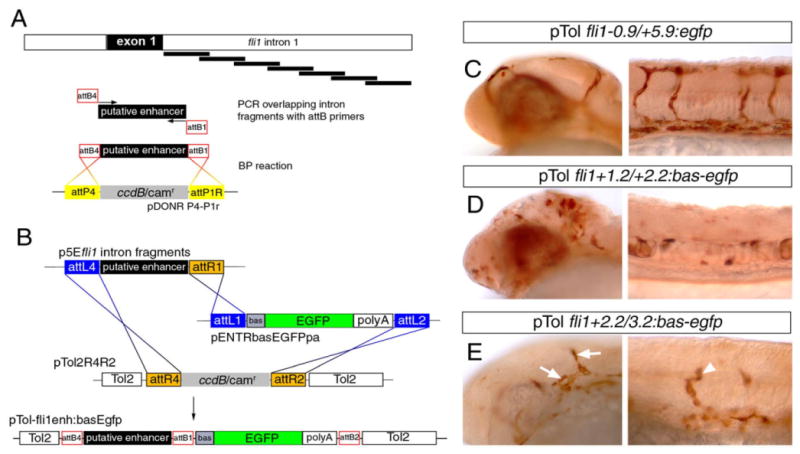
Identification of an endothelial enhancer in the fli1a first intron. A: Schematic description of polymerase chain reaction and BP cloning approach to generate overlapping 1-kb fragments for screening enhancer activity. B: Description of LR multisite cloning to generate injection constructs. C-E: Transmitted light images of embryos immunostained with an antibody against Egfp; embryos were co-injected with 25 pg of indicated Tol2 construct and 25 pg of Tol2 transposase mRNA. C: Expression of Egfp in blood vessels of the head and trunk in an embryo injected with pTol-fli1a-0.9/+5.9:egfp. D: Ectopic Egfp expression in the head and trunk in an embryo injected with pTol-fli1a+1.2/+2.2:egfp. E: Egfp expression in head vessels (arrows) and trunk vessels (arrowhead) in an embryo injected with pTolflia+2.2/+3.2:egfp.
To generate an endothelial cell-specific Tol2-Destination vector, we first constructed a hybrid fli1a enhancer/promoter fusion using overlapping PCR. This fragment contains the sequence between +2.2 and +3.2 kb of the fli1a first intron placed immediately upstream of a fragment containing the proximal 900 bp upstream of fli1a exon 1 (Fig. 9A). We refer to this chimeric fragment as “fli1ep” (fli1a enhancer/promoter). We transferred the fli1ep fragment upstream of the attR1/attR2 Gateway cassette in pTol2Dest to give the plasmid pTol2fli1epDest. To confirm the utility of this construct, we generated an injection construct by performing an LR reaction with pTol2fli1epDest and pENTRegfp2 to give pTol2fli1ep:egfp (Fig. 9A). After co-injection of pTol2fli1ep:egfp and Tol2 transposase mRNA into one-cell stage embryos, we observed robust fluorescence in blood vessels throughout zebrafish embryos at 26 hpf (Fig. 9B). These results demonstrate that it is possible to rapidly identify relevant cis regulatory elements in genes of interest using the Gateway system. In this case, we have identified an endothelial enhancer for the purpose of generating a useful reagent to drive gene expression in blood vessels. Accordingly, these Gateway compatible fli1ep constructs have been used to rapidly generate constructs for endothelial autonomous rescue experiments in the zebrafish redhead mutant (Buchner et al., 2007). We have also constructed Gateway compatible Tol2 vectors containing the fli1ep fragment for generating N-terminal fusions with 6xmyc, Egfp, and mCherry (data not shown and Table 1).
Fig. 9.

Construction and use of an endothelial Tol2 Destination vector. A: Description of the construction of a Tol2-based Destination vector for generating injection constructs to drive expression in endothelial cells. B: Embryo injected with pTolfli1ep:egfp showing robust Egfp expression throughout the trunk blood vessels at 26 hours postfertilization.
Table 1. Plasmids Used in this Study.
| Plasmid name | att sites | Eukaryotic promoter | Prokaryotic promoter |
|---|---|---|---|
| Destination vectors | |||
| pCSDest | R1-R2 | CMV | SP6 |
| pCSDest2 | R1-R3 | CMV | SP6 |
| pCSMTDest | R1-R2 | CMV | SP6 |
| pCSEgfpDest | R1-R2 | CMV | SP6 |
| pCSCherryDest | R1-R2 | CMV | SP6 |
| pTolDest | R1-R2 | none | none |
| pTolMTDest | R1-R2 | none | none |
| pTolEgfpDest | R1-R2 | none | none |
| pTolCherryDest | R1-R2 | none | none |
| pTollmo2Dest | R1-R2 | lmo2 | none |
| pTollmo2MTDest | R1-R2 | lmo2 | none |
| pTollmo2EgfpDest | R1-R2 | lmo2 | none |
| pTollmo2CherryDest | R1-R2 | lmo2 | none |
| pTolfli1epDest | R1-R2 | fli1a | none |
| pTolfli1epMTDest | R1-R2 | fli1a | none |
| pTolfli1epEgfpDest | R1-R2 | fli1a | none |
| pTolfli1epCherryDest | R1-R2 | fli1a | none |
| pTolR4-R2 | R4-R2 | none | none |
| pDestTol2pAa | R4-R3 | none | none |
| Entry vectors | |||
| pENTRnotch3icd | L1-L2 | NA | NA |
| pENTRegfp2 | L1-L2 | NA | NA |
| pENTRegfp3 | L1-L2 | NA | NA |
| p3Eegfp2 | R2-L3 | NA | NA |
| p3Emcherry | R2-L3 | NA | NA |
| p5Ebactin2a | L4-R1 | NA | NA |
| pENTRbasegfp | L1-L2 | e1b TATA | NA |
| Injection constructs | |||
| pCSmcherry-egfp | B1-B2 | CMV | SP6 |
| pCSmcherry-notch3icd | B1-B2 | CMV | SP6 |
| pCSegfp-notch3icd | B1-B2 | CMV | SP6 |
| pCSmyc-notch3icd | B1-B2 | CMV | SP6 |
| pTollmo2:egfp | B1-B2 | lmo2 | none |
| pTollmo2:egfp-dsredex | B1-B2 | lmo2 | none |
| pTollmo2:mcherry-egfp | B1-B2 | lmo2 | none |
| pCSegfp-mcherry | B1-B2-B3 | CMV | SP6 |
| pTolbactin:egfp-mcherry | B4-B1-B2-B3 | bactin | none |
Described in Kwan et al. (2007). Additional plasmid information, including downloadable annotated sequence files, can be found at http://lawsonlab.umassmed.edu/gateway.html.
In this study, we describe the construction and validation of several Gateway-based vectors (Table 1) that allow rapid cloning of ORFs of interest and subsequent functional analysis in zebrafish embryos. We also demonstrate the utility of Gateway cloning for the rapid identification of enhancer elements that are useful for directing tissue-specific gene expression. We believe the use of these plasmids will greatly facilitate and streamline standard day-to-day molecular cloning projects in most zebrafish labs. In other organisms, including mouse, human, and Caenorhabditis elegans, the Gateway cloning system has been adopted to generate large-scale collections of ORFs in a standard frame, and these are generally available as Entry clones flanked by attL1 and attL2 sites (Reboul et al., 2003; Dupuy et al., 2004). Additional efforts in some model organisms (e.g., C. elegans), have similarly established collections of promoter elements in which cis regulatory elements are available in Entry clones flanked by attL4 and attR1 (Hope et al., 2004). The availability of similar resources for zebrafish researchers would allow rapid functional and genetic analyses as injection constructions could be quickly and easily generated after receiving the appropriate Entry clones from a central resource. The use of appropriate epitope and/or promoter elements can then be determined in a matter of days with the ability to easily recombine ORFs into multiple Destination constructs simultaneously. The ultimate result is that much more time and effort can be directed toward functional analysis of genes rather than toward plasmid construction. With the availability of these Destination vectors, we strongly encourage that zebrafish researchers adopt the same Gateway standards as other model organisms. In addition, we hope that the widespread use of this technology by zebrafish labs would promote the generation of Gateway-compatible ORF and promoter collections, which would serve as a valuable resource to the zebrafish community.
Experimental Procedures
Fish lines
Adults and embryos were handled according to standard procedures (Westerfield, 1993). Wild-type (EK, Ekkwill farm derived) or golb1 embryos were used for injections.
Plasmid Construction
pCS-based plasmids
pCSDest: pCS2+ was digested with StuI and ligated with the Reading Frame B Gateway cassette (Invitrogen). pCSDest2: pCSDest was digested with PstI and XhoI to remove the attR2 site and was replaced with an attR3 site from pDESTR4-R3 (Invitrogen). pCSMTDest: pCS3+MT was digested with StuI and ligated with the Reading Frame A Gateway cassette fragment. pCSEGFPDest: pCS2efgp was digested with StuI and ligated with the Reading Frame A Gateway cassette fragment. pCSmCherryDest: a monomeric red fluorescent protein (mCherry) coding sequence was PCR amplified from pMEH2AmCherry (kindly provided by Chi-Bin Chien) using the following PCR primers: 5′-gatcgctagcgccaccatggtgagcaagggc, and 5′-gatcagatctcttgtacagctcgtccat. The resulting PCR product was digested sequentially with NheI and BglII and cloned into pCSEGFPDest in place of the EGFP coding sequence.
Tol2-based plasmids
The original Tol2 plasmid pT2K (Kawakami et al., 2004) was kindly provided by Koichi Kawakami. We modified pT2K by removing the Oryzias latipes tyrosinase 3′ untranslated region by digesting with Acc65I, followed by Klenow treatment and digestion with PshAI. The fragment containing Tol2 cis elements and the vector backbone was isolated by gel purification and re-ligated to generate pTol2005. To allow cloning of the epitope tag-Gateway-polyA cassettes from the pCS-Destination plasmids and subsequent cloning of promoter fragments, we further modified pTol2005 by adding several restriction sites. Overlapping oligonucleotides (5′-ggccGATATCGCTAGCATCGATgcatG and 5′-GGCCCatgcATCGATGCTAGCGATATC) were hybridized and cloned into pTol2005 cut with EagI and PspOMI to give pTol2005b. To generate each of the N-terminal tag and native Tol2-Destination vectors, cassettes were removed from the respective pCS-based plasmids listed above by digesting with ClaI and PspOMI. This digest releases a fragment that includes the 5′ epitope tag, Gateway cassette, and the poly adenylation signal. In each case, these fragments were cloned into pTol2005b digested with ClaI and PspOMI.
To generate the pTol-lmo2 Dest vectors, an lmo2 promoter fragment encompassing approximately 2,800 nucleotides upstream of the ATG was cloned into the SfiI and NheI sites upstream of the N-terminal tag and Gateway cassettes in each of the respective pTol-Dest vectors. The pDestTol2pA vector containing an attR4-attR3 cassette was constructed by Clemens Grabher and kindly provided by Chi-Bin Chien. The pTolR4-R2 Destination vector was constructed by excising an attR4-attR2 cassette from pDEST6 (kindly provided by Marian Walhout). This cassette was transferred to pTol2000 (kindly provided by Koichi Kawakami) that had been digested with XhoI and treated with Klenow. A description of the construction of the fli1ep containing vectors can be found elsewhere (Buchner et al., 2007). In all cases, plasmids containing Destination cassettes were grown in ccdB-tolerant bacterial strains (Invitrogen) and maintained in the presence of both ampicillin and chloramphenicol. Genbank file formatted text files for the Destination vectors constructed in this manuscript are available at http://lawsonlab.umassmed.edu/GWDestplasmids.htm.
Generation of Entry Clones
pDONR221 and pDONRP2r-P3 were obtained as part of the Multisite gateway Cloning kit (Invitrogen); pDONRP4-P1r was kindly provided by Marian Walhout. The pDONR plasmids were maintained in ccdB-tolerant bacteria and grown in the presence of kanamycin and chloramphenicol. To generate a notch3 intracellular domain (ICD) entry clone, we PCR amplified notch3 ICD from the pBSN3 plasmid (Lawson et al., 2001) using a forward primer containing an attB1 site (GGGGACAAGTTTGTACAAAAAAGCAGGCTtgatggttggcatgttgattg) and a reverse primer containing an attB2 site (GGGGACCACTTTGTACAAGAAAGCTGGGTctgcaggatgggctgctgg). The resulting fragment was included with pDONR221 in a BP recombination reaction to generate pENTR-notch3icd. To generate fluorescent protein entry clones, primers were designed that recognized common sequences in pCSGreen (Lawson et al., 2001) and pDsRedExpress-C1 (Clontech). The forward attB1 containing primer is GGGGACAAGTTTGTACAAAAAAGCAGGCTcgctaccggtcgccac, and the reverse attB2 primer is GGGGACCACTTTGTACAAGAAAGCTGGGTatcagttatctagatccggt. The forward primer lies immediately upstream of the Kozak consensus sequence for each of the fluorescent proteins listed above, while the reverse primer overlaps an in-frame stop codon. Each fluorescent protein coding sequence was amplified from the indicated template plasmid followed by BP recombination with pDONR221. The resulting plasmids are referred to pENTRegfp2 and pENTRDsRedEx2. To generate an Egfp entry clone (pENTRegfp3) to test C-terminal fusions, we amplified Egfp with the following primers: forward, attB1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTGGGCCACCATGGTGAGCAAGGGCGAG) and reverse attB2 (GGGGACCACTTTGTACAAGAAAGCTGGGTgTCTAGATCCGGTGGATCC). The reverse primer was designed in this case to not have a stop codon and to maintain the recommended reading frame to generate a C-terminal fusion by multisite Gateway cloning. To generate C-terminal epitope entry plasmids, we amplified Egfp and mCherry coding sequences using forward primers containing an attB2 site and a reverse primer containing an attB3 site. Egfp attB2 primers: GGGACAGCTTTCTTGTACAAAGTGGACATGGTGAGCAAGGGCGAG; Egfp attB3: GGGGACAACTTTGTATAATAAAGTTGGTCAAGATCCGGTGGATCC; mCherry attB2: GGGGACAGCTTTCTTGTACAAAGTGGACATGGTGAGCAAGGGCGAGG, mCherry attB3: GGGGACAACTTTGTATAATAAAGTTGGTCACGTTTCTCGTTCAGCTTT. In both cases, we included an in-frame stop codon in the reverse primer. The attB2-attB3 flanked PCR products were then mixed with pDONRP2R-P3 and subjected to a BP reaction. The pENTRbas-egfp-pa plasmid was generated by PCR amplification of a basal promoter-Egfp-polyA signal cassette from the plasmid pUAS:egfp (Lawson, unpublished data) using the following PCR primers: GGGGACAAGTTTGTACAAAAAAGCAGGCTagactctagagggtatataatgga and GGGGACCACTTTGTACAAGAAAGCTGGGTacgcgttaagatacattgatga. The basal promoter consists of a minimal adenovirus E1b promoter and the transcriptional start site of the carp bactin gene used previously in zebrafish UAS constructs (Scheer and Campos-Ortega, 1999). Al entry clones were confirmed by sequencing. An entry clone containing a fragment of the zebrafish bactin2 promoter flanked by attL4 and attR1 (pMEbactin2) was kindly provided by Chi-Bin Chien. Genbank file formatted text files for the Entry vectors constructed in this manuscript are available at http://lawsonlab.umassmed.edu/GWEntryplasmids.htm.
Generation of Injection Constructs
To generate plasmids for mRNA synthesis or injection into zebrafish embryos, we performed Gateway LR reactions according to manufacturer's recommendation (Invitrogen). For standard LR reactions (i.e., recombination between a single entry clone and Destination vector), we used LR II clonase and performed the reaction for 1 hr at room temperature. For multisite cloning (i.e., two or three entry plasmids with a single Destination vector), we used LR Plus clonase and performed the reaction overnight at room temperature. After LR reactions, the sample was diluted fivefold in water and 1 μl was electroporated into TOP10 bacteria. Clones were selected on ampicillin plates and screened by PCR and/or miniprep.
Injections
To generate mRNA for injection, pCS-based plasmids were digested with NotI, extracted once with phenol/chloroform and ethanol precipitated. The pCS2-TP plasmid encoding the Tol2 Transposase was kindly provided by Koichi Kawakami. Linearized plasmid was then used in a standard in vitro transcription reaction using SP6 polymerase (Message Machine, Ambion). We performed mRNA injections as described previously (Lawson et al., 2001). For injections using Tol2 constructs, we injected one-cell stage embryos with 25 pg of Tol2 transposase mRNA and 25 pg of indicated Tol2-containing plasmid.
Imaging
Fluorescent protein expression was monitored using a Leica MZFLIII microscope equipped with epifluorescence. The myc-epitope tag or GFP was detected using the ABC Vectastain kit as described previously (Lawson et al., 2001). Digital images were captured using an AxioCam MRc camera and Axiovision software. Confocal images were captured using a Leica SP2 confocal microscope.
Acknowledgments
We thank members of the Lawson Laboratory for helpful discussion and reading of the manuscript and Marian Walhout and Bart Deplancke for their initial discussions and ongoing support concerning Gateway cloning. We thank Chi-Bin Chien, Kristen Kwan, Tom Look, Clemens Grabher, and Koichi Kawakami for providing plasmids and sharing unpublished data in the course of this study. We also thank Tom Smith for valuable technical assistance. N.D.L. was funded by the NHLBI and J.A.V. received Ruth Kirchstein Minority Predoctoral Fellowship award.
Grant sponsor: National Heart, Blood, and Lung Institute; Grant number: R01HL079266; Grant number: F31 HL081927.
References
- Amsterdam A, Becker TS. Transgenes as screening tools to probe and manipulate the zebrafish genome. Dev Dyn. 2005;234:255–268. doi: 10.1002/dvdy.20541. [DOI] [PubMed] [Google Scholar]
- Beis D, Stainier DY. In vivo cell biology: following the zebrafish trend. Trends Cell Biol. 2006;16:105–112. doi: 10.1016/j.tcb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Buchner DA, Su F, Yamaoka JS, Kamei M, Shavit JA, McGee B, Hanosh AW, Kim S, Jagadeeswaran P, Amigo J, Lawson ND, Goldman D, Weinstein BM, Ginsburg D, Lyons SE. From the Cover: pak2a mutations cause cerebral hemorrhage in redhead zebrafish. Proc Natl Acad Sci U S A. 2007;104:13996–14001. doi: 10.1073/pnas.0700947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Dupuy D, Vidal M, Walhout AJ. A gateway-compatible yeast one-hybrid system. Genome Res. 2004;14:2093–2101. doi: 10.1101/gr.2445504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Mukhopadhyay A, Ao W, Elewa AM, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Reece-Hoyes JS, Hope IA, Tissenbaum HA, Mango SE, Walhout AJ. A gene-centered C. elegans protein-DNA interaction network. Cell. 2006;125:1193–1205. doi: 10.1016/j.cell.2006.04.038. [DOI] [PubMed] [Google Scholar]
- Dupuy D, Li QR, Deplancke B, Boxem M, Hao T, Lamesch P, Sequerra R, Bosak S, Doucette-Stamm L, Hope IA, Hill DE, Walhout AJ, Vidal M. A first version of the Caenorhabditis elegans Promoterome. Genome Res. 2004;14:2169–2175. doi: 10.1101/gr.2497604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope IA, Stevens J, Garner A, Hayes J, Cheo DL, Brasch MA, Vidal M. Feasibility of genome-scale construction of promoter::reporter gene fusions for expression in Caenorhabditis elegans using a multisite gateway recombination system. Genome Res. 2004;14:2070–2075. doi: 10.1101/gr.2463804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Shima A. Identification of the Tol2 transposase of the medaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2 element in zebrafish Danio rerio. Gene. 1999;240:239–244. doi: 10.1016/s0378-1119(99)00444-8. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N, Mishina M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell. 2004;7:133–144. doi: 10.1016/j.devcel.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G, Hu Z, Venkatesan K, Bethel G, Martin P, Rogers J, Lawlor S, McLaren S, Dricot A, Borick H, Cusick ME, Vandenhaute J, Dunham I, Hill DE, Vidal M. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, Horinouchi S, Yoshida M. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–847. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, Armstrong CM, Li S, Jacotot L, Bertin N, Janky R, Moore T, Hudson JR, Jr, Hartley JL, Brasch MA, Vandenhaute J, Boulton S, Endress GA, Jenna S, Chevet E, Papasotiropoulos V, Tolias PP, Ptacek J, Snyder M, Huang R, Chance MR, Lee H, Doucette-Stamm L, Hill DE, Vidal M. C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet. 2003;34:35–41. doi: 10.1038/ng1140. [DOI] [PubMed] [Google Scholar]
- Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, Garrity DM, Moon RT, Fishman MC, Lechleider RJ, Weinstein BM. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- Scheer N, Campos-Ortega JA. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech Dev. 1999;80:153–158. doi: 10.1016/s0925-4773(98)00209-3. [DOI] [PubMed] [Google Scholar]
- Scheer N, Groth A, Hans S, Campos-Ortega JA. An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development. 2001;128:1099–1107. doi: 10.1242/dev.128.7.1099. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Protein interaction mapping in C. elegans using proteins involvedinvulvaldevelopment. Science. 2000a;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames orORFeomes. MethodsEnzymol. 2000b;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Vidal M. High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods. 2001;24:297–306. doi: 10.1006/meth.2001.1190. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. Eugene, OR: University of Oregon Press; 1993. [Google Scholar]
- Yahata K, Kishine H, Sone T, Sasaki Y, Hotta J, Chesnut JD, Okabe M, Imamoto F. Multi-gene gateway clone design for expression of multiple heterologous genes in living cells: conditional gene expression at near physiological levels. J Biotechnol. 2005;118:123–134. doi: 10.1016/j.jbiotec.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, Svoboda K. Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci. 2006;9:283–291. doi: 10.1038/nn1635. [DOI] [PubMed] [Google Scholar]
- Zhang G, Gurtu V, Kain SR. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun. 1996;227:707–711. doi: 10.1006/bbrc.1996.1573. [DOI] [PubMed] [Google Scholar]
- Zhu H, Traver D, Davidson AJ, Dibiase A, Thisse C, Thisse B, Nimer S, Zon LI. Regulation of the lmo2 promoter during hematopoietic and vascular development in zebrafish. Dev Biol. 2005;281:256–269. doi: 10.1016/j.ydbio.2005.01.034. [DOI] [PubMed] [Google Scholar]