

Abstract
Glycosphingolipids (GSLs) are essential constituents of cell membranes and lipid rafts and can modulate signal transduction events. The contribution of GSLs in osteoclast (OC) activation and osteolytic bone diseases in malignancies such as the plasma cell dyscrasia multiple myeloma (MM) is not known. Here, we tested the hypothesis that pathological activation of OCs in MM requires de novo GSL synthesis and is further enhanced by myeloma cell–derived GSLs. Glucosylceramide synthase (GCS) inhibitors, including the clinically approved agent N-butyl-deoxynojirimycin (NB-DNJ), prevented OC development and activation by disrupting RANKL-induced localization of TRAF6 and c-SRC into lipid rafts and preventing nuclear accumulation of transcriptional activator NFATc1. GM3 was the prevailing GSL produced by patient-derived myeloma cells and MM cell lines, and exogenous addition of GM3 synergistically enhanced the ability of the pro-osteoclastogenic factors RANKL and insulin-like growth factor 1 (IGF-1) to induce osteoclastogenesis in precursors. In WT mice, administration of GM3 increased OC numbers and activity, an effect that was reversed by treatment with NB-DNJ. In a murine MM model, treatment with NB-DNJ markedly improved osteolytic bone disease symptoms. Together, these data demonstrate that both tumor-derived and de novo synthesized GSLs influence osteoclastogenesis and suggest that NB-DNJ may reduce pathological OC activation and bone destruction associated with MM.
Keywords: Bone Biology, Oncology
Introduction
Sphingolipids and their glycosylated derivatives, glycosphingolipids (GSLs), are essential structural components of mammalian cell membranes and are characterized by heterogenic composition among tissues and even in the same cell during different states of development (1). Quantitative and qualitative changes of the cellular GSL profile have long been recognized as a trait of malignant transformation (2). Surface-bound as well as shed GSLs have been shown to modulate cellular functions that promote tumor survival and growth, metastasis, and angiogenesis (2, 3).
In addition to conferring structural integrity to the cell membrane, GSLs agglomerated into cholesterol dense microdomains or lipid rafts participate in cellular recognition, adhesion processes, and modulation of signal transduction events. Signaling through lipid raft–associated GSLs has been shown to be an important process in many cell types, including myeloid cells, lymphocytes, and osteoclasts (OCs) (4). For example, GM3 has been shown to regulate lipid raft–mediated cell signaling and cellular adhesion in human lymphocytes (5–7). Increased synthesis of GM3 is also a defining characteristic that marks the differentiation and maturation of myeloid lineage precursors into monocytes and macrophages, a process that can be promoted by addition of GM3 (8).
OCs, the only cell type able to resorb bone, are also derived from myeloid precursors and form in response to stimulation with RANKL and M-CSF (9, 10). These cells play a crucial role in healthy bone homeostasis and are vital for the formation and maintenance of the BM hematopoietic stem cell (HSC) niche (11), as well as the regulation of hematopoiesis (12–14). The importance of OC activity in the HSC niche is best understood in various disease settings. BM modifications due to enhanced activation of OCs are involved in metastatic cancer (15, 16) and in multiple myeloma–related (MM-related) lytic bone lesions (17).
Previous in vitro studies suggest that de novo synthesized GSLs are required for OC development in vitro (18). Specifically, d-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (d-PDMP), a nonspecific inhibitor of glucose ceramide synthase (GCS) — an enzyme critical for GSL biosynthesis — prevents OC development (18). The role of GSL in OC biology is further highlighted by the development of OC activation–dependent crippling bone disease in lipid storage disorders (e.g., Gaucher disease) (19, 20), which are caused by impaired catabolism of GSLs. Notably, N-butyl-deoxynojirimycin (NB-DNJ; also known as Miglustat), an iminosugar inhibitor of GCS, is taken up by glucocerebroside-laden macrophages, thus ameliorating the clinical phenotype of Gaucher disease, as well as improving the osteoporotic bone phenotype (21, 22).
In MM, an incurable malignancy of plasma cells, osteolytic bone disease and skeletal complications occur in more than 80% of patients (23). Myelomatous osteolysis is localized to areas adjacent to tumor growth and is characterized by increased activity of OCs and suppression of osteoblastogenesis (23). Additionally, OCs can promote myeloma cell survival and protect the malignant cells from spontaneous and drug-induced apoptosis (24). Increased secretion of RANKL from stromal cells is the prime mechanism of OC activation in MM (25–27). In addition, increased secretion of the chemokine macrophage inflammatory protein–1α (MIP-1α) (28–30) and cytokines such as IL-3 (1), VEGF (2, 3), and IGF-1 (4) in the myeloma microenvironment also contribute to OC activation.
Whether myeloma-associated GSLs can modify the microenvironment and in particular contribute to OC activation is not known. Through a series of in vitro and in vivo assays, we delineate the cellular and molecular mechanisms of OC development and function in the presence of exogenous GM3, which we identify as the prevailing GSL in myeloma cells, and upon endogenous GSL inhibition with the clinically approved iminosugar NB-DNJ. Our results suggest that NB-DNJ may be an effective agent for the treatment of malignant bone loss associated with increased OC activity in MM and may be applicable to all cancer-induced bone diseases.
Results
Iminosugar agents inhibit RANKL-dependent OC development.
Previous work showed that the nonspecific GCS inhibitor d-PDMP inhibited OC development, suggesting that endogenous GSL synthesis by OCs themselves is required for their development (18). To test the role of de novo GSL synthesis in OC development in vitro, M-CSF– and RANKL-dependent murine BM OC assays were performed in the presence of NB-DNJ, N-butyl-deoxygalactonojirimycin (NB-DGJ), or N-octadecyl-deoxynojirimycin (N-OD-DNJ), three different iminosugar inhibitors of GCS, the enzyme that catalyzes the first and rate-limiting glycosylation step in GSL biosynthesis (31). Using a dose range that had previously been shown to inhibit glycolipid biosynthesis (32), we found that after 5 days of culture, d-PDMP, NB-DNJ, NB-DGJ, and N-OD-DNJ inhibited OC formation in a dose-dependent manner (Figure 1, A and B), without affecting cell viability (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI59987DS1). d-PDMP, NB-DNJ, and N-OD-DNJ almost completely inhibited OC development at the higher concentration of 500 μM, but NB-DGJ was less effective. A similar inhibitory effect was observed on human elutriated monocytes induced to form OC in vitro with increasing doses of NB-DNJ, thus confirming a direct effect of iminosugar agents on OC precursors and the involvement of de novo GSL production in OC formation (Supplemental Figure 1B).
Figure 1. GSL inhibitors inhibit osteoclastogenesis and bone resorption in vitro.

(A) Mouse BM cells were cultured in the presence of 25 ng/ml M-CSF with 50 ng/ml RANKL (Control) and with or without d-PDMP (5, 20, or 50 µM), NB-DNJ, NB-DGJ, or N-OD-DNJ (5, 50, or 500 µM) in 48-well plates for 5 days, and TRAP-positive OCs were counted and shown as cumulative results from 3 different experiments. (B) Cultures with iminosugar inhibitors at 500 µM and d-PDMP at 20 µM were photographed (original magnification, ×40). (C and D) Mouse BM cells were cultured with 25 ng/ml M-CSF and 50 ng/ml RANKL in 48-well plates. On day 3 d-PDMP (20 µM) or NB-DNJ, NB-DGJ, or N-OD-DNJ (each at 500 µM) was added. Cells were cultured for another 48 hours before being fixed and stained for TRAP and counted (C) and phalloidin stained to identify mature OCs by the formation of actin rings (D) (original magnification, ×40). (E and F) Mouse BM cells cultured with M-CSF and RANKL on dentine slices with 5 µM d-PDMP or 50 µM iminosugars added on day 1 were evaluated on day 14 when resorption lacunae on dentine slices were visualized with methylene blue (original magnification, ×4). (F) Resorption area is expressed as a percentage of the dentine surface area. A representative experiment of three, each performed in triplicate assays, is shown for C and F. Data are mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001 versus control).
To assess whether the iminosugar inhibitors would affect the process of OC fusion that is required for the development of mature OCs, we added these compounds into OC cultures 3 days after treatment with M-CSF and RANKL commenced and hence when mononuclear OCs had already formed. Mature OCs were identified by tartrate-resistant acid phosphatase (TRAP) activity as well as by phalloidin staining, reflecting F-actin polymerization and acquisition of resorptive activity. We found that d-PDMP, NB-DNJ, and N-OD-DNJ but not NB-DGJ effectively prevented the formation of mature OCs 2 days later (Figure 1, C and D). To determine the direct effect of iminosugar inhibitors on OC resorptive activity, we performed resorption pit assays on dentine. Inhibitors were added to murine BM cells 3 days after culture initiation with M-CSF and RANKL. After 14 days, d-PDMP, NB-DNJ and N-OD-DNJ but not NB-DGJ effectively reduced pit numbers and pit surface area as compared with untreated cultures, consistent with inhibition of OC maturation and activation (Figure 1, E and F).
Knockdown of Ugcg inhibits OC formation.
In order to test whether inhibition of GCS, and thus of the GSL biosynthesis pathway, by the iminosugar agents is responsible for the impaired osteoclastogenesis, we used two previously validated (33) shRNAs against UDP-glucose ceramide glucosyltransferase (Ugcg), i.e., the gene encoding GCS. First, using quantitative PCR (q-PCR) assay, we confirmed that as compared with scrambled control, sh783 and sh875 effectively reduced Ugcg mRNA levels (Supplemental Figure 2A). As shown in Figure 2, RAW264.7 cells transfected with sh783 and sh875 generated significantly fewer (Figure 2E) and also smaller OCs (Figure 2F) than cells transfected with scrambled shRNA. Importantly, inhibition of osteoclastogenesis by Ugcg knockdown was not due to changes in cell viability, as shown by MTT assay (Supplemental Figure 2B).
Figure 2. Knockdown of Ugcg inhibits OC formation.

OCs derived from non-transfected, PBS-treated (A) and transfected RAW264.7 cells (B–D) were TRAP stained and imaged using an Olympus inverted microscope with ×10 magnification. RAW264.7 cells were transfected with scrambled shRNA (shScr) (B) or with sh783 (C) or sh875 shRNA (D). OC count at ×10 fields from 2 different experiments, with each experiment performed in triplicate, was used to generate individual plots (E), and the mean size of OCs is expressed in pixels (F). Statistical differences in TRAP-positive cells (E) were analyzed with standard one-way ANOVA, and differences in mean size (F) were analyzed with the Kruskal-Wallis ANOVA test. Error bars correspond to SEM. n = 6; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
NB-DNJ perturbs OC lipid raft organization and NFATc1 nuclear localization.
The ganglioside GM1, also known as a glycosphingolipid-enriched microdomain marker, is invariably associated with lipid rafts — cell membrane signaling platforms rich in GSLs — with a subset of lipid rafts defined by the presence of caveolin-1 (34, 35). RANK, upon its engagement by its ligand RANKL, shifts into lipid rafts (4). RANK interacts with the raft-associated molecules TRAF6, an adaptor protein mediating activation of downstream signaling in developing OCs, including MAPK-dependent signaling; and c-SRC kinase, required for F-actin polymerization and OC resorptive activity (36). We tested whether disruption of GSL synthesis by NB-DNJ would perturb raft function and association of M-CSF receptor (M-CSFR), TRAF6, and c-SRC with the raft in response to RANKL.
As large numbers of OCs are required to obtain enough protein for this method, we used the murine macrophage cell line RAW264.7, which readily differentiates into OCs in response to RANKL (4, 37). First, we confirmed that OC formation was effectively inhibited by NB-DNJ when these cells were used as OC precursors, similar to both the primary mouse and human OCs (Supplemental Figure 1C). Next, the cell lysate was separated over a discontinuous sucrose gradient by ultracentrifugation in 9 fractions. Fractions 2–5 were enriched in lipid rafts, as indicated by the presence of GM1 (Figure 3A). In untreated RAW264.7 cells (Figure 3A), M-CSFR was found to reside primarily in the non-raft part of the cell membrane, with only a small proportion associated with the rafts; likewise, TRAF6 was found almost exclusively outside the rafts, while as previously reported (4), a significant proportion of c-SRC resided in the rafts. Upon RANKL treatment (Figure 3A), a clear shift of M-CSFR, TRAF6, and caveolin-1 into the raft fraction was observed, with almost all c-SRC localized in the rafts. In the presence of NB-DNJ (Figure 3A), RANKL-induced movement of TRAF6 and M-CSFR into rafts was not observed, while c-SRC and caveolin-1 only existed outside the raft fraction. Taken together, these findings show that iminosugar inhibitors perturb association of molecules critical for OC differentiation and activation with lipid rafts.
Figure 3. GSL inhibitors perturb the association of SRC and TRAF6 with lipid rafts during osteoclastogenesis.

(A) Localization of M-CSFR, TRAF6, c-SRC, and GM1 ganglioside in the lipid raft (fractions 3–5) or non-raft fractions (fractions 7–9) in RAW 264.7 cells cultured in medium only (control) or in the presence of 50 ng/ml RANKL or RANKL+NB-DNJ in 48-well plates for 4 days. Representative blots from 3 separate experiments are shown. (B) Overnight serum-starved OCs stained with anti-NFATc1 and anti-mouse–Alexa Fluor 546 and viewed by immunofluorescence microscopy.
Next we investigated the effect of NB-DNJ on OC signaling. In response to RANKL, signaling occurs via the MAPK and NF-κB pathways, leading to the activation of OC-specific genes via the binding of transcription factors such as NFATc1 and AP1 (38, 39). When we investigated MAPK signaling in RAW264.7 cells in response to RANKL or M-CSF treatment, there was the expected upregulation of the three MAPK pathways; however, there was no significant change in the phosphorylation of p38, ERK, or JNK following NB-DNJ treatment (Supplemental Figure 3, A and B). One of the key transcription factors in osteoclastogenesis is NFATc1, which is induced via TRAF6/NF-κB and c-Fos pathways and autoamplifies to give robust activation of OC-specific genes such as TRAP and cathepsin K (38, 40, 41). Using immunofluorescence microscopy, we observed nuclear localization and accumulation of NFATc1 in response to RANKL treatment; the addition of NB-DNJ prevented this nuclear accumulation of NFATc1 (Figure 3B).
GM3 and GM2 are the dominant polar GSLs in MM cells.
As a first step in investigating the potential role of myeloma-derived GSL in modifying the tumor microenvironment and specifically contributing to OC activation, we determined the GSL profiles of primary myeloma cells from four patients and human MM cell lines by mass spectrometry (42). As shown in Table 1 and Figure 4, A and B, MALDI-TOF mass spectra of GSLs showed that GM3 was the most abundant GSL in primary myeloma CD138+ tumor cells (n = 1; the cluster of molecular ion peaks between m/z 1,372 and 1,482), as well as in all of the human MM cell lines tested. GM2 was also abundant in MM cell lines but not in primary myeloma cells. Neither GM3 nor GM2 was present in myeloma patient–derived, CD138– non-malignant BM cells (n = 3); instead, lactosylceramide (m/z 1,011) and GA1 (cluster of molecular ion peaks between m/z 1,460 and 1,570) were the predominant GSLs in these cells. The preponderance of lactosylceramide over GM3 and GM2 was also observed in bone marrow samples from healthy donors, as determined by HPLC (Supplemental Table 1). Finally, analysis of purified malignant CD138+ plasma cells by HPLC confirmed GM3 as one of the most abundant polar GSLs in 4 of 6 patients and GM2 in 1 of 6 patients (Figure 4C). Thus, GM3 and GM2 were the predominant GSL constituents of patient myeloma cells and MM cell lines (43) (Figure 4 and Table 1).
Table 1. Mass spectrometry profiling of glycan moiety of GSLs from patient BM cells and human myeloma cells lines.
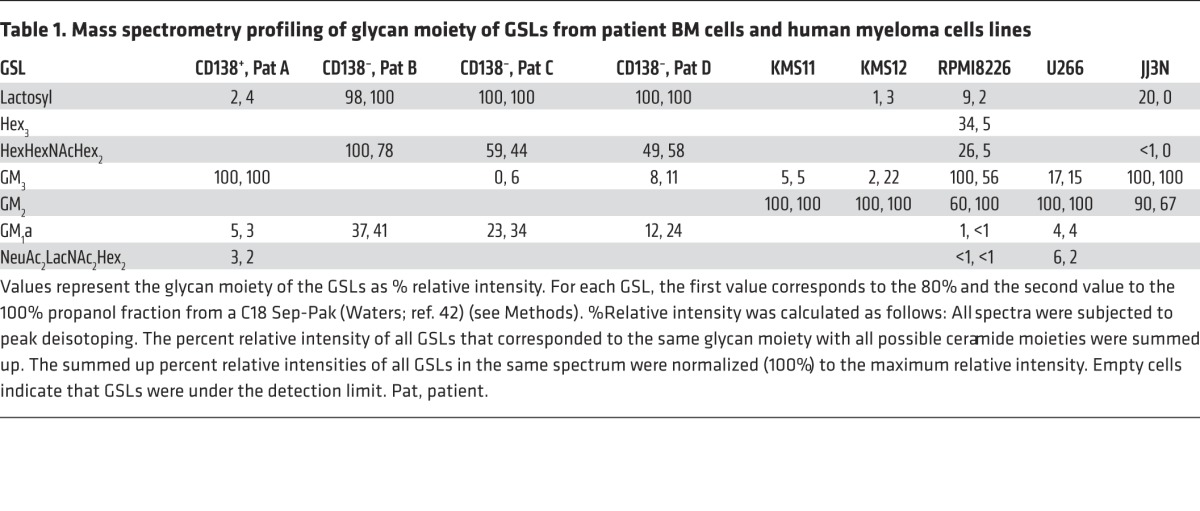
Figure 4. GSL profiles of primary human myeloma cells and human MM cell lines.

(A and B) MALDI-TOF mass spectra of permethylated GSLs from human primary cells. Upper-phase GSLs from (A) MM patient CD138+ and (B) MM patient CD138– BM cells. Profiles of GSLs are from the 80% (left panels) and 100% propanol (right panels) fractions from a C18 Sep-Pak (Waters; ref. 42). The inset corresponds to a zoomed scan of the GM3 cluster area. GSLs are indicated as cartoon structures for the glycan moiety and composition of the fatty acid for the lipoform moiety, with d-erythro-sphingosine considered as the sphingosine base. Cartoon structures are according to Consortium for Functional Glycomics (http://www.functionalglycomics.org) guidelines. Fatty acid composition is indicated below each cartoon structure. Unassigned peaks correspond to chemical derivatization artifacts and/or to structures not corresponding to GSLs. All molecular ions are [M+Na]+. Structural assignments of the glycan moieties are based on monosaccharide composition, tandem mass spectrometry, and known biosynthetic pathways. (C) HPLC analysis of CD138+ selected plasma cells from 6 patients with MM. In 4 of 6 cases, GM3 is the predominant GSL, followed by GM2. Absolute quantification of GM3 is also shown. The top panel shows the various marker GSLs used for identification. GalNAc, N-acetylgalactosamine; HexNAc, N-acetylhexoseamine; NeuAc, N-acetylneuraminic acid.
GM3 cooperates with RANKL and IGF-1 to promote OC maturation.
Given its abundance in myeloma cells, we next tested the hypothesis that exogenous GM3 can modify the BM environment and specifically contribute to OC activation in a paracrine fashion. For this purpose, murine BM OCs were generated in the presence of conventional M-CSF concentration (25 ng/ml) but a limiting concentration of RANKL (5 ng/ml); multinucleated (≥3 nuclei) OCs were enumerated after TRAP staining. In this system, the addition of GM3 to RANKL and M-CSF further enhanced the formation of mature OCs in a dose-dependent manner by day 7 (Figure 5A). Cells cultured with GM3 and M-CSF in the absence of RANKL did not form OCs (data not shown). The pro-osteoclastogenic effect of GM3 was also observed in human OC assays, in which elutriated PBMCs were differentiated into OCs in the presence of M-CSF and a suboptimal concentration of RANKL (Supplemental Figure 4); this demonstrated a direct effect of GM3 on OCs, as there are no contaminating stromal cells present in the human cell preparation. Taken together, these results show that the pro-osteoclastogenic effect of GM3 occurs via direct action on OC precursors under permissive conditions, i.e., in the presence of a source of RANKL.
Figure 5. GM3 cooperates with RANKL and IGF-I to promote osteoclastogenesis.

(A) Mouse BM cells were cultured in the presence of 25 ng/ml M-CSF and only 5 ng/ml RANKL (control) and GM3 (0.05, 0.5, or 5 μM), (B) with IGF-1 (25, 50, or 100 ng/ml), or (C) with IGF-1+ GM3 in the presence of RANKL and M-CSF in 96-well plates for 7 days, and OC numbers were evaluated by TRAP staining. A representative experiment of 3, each performed in triplicate assays, is shown (n = 3). Error bars correspond to SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001 versus control).
Since previous work in Igf1–/– mice suggested that IGF-1 is a pro-osteoclastogenic factor and it plays a significant role in the biology of MM (44), we tested whether GM3, as well as RANKL, could also cooperate with IGF-1 to promote osteoclastogenesis. First, in line with a direct pro-osteoclastogenic effect, we found that IGF-1 in the presence of a limiting concentration of RANKL enhanced murine OC formation in a dose-dependent manner (Figure 5B), while GM3 and IGF-1 in combination with RANKL additively enhanced OC maturation (Figure 5C). Thus, GM3 can cooperate with both RANKL and IGF-1 to promote OC maturation.
GM3-mediated OC activation and effect of NB-DNJ in vivo.
Next we tested whether GM3 would promote OC development and activation in vivo and the effect of NB-DNJ on this process. To determine the optimal in vivo dose of NB-DNJ, we treated five groups of mice with 0–100 mg/mouse/d of inhibitor for 6 days by oral gavage. The overall biological activity of NB-DNJ was assessed by HPLC in a surrogate assay that tracks the levels of free plasma glucose–containing oligosaccharides (45). These are generated as a result of inhibition of ER-resident α-glucosidases I and II by NB-DNJ, leading to glycoprotein misfolding and eventually — as misfolded proteins are degraded in the proteasome — release of free oligosaccharides (45). We found that detection of plasma oligosaccharides was dose dependent and highest at the largest dose used (100 mg/mouse/d), corresponding to a serum level of NB-DNJ of 109.3 ± 5.3 μM (Figure 6A). Analysis of BM cells also confirmed GSL depletion in a dose-dependent manner (Figure 6B). Furthermore, in vitro assays using BM from mice treated for 6 days with NB-DNJ showed a dose-dependent reduction in OC numbers at a dose of 75–100 mg/mouse/d, resulting in an approximately 30% reduction as compared with PBS-treated controls (Figure 6, C and D), suggesting that NB-DNJ has an anti-osteoclastogenic effect in vivo.
Figure 6. NB-DNJ inhibits ex vivo osteoclastogenesis in a dose-dependent manner.

(A) Quantification of free serum oligosaccharides recovered by HPLC from NB-DNJ–treated mice. (B) GSL analysis of BM cells following administration of 0, 25, 50, 75, and 100 mg NB-DNJ. The representative species of oligosaccharides — GM1b, GA1, and GD2 — were identified in the BM of NB-DNJ–treated mice. (C) BM cells obtained from mice treated with different doses of NB-DNJ were induced to form OCs (with 25 ng/ml M-CSF and RANKL) in 96-well plates and were identified by TRAP staining (original magnification, ×20). (D) Enumeration of TRAP-positive multinucleated cells/well differentiated from NB-DNJ–treated mouse BM (n = 5 mice/treatment; BM from each mouse was evaluated in triplicate). Error bars correspond to SEM (*P < 0.05, **P < 0.01, ***P < 0.001, versus 0 mg NB-DNJ); x axis represents NB-DNJ dose in mg/mouse/d in A, B and D. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test.
To address whether GM3 exerts a pro-osteoclastogenic effect in vivo and how such an effect might be modified by NB-DNJ, we studied ex vivo OC formation and activity using BM cells from mice treated with PBS, GM3+PBS, NB-DNJ, or GM3+NB-DNJ. In line with the in vitro results, assessment on day 7 of the experiment when the mice were sacrificed showed that as compared with PBS treatment, a single injection of GM3 on day 4 resulted in a significant increase in the number of in vitro generated OCs. However, in mice treated with NB-DNJ, the enhanced osteoclastogenesis observed after GM3 injection was abrogated (Figure 7A). Furthermore, OCs from mice treated with GM3 displayed greater resorptive activity in ex vivo pit assays than OCs from PBS-treated mice, and again this effect was reversed in mice receiving GM3+NB-DNJ (Figure 7B).
Figure 7. NB-DNJ inhibits GM3-mediated enhanced OC development and activity in vivo.

(A) TRAP staining analysis of mouse OCs from different groups treated with GM3 and/or NB-DNJ as indicated in Methods after 7 days of culture. (B) Representative images of OC resorption pits on dentine (original magnification, ×10). (C) Representative histological sections of mouse tibiae stained for OC TRAP activity and counterstained with 0.25% methyl green solution (original magnification, ×40). For A, B, and C, a representative experiment of 3 is shown, with n = 5 mice in each treatment group and with each individual BM sample evaluated in triplicate. (D and E) Bone histomorphometry parameters obtained by analyzing cumulatively the histological sections of two different experiments of 3 (n = 10). Number of OCs/total bone area (N.Oc/T.Ar); OC surface is expressed as percentage of total bone surface (Oc.S/BS). (F) Serum values of CTX (ng/ml) combined from two different experiments of 3 (n = 10). Error bars correspond to SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001 versus control).
Analysis of in situ OC development on histological bone sections stained for TRAP (Figure 7C) showed a significantly higher number of OCs (Figure 7D), with increased surface area (Figure 7E) in mice treated with GM3 plus PBS, an effect that was almost entirely reversed in GM3+NB-DNJ–treated mice. In vivo resorptive OC activity as assessed by serum C-terminal telopeptides of collagen type I (CTX) levels also showed a significant increase in response to GM3 and reversal of this effect in GM3+NB-DNJ–treated mice (Figure 7F). Finally, the same inhibitory effect of NB-DNJ as assessed by serum CTX levels was observed when osteoclastogenesis was activated by α-galactosylceramide, a glycolipid that, as we previously demonstrated, promotes differentiation and maturation of OCs in vivo (46) (Supplemental Figure 5). Therefore, in line with the in vitro studies, exogenous GM3 exerted significant pro-osteoclastogenic activity in vivo, while treatment of mice with NB-DNJ effectively inhibited baseline as well as GM3-induced OC development and function.
NB-DNJ effectively reduces osteolytic bone lesions in the 5TGM1 murine myeloma model.
To assess the effectiveness of NB-DNJ on MM disease progression, we employed the murine myeloma 5TGM1 model (47, 48). First we determined that in vitro, NB-DNJ had a modest inhibitory effect (up to 25%) on the viability of 5GTM1-GFP cells (Supplemental Figure 6A) and that as in human myeloma cells, GM3 was the most abundant GSL (64.8% ± 1.2%; n = 3) in 5GTM1-GFP cells. Next, C57BL/KaLwRijHsd mice receiving transplants of 5GTM1-GFP cells were treated with PBS (n = 6) or NB-DNJ (50 mg/d, n = 6) starting day 4 after transplant. Non-transplanted and PBS-treated mice (n = 6) served as naive controls. On day 26 all of the mice were killed, and the tumor burden was assessed via GFP expression by flow cytometry. In both the BM and spleen, NB-DNJ treatment reduced the percentages of 5TGM1-GFP cells by 30%–40% when compared with the PBS-treated groups; however, this was not statistically significant (Supplemental Figure 6B).
To determine the consequences of GSL inhibition on OC activity in a pathologic setting, we performed histomorphometric evaluation of TRAP-stained longitudinal cross sections of tibiae. MM-diseased and PBS-treated mouse tibiae showed a significantly higher number of OCs with increased surface area when compared with naive control mice (Figure 8A). However, NB-DNJ treatment of MM-diseased mice reduced OC number and surface area (Figure 8A). Further, tibiae were evaluated for osteolytic bone lesions by micro-CT analysis. Treatment with NB-DNJ decreased bone loss in vivo and reduced myeloma-induced lytic bone lesions, as shown by a significant increase in bone parameters such as bone volume (BV/TV) and trabecular thickness (Tb.Th), and a consequent decrease in trabecular pattern factor (Tb.Pf) (Figure 8, B and C, and Supplemental Figure 7). All bone parameters from myeloma diseased and NB-DNJ–treated mice were either similar (bone surface density [BS/BV], number of trabeculae [Tb.N], Tb.Pf, structure model index [SMI]) or close (BV/TV, Tb.Th) to the values in the naive control animals, as shown in Figure 8C. Taken together, these findings are consistent with the in vitro results, i.e., that NB-DNJ could inhibit the function of OCs activated by either exogenous GSLs such as GM3 or by de novo synthesized, endogenous GSLs.
Figure 8. NB-DNJ effectively inhibits 5TGM1 myeloma cell–caused bone lesions in C57BL/KaLwRijHsd mice.

(A) Representative histological sections of TRAP-stained naive control (Ctr), myeloma-diseased and PBS-treated, or myeloma-diseased and NB-DNJ–treated (2,500 mg/kg/d) mouse tibiae (original magnification, ×20) and tibial histomorphometric evaluation: number of OCs reported to total area (N.Oc/T.Ar) and OC surface reported to bone surface (Oc.S/BS). (B) Longitudinal cross section of diseased and PBS- or NB-DNJ–treated (2,500 mg/kg/d) mouse tibiae evaluated by micro-CT reconstruction. (C) Micro-CT analysis parameters of tibiae: BV/TV, BS/BV, Tb.Th, bone surface (BS), Tb.N, Tb.Pf, SMI, and fractal dimension (FD). n = 6 mice/treatment; *P < 0.05, **P < 0.01, ***P < 0.001 versus 5GTM1 cell–transplanted and PBS-treated mice; #P < 0.05 versus non-transplanted naive controls. Error bars correspond to SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test.
Discussion
We demonstrate here that GSLs, and in particular GM3, play an important role in OC activation. We have further shown by mass spectrometry and HPLC analysis that GM3 is the dominant polar GSL present in myeloma cells. Importantly, the administration of the GSL inhibitor NB-DNJ was able to reduce osteolytic lesions in a mouse model of MM (47). In vitro, GM3 promoted osteoclastogenesis in the presence of a limiting RANKL concentration in a cooperative manner. This was demonstrated by culturing BM or elutriated PBMCs with a suboptimal dose of RANKL; this allows for the measurement of further OC differentiation following GM3 addition.
It is not yet clear how exogenous GM3 enhances the effect of RANKL and M-CSF in vitro. It was previously shown that GSLs can directly incorporate into the cell membrane, and through lateral interaction with signaling proteins such as the EGF and VEGF receptors, it can modulate downstream signaling (49). It is possible that in an analogous way, exogenous GM3, by incorporating into the cell membrane of developing OCs, can increase the responsiveness of RANK and M-CSFR to their ligands, RANKL and M-CSF, respectively. In support of such mechanism, stromal cell–derived GM3 was previously shown to incorporate into the cell membrane rafts of the myeloid cell progenitor line FDCP1 and, through interaction with GM-CSF receptor, to promote cell proliferation in response to GM-CSF (50, 51). Alternatively, exogenous GM3 might act indirectly by promoting secretion of pro-osteoclastogenic factors from other BM cells, such as osteoblasts, monocytes/macrophages, and T cells. However, the pro-osteoclastogenic effect of GM3 in our ex vivo experiments supports a direct role for GM3 and GSL on OC precursor differentiation, as GM3 participates in RANKL-induced osteoclastogenesis, and reduced OC fusion and activation are observed in the absence of GSL biosynthesis. Although not directly tested, it is likely that GM2, another prominent GSL in some myeloma cells, exerts a similar direct pre-osteoclastogenic effect.
We show inhibition of OC development and maturation by pharmacological and genetic inhibition of Ugcg, the gene encoding for GCS, the rate-limiting enzyme in GSL synthesis. This suggests an important role for de novo synthesized GSL in baseline osteoclastogenesis. Among the iminosugar inhibitors tested, NB-DNJ has been therapeutically successful in patients with lysosomal storage diseases by reducing the overall burden of unprocessed GSLs through inhibition of de novo synthesis of GSLs (31, 52). In these patients, increased OC activation and osteoporosis occur as a consequence of the lysosomal storage disease, and treatment with NB-DNJ results in a significant restoration of bone mass (22).
We identify lipid rafts as an important target for the biological effect of NB-DNJ (53). These GSL-rich membrane structures are required for the clustering of growth factor receptors, including RANK (4) and IGF1R (54), and subsequent activation of downstream signaling, including the MAPK-dependent pathways (55). We show that the reduced presence of GM1 in the rafts in response to NB-DNJ is associated with a significant reduction in raft-resident TRAF6 and c-SRC. TRAF6 is a pivotal adaptor that upon RANKL ligation binds to RANK and promotes OC differentiation as well as maturation (56, 57), while the presence of c-SRC in rafts is required for its association with β3 integrin and subsequent Rho GTPase–dependent polymerization of F-actin (58). Mechanistically, by using NB-DNJ, we were able to show that GSLs are essential for clustering of TRAF6 in lipid rafts during RANKL signaling in OCs. Furthermore, the presence of NB-DNJ prevented the accumulation of NFATc1 in the nucleus. This transcription factor is essential for the expression of osteoclastogenic genes, and in its absence OC formation and activity would be impaired (40, 41), thus describing one of the consequences of NB-DNJ administration.
The in vitro effects of GM3 as a pro-osteoclastogenic factor were supported by its effects in vivo. GM3 significantly promoted OC development and function, as shown by increased OC numbers and surface in situ and serum CTX levels, and both of these effects were effectively inhibited by NB-DNJ. Exogenous GM3 has been shown previously to incorporate into cell membranes and lipid rafts of myeloid cells and to promote downstream cell signaling (50, 51). It is possible, therefore, that disruption of lipid rafts in OC precursors by NB-DNJ prevents the OC-activating effect of exogenous GM3.
Increased GSL biosynthesis (59, 60) and GSL-mediated promotion of angiogenesis and metastatic tumor growth (6) have been demonstrated in different types of cancers. We have shown that GM3 is the predominant polar GSL produced by both myeloma cell lines and primary myeloma cells from patients with MM. Furthermore, the administration of NB-DNJ to mice susceptible to developing MM following transplantation of 5TGM1 myeloma cells demonstrated the effectiveness of this GSL inhibitor in limiting osteolytic bone lesion development. Although the reduction in myeloma cell burden of 30%–40% did not reach statistical significance, our data suggest direct antimyeloma activity of NB-DNJ. Hence, it seems likely that the combination of reduced myeloma cell burden along with substantial inhibitory effects of NB-DNJ on OCs prevents the development of osteolytic bone lesions. It is possible that the well-established murine 5TGM1 MM model with its short duration of only 26 days does not reveal NB-DNJ’s full effects on myeloma cell burden; however, our in vitro data confirm an NB-DNJ inhibitory effect on 5TGM1 cells.
Taken together, our in vitro and in vivo data suggest that this salutary effect of NB-DNJ on myeloma-induced bone disease is multifactorial and the result of reduced pro-osteoclastogenic GM3 production by the tumor cells, as well as inhibition of OC development and maturation through inhibition of de novo GSL synthesis. Therefore, since nearly all MM patients experience the consequences of MM-related bone disease, we propose that the clinically approved NB-DNJ and possibly other GCS inhibitors can be used as clinical agents to abrogate pathological OC activation in MM.
In summary, we showed here that GM3, the main GSL constituent of myeloma plasma cells, is a pro-osteoclastogenic factor in vitro as well as in vivo. The inhibitory effect of NB-DNJ on de novo and exogenous GM3-mediated OC activation and its ability to limit myeloma-induced bone loss in an animal model of disease provides a rational basis for a clinical study aiming at reducing OC activation and bone destruction in patients with MM.
Methods
GSL inhibitors.
d-PDMP was purchased from Matreya LLC, NB-DNJ was provided by Sigma-Aldrich, N-OD-DNJ was a gift from United Therapeutics, and NB-DGJ was purchased from Toronto Research Chemicals Inc. They were dissolved in water at 100 mM and stored at –20°C until use. For each experiment, they were diluted immediately before use in culture medium (5–500 μM).
In vivo assays.
For in vivo experiments 8- to 10-week-old female WT C57BL/6 mice (Charles River) and 10-week-old female C57BL/KaLwRijHsd mice (Harlan) were maintained in the Biological Services Unit, Kennedy Institute of Rheumatology, University of Oxford. Animals were administered PBS or 0–5,000 mg/kg/d NB-DNJ (diluted in water) once a day by gavage in a volume of 200 μl for 6 days (n = 5 mice/group). On day 4, PBS or 10 μg/mouse of GM3 (Sigma-Aldrich or Avanti Polar Lipids Inc.) was injected i.v. in a volume of 200 μl. α-Galactosylceramide (a gift from S. Gadola, University of Southampton, Southampton, UK) or PBS was administered by a single i.p. injection of 2 μg on day 3. Serum was collected on day 7, by intracardiac puncture. Bones were either fixed for histology or flushed to obtain live BM cells. In order to establish the 5TGM1 mouse myeloma model (48), C57BL/KaLwRij mice were i.v. transplanted with 106 5TGM1-GFP cells (gift from Claire Edwards, University of Oxford, and Kay Oyajobi, University of Texas, San Antonio, Texas) in 100 μl PBS in the tail vein. Non-transplanted mice served as naive controls (n = 6 mice/group). Starting from day 4 after transplantation, mice were gavaged daily with PBS or NB-DNJ (2,500 mg/kg/d) for another 21 days.
HPLC profiling of GSL and free plasma glucose–containing oligosaccharide assay.
As previously described (61), GSLs were extracted from cells and the oligosaccharide moiety removed using ceramide glycanase purified from Hirudo medicinalis and labeled with anthranilic acid. Following separation of labeled GSL-derived oligosaccharides by normal-phase HPLC, peak areas were measured using Empower software (Waters), converted to molar amounts using an experimentally derived factor, and normalized to cellular protein. Levels of serum NB-DNJ were determined as previously published (62).
In vitro OC formation and TRAP assay.
Cells obtained from the BM of 8- to 10-week-old C57BL/6 mice were cultured at 37°C in 5% CO2 in complete medium — α-MEM with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) at a density of 5 × 105 cells/well in 96-well or 48-well plastic plates — and OC precursors were induced to proliferate and differentiate with 20–25 ng/ml murine M-CSF (Peprotech) and 5–50 ng/ml RANKL (PeproTech or R&D Systems) supplements as indicated. Medium was changed every 3 days, and after 5 or 7 days cells were washed with PBS and fixed with 4% paraformaldehyde, then permeabilized with acetone/ethanol 1:1 solution and stained for TRAP activity for 15 minutes at 37°C (0.1 mg/ml Naphthol AS-MX phosphate, 0.4 mg/ml Fast Red Violet LB Salt, 0.5 ml dimethylformamide in 50 ml TRAP buffer; all from Sigma-Aldrich). Multinucleated (≥3 nuclei) TRAP-positive cells were considered to be OCs. In all the experiments, BM cells were also cultured in complete medium with M-CSF but no RANKL, serving as a negative control for OC formation.
In vitro OC resorption assay.
Resorptive activity of OCs was determined by a lacunar resorption assay on dentine disks placed in 96-well plates according to regular techniques. Cells were cultured in complete medium as in the TRAP assay on dentine for 14 days with media changes every 3 days. Dentine disks were rinsed in dH2O to lyse and remove the OCs. Resorption pits were stained with toluidine blue or marked with carbon, and resorption areas were analyzed by using ImageJ software (http://imagej.nih.gov/ij/) and expressed as the percentage of resorption area per dentine.
RAW264.7 cell transfection with shRNA against mouse Ugcg.
shRNAs targeting mouse Ugcg (sh783, 5′-CTCAGTACATTGCTGAAGATT, and sh875, 5′-TCTGGTTCGTACTCAATTTCT) were prepared as previously described (33). RAW264.7 cells were transfected with scrambled shRNA, sh783, and sh875 using FuGENE HD (E2311, Promega). Seventy-two hours after transfection, GFP-positive cells were sorted and plated at a density of 0.1 × 105 cells per well in 24-well plates. OC differentiation was induced on both non-transfected RAW264.7 and sorted transfected RAW264.7 cells, using 50 ng/ml M-CSF plus 75 ng/ml RANKL. After 7 days, cells were fixed and TRAP stained. TRAP-positive cells were counted, and the size of the OCs was measured using Fiji image analysis software (63). Data analysis was performed using Prism software (GraphPad Software). Mean values for each experimental condition were calculated.
Patient samples.
BM cells from myeloma patients were subjected to red blood cell lysis using BD FACS lysing solution (BD Pharmingen). BM myeloma cells were subsequently selected using an EasySep Human CD138 Positive Selection Cocktail Kit (Stemcell Technologies) following the manufacturer’s instructions. Plasma cell purity was greater than 90%, as evaluated by FACS analysis using anti-CD138–FITC antibody (BD Pharmingen).
Glycolipid extraction, MALDI-TOF mass spectrometry, and tandem mass spectrometry analysis.
Glycolipid extraction was performed as previously described (42). MS and tandem mass spectrometry (MS/MS) data were acquired using a 4800 MALDI-TOF/TOF (Applied Biosystems) mass spectrometer. Purified permethylated GSLs were dissolved in 10 μl methanol, and 1 μl of dissolved sample was premixed with 1 μl of matrix (10 mg/ml 2,5-dihydroxybenzoic acid [DHB] in 70% [vol/vol] aqueous methanol), spotted onto a target plate (2 × 0.5 μl), and dried under vacuum. The collision energy of the MALDI-TOF/TOF was set to 1 kV, and argon was used as a collision gas. The 4700 Calibration standard kit, Cal Mix (Applied Biosystems), was used as the external calibrant for the MS mode and [Glu1]-Fibrinopeptide B human (Sigma-Aldrich) was used as an external calibrant for the MS/MS mode.
The MS and MS/MS data were processed using Data Explorer 4.9 Software (Applied Biosystems). The spectra were subjected to manual assignment and annotation with the aid of the glycobioinformatics tool GlycoWorkBench (http://www.glycoworkbench.org). The proposed assignments for the selected peaks were based on 12C isotopic composition together with the known biosynthetic pathway. The proposed structures were confirmed by data obtained from MS/MS.
Decalcified bone histology.
Mouse tibiae were fixed in 4% formaldehyde at 4°C. After 24 hours of fixation, tibiae were transferred into 10% EDTA for 21 days. To ensure proper decalcification, every 3–4 days the EDTA solution was changed. After paraffin embedding following standard techniques, the bones were sectioned with a Sakura Accu-Cut SRM 200 microtome. Sections (4 μm) were stained for TRAP activity and counterstained with methyl green following standard procedures. Slides were analyzed using an Olympus Bx51 microscope, and pictures were taken with an Olympus DP71 camera.
Bone histomorphometric analysis.
Analysis of bone volume and bone surface and determination of OC numbers and OC surface were carried out using the OsteoMeasure histomorphometry system (OsteoMetrics) in accordance with American Society for Bone and Mineral Research (ASBMR) standard nomenclature (http://www.asbmr.org/standardizationofbonestructureanddensitynomenclature.aspx) on TRAP-stained and 0.25% methyl green counterstained 4-μm-thick histological sections, as previously described (64, 65).
Micro-CT analysis.
Mouse tibiae were harvested 26 days after 5TGM1-eGFP myeloma cell transplant and fixed in 70% ethanol. Bones were scanned with a SkyScan 1174 scanner: 50 kV, 800 μA, 8.3-μm isometric voxel resolution, 0.7-degree rotation step. Images were analyzed using SkyScan CT Analyzer software version 1.9.3.0. by selecting the trabecular region volume of interest commencing immediately after the growth plate and preceding distally for 208 slices (1.72 mm). A threshold of 80 density units was selected to distinguish mineralized from nonmineralized tissue.
Supplemental methods.
Reagents and cell lines, Western blots, lipid raft isolation, phalloidin staining, ELISA, elutriation, cell viability assays, and FACS analysis for myeloma burden are described in Supplemental Methods.
Statistics.
One-way ANOVA was used for statistical analysis of all the experiments with more than three experimental groups, and a Tukey post hoc test, as described in the figure legends, was applied to determine the differences between the specific experimental groups. For comparing only two experimental groups, we used two-tailed t tests. In all cases, data are presented as mean ± SEM, and a P value of 0.05 was considered significant. For the statistical analysis of free oligosaccharides recovered by HPLC from mice serum, second-order polynomial regression was used.
Study approval.
Animal care and experimental procedures were approved by the University of Oxford Local Ethical Review Committee under PPL 7161 and were conducted in accordance with the UK Home Office Animals Act of 1986. BM from myeloma patients was obtained after receipt of informed consent in accordance with the Declaration of Helsinki and the study was approved by Hammersmith Hospital NHS Trust Research Ethics Committee, London.
Supplementary Material
Acknowledgments
We thank Claire Edwards for constructive comments on the final manuscript — it is greatly appreciated. This work was supported by the Multiple Myeloma Research Foundation (A. Ersek, N.J. Horwood, A. Karadimitris), Leukaemia and Lymphoma Research (K. Xu, A. Ersek, A. Chaidos, E. Spanoudakis, M. Hu, A. Rahemtulla, I. Roberts, D.S. Alonzi, N. J. Horwood, and A. Karadimitris), Arthritis Research UK (N.J. Horwood, L. Danks), Kennedy Institute Trustees (Y. Vattakuzhi), the Glycobiology Institute, University of Oxford (G. Twigg, R.A. Dwek, and T.D. Butters), and the National Institute for Health Research (NIHR) Biomedical Research Centre at Imperial College.
Footnotes
Conflict of interest: Raymond A. Dwek is a Director of United Therapeutics (UT) and Chairman of its Scientific Committee. UT supports research at the Glycobiology Institute. Terry D. Butters is also in receipt of a research contract and consultancy from UT.
Reference information:J Clin Invest. 2015;125(6):2279–2292. doi:10.1172/JCI59987.
References
- 1.Degroote S, Wolthoorn J, van Meer G. The cell biology of glycosphingolipids. Semin Cell Dev Biol. 2004;15(4):375–387. doi: 10.1016/j.semcdb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Hakomori S. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. 2001;491:369–402. doi: 10.1007/978-1-4615-1267-7_24. [DOI] [PubMed] [Google Scholar]
- 3.Birkle S, Zeng G, Gao L, Yu RK, Aubry J. Role of tumor-associated gangliosides in cancer progression. Biochimie. 2003;85(3):455–463. doi: 10.1016/s0300-9084(03)00006-3. [DOI] [PubMed] [Google Scholar]
- 4.Ha H, et al. Membrane rafts Play a crucial role in receptor activator of nuclear factor kB signaling and osteoclast function. J Biol Chem. 2003;278(20):18573–18580. doi: 10.1074/jbc.M212626200. [DOI] [PubMed] [Google Scholar]
- 5.Sorice M, Longo A, Garofalo T, Mattei V, Misasi R, Pavan A. Role of GM3-enriched microdomains in signal transduction regulation in T lymphocytes. Glycoconj J. 2003;20(1):63–70. doi: 10.1023/B:GLYC.0000018018.29488.c6. [DOI] [PubMed] [Google Scholar]
- 6.Hashiramoto A, Mizukami H, Yamashita T. Ganglioside GM3 promotes cell migration by regulating MAPK and c-Fos/AP-1. Oncogene. 2006;25(28):3948–3955. doi: 10.1038/sj.onc.1209416. [DOI] [PubMed] [Google Scholar]
- 7.Garofalo T, et al. Ganglioside GM3 activates ERKs in human lymphocytic cells. J Lipid Res. 2002;43(6):971–978. [PubMed] [Google Scholar]
- 8.Nojiri H, Takaku F, Terui Y, Miura Y, Saito M. Ganglioside GM3: an acidic membrane component that increases during macrophage-like cell differentiation can induce monocytic differentiation of human myeloid and monocytoid leukemic cell lines HL-60 and U937. PNAS. 1986;83(3):782–786. doi: 10.1073/pnas.83.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horwood NJ, Elliott J, Martin TJ, Gillespie MT. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology. 1998;139(11):4743–4746. doi: 10.1210/endo.139.11.6433. [DOI] [PubMed] [Google Scholar]
- 10.Mellis DJ, Itzstein C, Helfrich MH, Crockett JC. The skeleton: a multi-functional complex organ: the role of key signalling pathways in osteoclast differentiation and in bone resorption. J Endocrinol. 2011;211(2):131–143. doi: 10.1530/JOE-11-0212. [DOI] [PubMed] [Google Scholar]
- 11.Mansour A, et al. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med. 2012;209(3):537–549. doi: 10.1084/jem.20110994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teti A. 2012. Osteoclasts and hematopoiesis. Bonekey Rep. 2012;1:46. doi: 10.1038/bonekey.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood. 2011;117(5):1540–1549. doi: 10.1182/blood-2010-05-282855. [DOI] [PubMed] [Google Scholar]
- 14.Kollet O, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12(6):657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Kong D, Ahmad A, Bao B, Sarkar FH. Targeting bone remodeling by isoflavone and 3,3′-diindolylmethane in the context of prostate cancer bone metastasis. PLoS ONE. 2012;7(3):e33011. doi: 10.1371/journal.pone.0033011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, et al. Inhibition of bone resorption, rather than direct cytotoxicity, mediates the anti-tumour actions of ibandronate and osteoprotegerin in a murine model of breast cancer bone metastasis. Bone. 2007;40(2):471–478. doi: 10.1016/j.bone.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Vallet S, et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007;110(10):3744–3752. doi: 10.1182/blood-2007-05-093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwamoto T, et al. Lactosylceramide is essential for the osteoclastogenesis mediated by macrophage-colony-stimulating factor and receptor activator of nuclear factor-κB ligand. J Biol Chem. 2001;276(49):46031–46038. doi: 10.1074/jbc.M104464200. [DOI] [PubMed] [Google Scholar]
- 19.Butters TD. Gaucher disease. Curr Opin Chem Biol. 2007;11(4):412–418. doi: 10.1016/j.cbpa.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 20.Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta. 2006;1758(12):2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 21.El-Beshlawy A, et al. Enzyme replacement therapy and bony changes in Egyptian paediatric Gaucher disease patients. J Inherit Metab Dis. 2006;29(1):92–98. doi: 10.1007/s10545-006-0121-6. [DOI] [PubMed] [Google Scholar]
- 22.Pastores GM, Elstein D, Hrebicek M, Zimran A. Effect of Miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin Ther. 2007;29(8):1645–1654. doi: 10.1016/j.clinthera.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Edwards CM, Zhuang JL, Mundy GR. The pathogenesis of the bone disease of multiple myeloma. Bone. 2008;42(6):1007–1013. doi: 10.1016/j.bone.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fowler JA, Edwards CM, Croucher PI. Tumor-host cell interactions in the bone disease of myeloma. Bone. 2011;48(1):121–128. doi: 10.1016/j.bone.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terpos E, Politou M, Viniou N, Rahemtulla A. Significance of macrophage inflammatory protein-1 alpha (MIP-1alpha) in multiple myeloma. Leuk Lymphoma. 2005;46(12):1699–1707. doi: 10.1080/10428190500175049. [DOI] [PubMed] [Google Scholar]
- 26.Terpos E, Politou M, Szydlo R, Goldman JM, Apperley JF, Rahemtulla A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br J Haematol. 2003;123(1):106–109. doi: 10.1046/j.1365-2141.2003.04561.x. [DOI] [PubMed] [Google Scholar]
- 27.Han JH, Choi SJ, Kurihara N, Koide M, Oba Y, Roodman GD. Macrophage inflammatory protein-1alpha is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor kappaB ligand. Blood. 2001;97(11):3349–3353. doi: 10.1182/blood.V97.11.3349. [DOI] [PubMed] [Google Scholar]
- 28.Choi SJ, et al. Antisense inhibition of macrophage inflammatory protein 1-alpha blocks bone destruction in a model of myeloma bone disease. J Clin Invest. 2001;108(12):1833–1841. doi: 10.1172/JCI13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi SJ, et al. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood. 2000;96(2):671–675. [PubMed] [Google Scholar]
- 30.Roussou M, et al. Increased expression of macrophage inflammatory protein-1alpha on trephine biopsies correlates with extensive bone disease, increased angiogenesis and advanced stage in newly diagnosed patients with multiple myeloma. Leukemia. 2009;23(11):2177–2181. doi: 10.1038/leu.2009.130. [DOI] [PubMed] [Google Scholar]
- 31.Butters TD, Dwek RA, Platt FM. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology. 2005;15(10):43R–52R. doi: 10.1093/glycob/cwi076. [DOI] [PubMed] [Google Scholar]
- 32.Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269(11):8362–8365. [PubMed] [Google Scholar]
- 33.Jung J-U, Ko K, Lee D-H, Ko K, Chang K-T, Choo Y-K. The roles of glycosphingolipids in the proliferation and neural differentiation of mouse embryonic stem cells. Exp Mol Med. 2009;41(12):935–945. doi: 10.3858/emm.2009.41.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betz J, et al. Shiga toxin glycosphingolipid receptors in microvascular and macrovascular endothelial cells: differential association with membrane lipid raft microdomains. J Lipid Res. 2011;52(4):618–634. doi: 10.1194/jlr.M010819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014;1838(2):532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong BR, et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4(6):1041–1049. doi: 10.1016/S1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- 37.Quinn JM, et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res. 2001;16(10):1787–1794. doi: 10.1359/jbmr.2001.16.10.1787. [DOI] [PubMed] [Google Scholar]
- 38.Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23(11):582–590. doi: 10.1016/j.tem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends in Molecular Medicine. 2006;12(1):17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Takayanagi H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. doi: 10.1016/S1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 41.Asagiri M, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202(9):1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parry S, et al. Integrated mass spectrometric strategy for characterizing the glycans from glycosphingolipids and glycoproteins: direct identification of sialyl Le(x) in mice. Glycobiology. 2007;17(6):646–654. doi: 10.1093/glycob/cwm024. [DOI] [PubMed] [Google Scholar]
- 43.Kalisiak A, Minniti JG, Oosterwijk E, Old LJ, Scheinberg DA. Neutral glycosphingolipid expression in B-cell neoplasms. Int J Cancer. 1991;49(6):837–845. doi: 10.1002/ijc.2910490607. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res. 2006;21(9):1350–1358. doi: 10.1359/jbmr.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alonzi DS, Neville DCA, Lachmann RH, Dwek RA, Butters TD. Glucosylated free oligosaccharides are biomarkers of endoplasmic-reticulum α-glucosidase inhibition. Biochem J. 2008;409(2):571–580. doi: 10.1042/BJ20070748. [DOI] [PubMed] [Google Scholar]
- 46.Hu M, et al. Activated invariant NKT cells regulate osteoclast development and function. J Immunol. 2011;186(5):2910–2917. doi: 10.4049/jimmunol.1002353. [DOI] [PubMed] [Google Scholar]
- 47.Edwards CM, et al. Increasing Wnt signaling in the bone marrow microenvironment inhibits the development of myeloma bone disease and reduces tumor burden in bone in vivo. Blood. 2008;111(5):2833–2842. doi: 10.1182/blood-2007-03-077685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyajobi BO, et al. Detection of myeloma in skeleton of mice by whole-body optical fluorescence imaging. Mol Cancer Ther. 2007;6(6):1701–1708. doi: 10.1158/1535-7163.MCT-07-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasahara K, Sanai Y. Functional roles of glycosphingolipids in signal transduction via lipid rafts. Glycoconj J. 2000;17(3):153–162. doi: 10.1023/a:1026576804247. [DOI] [PubMed] [Google Scholar]
- 50.Ziulkoski AL, et al. Gangliosides of myelosupportive stroma cells are transferred to myeloid progenitors and are required for their survival and proliferation. Biochem J. 2006;394:1–9. doi: 10.1042/BJ20051189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ziulkoski AL, et al. Anchored and soluble gangliosides contribute to myelosupportivity of stromal cells. Biochem Biophys Res Commun. 2009;388(1):17–20. doi: 10.1016/j.bbrc.2009.07.092. [DOI] [PubMed] [Google Scholar]
- 52.Cox TM, et al. The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26(6):513–526. doi: 10.1023/A:1025902113005. [DOI] [PubMed] [Google Scholar]
- 53.Ersek A, Karadimitris A, Horwood NJ. Effect of glycosphingolipids on osteoclastogenesis and osteolytic bone diseases. Front Endocrinol (Lausanne). 2012;3:106. doi: 10.3389/fendo.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tai YT, et al. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3’-kinase/AKT signaling. Cancer Res. 2003;63(18):5850–5858. [PubMed] [Google Scholar]
- 55.Tai YT, et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood. 2007;110(5):1656–1663. doi: 10.1182/blood-2007-03-081240. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Kobayashi N, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20(6):1271–1280. doi: 10.1093/emboj/20.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13(8):1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 59.Nojiri H, Takaku F, Tetsuka T, Motoyoshi K, Miura Y, Saito M. Characteristic expression of glycosphingolipid profiles in the bipotential cell differentiation of human promyelocytic leukemia cell line HL-60. Blood. 1984;64(2):534–541. [PubMed] [Google Scholar]
- 60.Nojiri H, Takaku F, Ohta M, Miura Y, Saito M. Changes in glycosphingolipid composition during differentiation of human leukemic granulocytes in chronic myelogenous leukemia compared with in vitro granulocytic differentiation of human promyelocytic leukemia cell line HL-60. Cancer Research. 1985;45(12):6100–6106. [PubMed] [Google Scholar]
- 61.Neville DC, et al. Analysis of fluorescently labeled glycosphingolipid-derived oligosaccharides following ceramide glycanase digestion and anthranilic acid labeling. Anal Biochem. 2004;331(2):275–282. doi: 10.1016/j.ab.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 62.Mellor HR, Adam A, Platt FM, Dwek RA, Butters TD. High-performance cation-exchange chromatography and pulsed amperometric detection for the separation, detection, and quantitation of N-alkylated imino sugars in biological samples. Anal Biochem. 2000;284(1):136–142. doi: 10.1006/abio.2000.4678. [DOI] [PubMed] [Google Scholar]
- 63.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Meth. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, Dazzi F. Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood. 2008;111(3):1173–1181. doi: 10.1182/blood-2007-03-082800. [DOI] [PubMed] [Google Scholar]
- 65.Parfitt AM, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2(6):595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.