

Abstract
Autoimmune reactions reflect an imbalance between effector and regulatory immune responses, typically develop through stages of initiation and propagation, and often show phases of resolution (indicated by clinical remissions) and exacerbations (indicated by symptomatic flares). The fundamental underlying mechanism of autoimmunity is defective elimination and/or control of self-reactive lymphocytes. Studies in humans and experimental animal models are revealing the genetic and environmental factors that contribute to autoimmunity. A major goal of research in this area is to exploit this knowledge to better understand the pathogenesis of autoimmune diseases and to develop strategies for reestablishing the normal balance between effector and regulatory immune responses.
Introduction
Autoimmune diseases are a significant clinical problem because of their chronic nature, the associated healthcare cost, and their prevalence in young populations during the prime of their working and peak reproductive years. Current therapies, such as cytokine antagonists, have shown great promise in treating many of these diseases. TNF-α antagonists have changed the course of rheumatoid arthritis, and other cytokine antagonists are showing impressive efficacy in various other diseases (1, 2). However, most of the current therapeutic agents target the terminal phase of inflammation and do not address the fundamental problems that are responsible for the initiation and progression of the autoimmune process. In most cases, this necessitates continued and sometimes life-long therapy, resulting in an increased risk of malignant and infectious complications. Tackling these diseases at their source will require an understanding of how the abnormal immune reactions arise, how they are sustained, and the intrinsic mechanisms used to suppress these responses in healthy individuals.
Autoimmune diseases vary greatly in the organs they affect and in their clinical manifestations, with some being limited to particular tissues and others being systemic or disseminated. Despite these variations, all autoimmune diseases are believed to go through sequential phases of initiation, propagation, and resolution (Figure 1). All stages of autoimmune disease are thought to be associated with a failure of regulatory mechanisms, with the resolution phase defined by a partial, and in most cases, short-term ability to restore the balance of effector and regulatory responses. Augmenting regulatory mechanisms and establishing robust and long-lived disease resolution is a goal of new therapeutic strategies.
Figure 1. Three major phases of autoimmune disease.
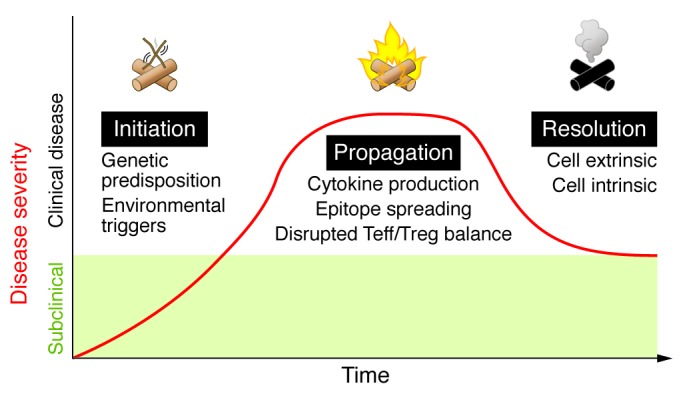
Autoimmunity is initiated by a combination of genetic predisposition and environmental triggers. Patients in the initiation phase of disease are typically unaware of clinical symptoms (subclinical). Patients present with clinical disease during the propagation phase, which is characterized by self-perpetuating inflammation and tissue damage due to cytokine production, epitope spreading, and a disrupted effector T cell/Treg (Teff/Treg) balance. Autoimmune reactions resolve with the activation of cell-intrinsic (inhibitory pathways) and cell-extrinsic (Tregs) mechanisms to limit effector responses and restore the Teff/Treg balance. Patients in this phase often suffer from relapsing and remitting disease as a result of a persistent struggle between pathogenic effector responses and regulation.
The initiation of autoimmunity: lighting the match
Because most patients with autoimmune disease develop symptoms well after the abnormal immune reactions begin, it is often difficult to pinpoint the factors responsible for the initiation of disease. Although animal models are informative, there are in fact few models of spontaneous autoimmunity that reliably mimic the human disorders. Nevertheless, studies using existing models, as well as genetic and other analyses, are beginning to reveal some of the abnormalities that account for the early steps in the autoimmune reaction.
Autoimmune diseases, like many other complex disorders, are believed to arise from a combination of genetic and environmental factors. A simple hypothesis is that polymorphisms in various genes result in defective regulation or reduced threshold for lymphocyte activation, and environmental factors initiate or augment activation of self-reactive lymphocytes that have escaped control and are poised to react against self-constituents (Figure 2). Some of these genetic factors and environmental influences are beginning to be identified.
Figure 2. Genetic susceptibility, environmental stimuli, and defective regulation are responsible for initiating autoimmunity.
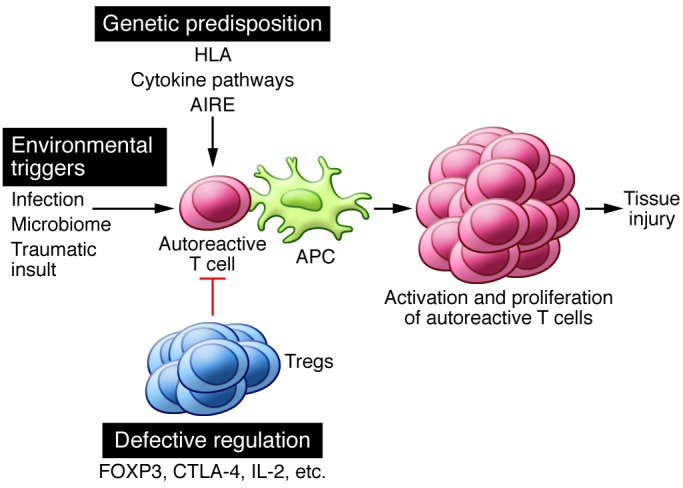
Genetic polymorphisms in immune-related genes (including HLA, cytokines/receptors, and those involved in central tolerance) may lower the threshold for the activation of autoreactive T cells. Environmental triggers such as infection, the microbiome, and tissue injury generate a proinflammatory environment that supports the activation of autoreactive lymphocytes. Tregs normally function to suppress autoreactive T cells, but defects in development, stability, or function may render these cells dysfunctional and unable to control autoreactive T cell responses. Alone or in combination, these factors can contribute to the escape, activation, and proliferation of autoreactive lymphocytes that result in tissue injury and clinical disease.
Genetic susceptibility to autoimmune diseases.
A large number of genome-wide association studies have suggested a role for numerous genetic polymorphisms in different autoimmune diseases (3, 4). Most of the polymorphisms are located in regulatory regions of genes whose products are believed to play roles in immune responses. The contribution of each gene to a particular disease, as indicated by the odds ratio, is small, and it is likely that multiple polymorphisms contribute to disease development in individual patients. However, it has proved difficult to define the role of most of these polymorphisms in the breakdown of tolerance to self-antigens and the development of autoimmunity. For instance, of all the genes associated with autoimmune diseases, the strongest associations, and the ones that have been known for the longest time, are with particular HLA alleles (5). Nevertheless, it is still not definitively known how different HLA alleles contribute to any autoimmune disease. It is unlikely that a disease-associated allele is especially efficient at displaying the autoantigens targeted by self-reactive T cells because most HLA alleles are capable of presenting self-antigens even in healthy individuals. Additionally, most healthy individuals have autoreactive T cells that escape thymic deletion (6, 7). The problem of using knowledge of the genes involved to elucidate the pathogenesis of autoimmune diseases is much more daunting for other polymorphisms with odds ratios far lower than those for HLA alleles.
Cytokine and cytokine receptor genetic polymorphisms have been linked to many different autoimmune diseases. Perhaps the best example of this is IL23R. IL-23 is a cytokine that augments the proinflammatory capacity of Th17 cells (8). Genetic polymorphisms in IL23R have been discovered in ankylosing spondylitis, Behcet’s disease, Crohn’s disease, psoriasis, and ulcerative colitis (9). Accordingly, inflammatory Th17 cells have been associated with tissue damage in all of these diseases, and targeting these pathways with monoclonal antibodies specific for either p40 (a subunit of IL-23) or IL-17A has shown efficacy in almost all of these disorders (10, 11). Thus, genetic polymorphisms in IL23R have in some cases correlated quite nicely with responses to targeted anti-cytokine therapies.
Whereas a predisposition to develop most human autoimmune diseases is thought to result from polymorphisms of multiple genes involved in immune function, there are rare examples in which genetic alterations in a single gene result in fulminant autoimmunity. Perhaps the two best examples of monogenetic autoimmune diseases are autoimmune polyendocrine syndrome (APS) and immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. These diseases directly result from mutations in AIRE and FOXP3, respectively (12, 13), leading to catastrophic dysfunction in central (APS) and peripheral (IPEX) tolerance. Another example is autoimmune lymphoproliferative syndrome, a rare lymphoproliferative disorder caused by mutations in Fas or Fas ligand, or in caspases downstream of Fas signaling. These mutations result in a defective Fas-mediated apoptotic pathway and chronic lymphoproliferation causing lymphadenopathy, splenomegaly, and autoimmune cytopenias (14). Discovery of the single genes responsible for these disorders has greatly contributed to our understanding of the cellular and molecular pathways that are dysfunctional in many autoimmune diseases.
Environmental triggers for autoimmunity.
Infections have long been suspected to trigger autoimmune reactions (15, 16). Multiple theories have been proposed to explain this association, including epitope spreading, antigenic complementarity, and excessive innate/pattern recognition receptor activation. For example, evidence of EBV infection in postmortem brain tissue has been associated with MS but not other inflammatory disorders (17). Additionally, systemic infections have been reported to trigger relapses in patients with relapsing-remitting MS through enhancement of myelin-specific T cell responses (15). Another example of the association of infections with autoimmunity is that of periodontal infections and rheumatoid arthritis (18). In contrast, infections are also postulated to protect against some autoimmune diseases. For example, infection of germ-free mice with Bacteroides fragilis has been reported to protect against experimental autoimmune encephalomyelitis, the mouse model of MS, through induction of Treg cells (19). Additionally, a higher incidence of MS and type 1 diabetes is correlated with a decreased number of infections in developed countries (20).
Recent interest has focused on the possible role of the microbiome in influencing local and systemic immune responses. Much of the emphasis has been on the gut microbiome. It is now believed that inflammatory bowel disease (IBD) is initiated by dysregulated and exaggerated immune responses to intestinal commensal microbes. In fact, the major manifestations of IBD may be caused by antimicrobial immune reactions and not by true autoimmunity (i.e., directed at tissue self-antigens). There are also several studies in mice that implicate commensal microbes in autoimmune disease, including type 1 diabetes (reviewed in ref. 21).
A well-recognized nonmicrobial environmental trigger is UV irradiation for cutaneous lupus. A possible explanation for this connection is that UV radiation induces apoptotic death of many cell types and increases the burden of nuclear antigens, especially if the dead cells cannot be efficiently cleared (22). It has been suggested that low-level natural cell death in tissues is a mechanism for maintaining peripheral tolerance to tissue antigens through tolerance-promoting dendritic cell populations (23). It is plausible that lupus patients have a genetic predisposition for this system to become easily overwhelmed and are thus unable to maintain tolerance in the presence of continual UV exposure.
Defective regulation as the cause of autoimmunity.
If failure of self-tolerance is the fundamental abnormality in autoimmune diseases, the central question becomes — which mechanisms of tolerance fail in specific diseases, and why? In patients with SLE, defects in deletion of immature B cells in the bone marrow, in receptor editing, and in control of mature B cells in peripheral tissues have all been proposed (24). In humans with SLE, mature naive B cells can produce autoantibodies even before encounter with antigen, suggesting that defects in early B cell tolerance checkpoints may contribute to disease development (24).
In T cell–dependent inflammatory autoimmune diseases, there is likely an imbalance between effector T cells and functional Treg cells. Mouse models of autoimmunity support the pathogenic importance of such an imbalance (25). It also appears likely that decreases in the number of functional Tregs, or resistance of effector T cells to regulation, play a role in the initiation of human autoimmune disease. However, the data from patients with different autoimmune diseases tend to be variable and often inconsistent. This is most likely due to the fact that for most autoimmune diseases, Treg cells function in the tissue targeted by the autoimmune process (not in the blood) and mechanistic studies are difficult to perform with these cell populations, given the limited accessibility of human tissue. Furthermore, when adequate numbers of these cells are obtained, their function (or lack thereof) is usually assessed by in vitro assays that may not accurately recapitulate their functional capacity in vivo. Longitudinal studies of effector and Treg cells specific for target self-antigens in human disease remain a considerable technical challenge. It is not obvious that population assays will uncover defects that, almost by definition, must affect only a small fraction of lymphocyte clones, particularly those that are specific for self-antigens involved in specific diseases.
In addition to a failure of regulation underlying the development of autoimmunity, other factors have been proposed. There is experimental evidence that autoimmune reactions are associated with abnormalities in the types of self-antigens that are displayed to the immune system. For instance, atypical presentation of extracellularly derived peptides or denatured proteins by antigen-presenting cells (APCs) can give rise to peptide/MHC complexes that are distinct from those generated normally inside APCs and are thus capable of activating potentially pathogenic T cells (26). This unconventional activation of potentially self-reactive T cells may arise from either the recognition of conformational isomers of peptide/MHC or from a differential binding register of a peptide within the groove of the MHC molecule (27, 28). It is, however, unknown whether such altered self-antigens are triggers for autoimmunity in human diseases.
In addition to altered peptide/MHC recognition, an early innate immune response can be a trigger for autoimmunity (29). For example, mice that lack the ubiquitin modifying enzyme A20 develop lethal autoimmunity due to unregulated MyD88-independent TLR signals (30). Additionally, Yaa mice, which have a genetic duplication of Tlr7, develop a spontaneous SLE-like syndrome due to increased B cell recognition of nucleolar antigens (31). The contribution of excessive or aberrant innate immune responses to human autoimmune diseases remains to be defined.
Despite our current lack of understanding of the initiation of autoimmune disease in humans, models that attempt to recapitulate this process with human tissue in vivo are actively being pursued and constitute an area of exciting new investigation. Indeed, transplantation of apparently normal skin from psoriasis patients onto immunodeficient mice (i.e., humanized mice) has been shown to induce skin changes consistent with the initiation of psoriasis (32), and this type of model is currently being explored in other human autoimmune diseases.
The propagation of autoimmune reactions: fanning the flames
Most patients present with clinical disease during the propagation phase, which is characterized by progressive inflammation and tissue damage. The self-perpetuating nature of autoimmune diseases may help to explain why these conditions reach the propagation phase. First, the self-antigens that drive the reaction can obviously not be eliminated. This problem is compounded by the emergence of new antigenic epitopes as a result of tissue damage and alterations in self-proteins, the phenomenon known as epitope spreading. Epitope spreading sets up a vicious cycle in which newly created antigenic epitopes activate more lymphocytes of different specificities and recruit these cells into the reaction, leading to more tissue damage and the emergence of even more novel epitopes targeted by autoreactive lymphocytes. Second, the autoimmune reaction creates an inflammatory environment in which multiple immune cells interact to produce cytokines and other mediators that amplify the reaction, creating a catastrophic inflammatory loop. Consistent with this notion is the finding that type I interferons, a product of plasmacytoid dendritic cells that is produced during inflammatory reactions, is a biomarker for the progression of SLE and may be involved in the propagation of this disease (33).
Possibly the best evidence of the dominant drivers of disease propagation in humans is clinical data showing that targeting of specific cytokines can result in a complete amelioration of the propagation phase of disease. TNF-α inhibitors are highly efficacious for the treatment of both rheumatoid arthritis and psoriatic arthritis, which suggests a major role for this cytokine in the propagation of inflammation in these diseases (34, 35). However, despite the success of TNF-targeted therapies, one-third of patients do not respond and up to 50% of patients will eventually discontinue previously effective therapy due to adverse effects or lack of efficacy (36). Ustekinumab, a human monoclonal antibody that blocks the cytokine subunit p40, has been shown to be efficacious for the treatment of both psoriasis and IBD (10). The cytokines IL-12 (a powerful inducer of Th1 responses) and IL-23 (promoter of Th17 differentiation) share p40. The efficacy of p40 blockade indicates that Th1 cells, Th17 cells, or both play major roles in propagating these diseases. Consistent with the role of Th17 cells in mediating disease pathology in psoriasis, the human anti–IL-17A monoclonal antibody brodalumab has recently shown excellent results in a phase II trial (11). IL-17 antagonists are also showing efficacy in ankylosing spondylitis and rheumatoid arthritis and are being tested in several other inflammatory diseases (37, 38). Although results of cytokine antagonism for many patients have been encouraging, not all patients respond to a given class of therapy; this suggests that subgroups of patients may differ with respect to the dominant cytokines that drive their disease.
Finally, and perhaps most importantly, the propagation of autoimmunity may be related to the progressively increasing ratio of effector to regulatory cells. Once a pathologic immune reaction starts, there is increasing accumulation of effector T cells in the tissues that are the main drivers of the autoimmune reaction. This may be accompanied by a relative decline in Treg number, or increased numbers of dysfunctional Tregs (39–41). Paradoxically, the inflamed skin of patients with psoriasis has increased numbers of Tregs; however, these cells appear to function abnormally, in that they are capable of producing increased amounts of IL-17 (42, 43). It is not clear whether production of seemingly pathogenic cytokines from Tregs in inflamed tissues significantly contributes to disease pathology. Regardless of the etiology, numerous animal models have shown that an imbalance between these injurious and protective cell populations will inevitably lead to worsening inflammation and tissue damage (44–46). It follows that therapeutic strategies to reset the effector T cell/Treg balance are an exciting new approach to treat autoimmune disease. These studies provide proof of principle in humans that this imbalance, at least in part, enables disease propagation. Low-dose IL-2 therapy is thought to preferentially augment Treg numbers and/or function and has been shown to be of clinical benefit in patients with chronic graft-versus-host disease (GVHD), autoimmune vasculitis, and alopecia areata (47–49). In addition, adoptive transfer of large numbers of ex vivo expanded Tregs has been shown to be efficacious in preventing acute GVHD and is currently being tested for the treatment of type 1 diabetes (50).
The resolution of autoimmunity: putting out the fire
The control of autoimmune reactions likely involves the induction and activation of regulatory mechanisms that limit the effector response and restore the effector/regulatory balance. The most important of these control mechanisms appear to be Tregs. Tregs, which are best identified by expression of the transcription factor FOXP3, develop in the thymus and peripheral (secondary) lymphoid organs. Both populations of Tregs likely acquire their potent suppressive function only after encountering their target antigen, which in most cases is likely to be a self-antigen. Some of these activated Tregs may survive in tissues as long-lived populations that are capable of controlling autoimmune reactions in that tissue, termed “effector Tregs” or “effector memory Tregs” (43, 46, 51, 52). In mouse models, there is convincing evidence that the induction of systemic or tissue-specific autoimmune inflammation is followed by the activation of Tregs and control of the inflammation, resulting in a cycle of disease and resolution. Mechanistically, it has been shown that IL-2 produced by effector T cells in the initial phases of the autoimmune response drives the activation and expansion of preexisting Tregs and also plays a role in the development of new Tregs, explaining at least in part why effector responses may be followed closely by a wave of Treg-mediated suppression, as seen in many animal models of autoimmunity (53). If the generation or maintenance of these Tregs is defective, the inflammation fails to resolve, resulting in progressive, nonresolving disease.
In addition to Tregs, other mechanisms that have been proposed to limit autoimmune reactions include the activation of various inhibitory receptors. For example, following activation, T cells begin to express two receptors of the CD28 family, CTLA-4 and PD-1, which function to suppress various immune responses. Genetic deletion of these receptors in mice and blockade in humans (for cancer immunotherapy) are associated with autoimmune inflammation of variable severity. Two recent studies have described immune dysregulation in humans who inherit one mutant allele of CTLA-4 (54, 55). These patients developed multiple autoimmune clinical features and extensive CD4+ T cell infiltration into different organs. Although Treg numbers were elevated in these patients, CTLA-4 protein expression in Tregs was markedly decreased and suppressive function was impaired. It is currently debated whether CTLA-4 and other inhibitory receptors exert their inhibitory effects primarily in a cell intrinsic (Teffs) or extrinsic (Tregs) manner, as both Tregs and Teffs express these receptors. Whether such checkpoints become operational in autoimmune reactions or fail in progressive autoimmune diseases are open questions. B lymphocytes also express inhibitory receptors, notably CD22 and FcγRII, and deletion of either in mice results in lupus-like autoimmunity (56, 57). To what extent these pathways and their deficiencies contribute to B cell autoimmunity in humans is also not established. Nonetheless, targeted therapies that specifically enhance inhibitory pathways in lymphocytes remain a conceptually attractive approach to augment disease resolution in an attempt to effectively treat human autoimmune disease.
Future prospects
Solving the enigma of human autoimmunity will require translating the vast amount of knowledge that has accumulated from animal models for the clinic. Several challenges inherent in this translation to the clinic have become apparent. First, there needs to be a renewed emphasis on studying possible defects in autoantigen-specific T and B cells in humans. Analyses of bulk lymphocyte populations are unlikely to reveal defects that largely affect cells of only a few specificities. This may be especially true in organ-specific autoimmune diseases. Studying antigen-specific lymphocytes will require new technologies that can be readily applied to humans. Second, the definitions of biological aberrations need to be complemented by analysis of molecular pathways. Although our understanding of the molecular basis of immune regulation is still incomplete, the progress in this area in the last decade has been remarkable, especially the elucidation of Tregs and checkpoints associated with inhibitory receptors. Applying this expanding knowledge to humans should be a high priority. Finally, high-throughput, large-scale approaches should be combined with more targeted analyses of individual pathways. Although modern technology has ushered in the era of “big data” and made it feasible to collect such data from large cohorts of patients, discovery of new mechanisms and paradigms has largely relied on studying candidate molecules and their functions in depth.
Understanding and successfully treating human autoimmune disease continues to be a significant challenge, but we have made incredible progress. Targeted cytokine inhibition has launched the age of biologic therapy for autoimmune diseases, and it seems that we are still in the early stages with many more exciting new treatments being developed. Although we have yet to be able to successfully prevent the match from being lit, we are making serious headway in dampening the flames and in some cases extinguishing the fire.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(6):2228–2233. doi:10.1172/JCI78088.
References
- 1.Kim SY, Solomon DH. Tumor necrosis factor blockade and the risk of viral infection. Nat Rev Rheumatol. 2010;6(3):165–174. doi: 10.1038/nrrheum.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Targownik LE, Bernstein CN. Infectious and malignant complications of TNF inhibitor therapy in IBD. Am J Gastroenterol. 2013;108(12):1835–1842. doi: 10.1038/ajg.2013.294. [DOI] [PubMed] [Google Scholar]
- 3.Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell. 2010;140(6):791–797. doi: 10.1016/j.cell.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hafler D, Housley W, Marson A. Genetics of autoimmunity. J Clin Invest. 2015;125(6):2234–2241. doi: 10.1172/JCI78086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goris A, Liston A. The immunogenetic architecture of autoimmune disease. Cold Spring Harb Perspect Biol. 2012;4(3):a007260. doi: 10.1101/cshperspect.a007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liblau R, et al. T cell response to myelin basic protein epitopes in multiple sclerosis patients and healthy subjects. Eur J Immunol. 1991;21(6):1391–1395. doi: 10.1002/eji.1830210610. [DOI] [PubMed] [Google Scholar]
- 7.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immuno-dominant myelin basic protein epitope in multiple sclerosis. Nature. 1990;346(6280):183–187. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]
- 8.Ghoreschi K, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467(7318):967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandenbroeck K. Cytokine gene polymorphisms and human autoimmune disease in the era of genome-wide association studies. J Interferon Cytokine Res. 2012;32(4):139–151. doi: 10.1089/jir.2011.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott M, et al. Ustekinumab: lessons learned from targeting interleukin-12/23p40 in immune-mediated diseases. Ann N Y Acad Sci. 2009;1182:97–110. doi: 10.1111/j.1749-6632.2009.05070.x. [DOI] [PubMed] [Google Scholar]
- 11.Papp KA, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 12.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 13.Finnish-German APECED Consortium An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. 1997;17(4):399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 14.Fisher GH, et al. Dominant interfering fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 15.Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11(12):807–822. doi: 10.1038/nri3095. [DOI] [PubMed] [Google Scholar]
- 16.Root-Bernstein R, Fairweather D. Complexities in the relationship between infection and autoimmunity. Curr Allergy Asthma Rep. 2014;14(1):407. doi: 10.1007/s11882-013-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serafini B, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204(12):2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikuls TR, et al. Periodontitis and porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(5):1090–1100. doi: 10.1002/art.38348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochoa-Repáraz J, et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol. 2010;185(7):4101–4108. doi: 10.4049/jimmunol.1001443. [DOI] [PubMed] [Google Scholar]
- 20.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathis D, Benoist C. Microbiota and autoimmune disease: the hosted self. Cell Host Microbe. 2011;10(4):297–301. doi: 10.1016/j.chom.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. 2014;47(2):148–162. doi: 10.1007/s12016-013-8403-x. [DOI] [PubMed] [Google Scholar]
- 23.Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191(3):411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yurasov S, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bluestone JA, Tang Q, Sedwick CE. T regulatory cells in autoimmune diabetes: past challenges, future prospects. J Clin Immunol. 2008;28(6):677–684. doi: 10.1007/s10875-008-9242-z. [DOI] [PubMed] [Google Scholar]
- 26.Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol. 2012;12(10):721–728. doi: 10.1038/nri3294. [DOI] [PubMed] [Google Scholar]
- 27.Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 2011;208(12):2375–2383. doi: 10.1084/jem.20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11(4):350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. 2011;243(1):61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med. 2008;205(2):451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 32.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-α. J Exp Med. 2004;199(5):731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Papoutsaki M, Costanzo A. Treatment of psoriasis and psoriatic arthritis. BioDrugs. 2013;27(suppl 1):3–12. doi: 10.1007/BF03325637. [DOI] [PubMed] [Google Scholar]
- 35.Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):578–582. doi: 10.1038/nrrheum.2009.181. [DOI] [PubMed] [Google Scholar]
- 36.Thalayasingam N, Isaacs JD. Anti-TNF therapy. Best Pract Res Clin Rheumatol. 2011;25(4):549–567. doi: 10.1016/j.berh.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 37.Baeten D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9906):1705–1713. doi: 10.1016/S0140-6736(13)61134-4. [DOI] [PubMed] [Google Scholar]
- 38.Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72(suppl 2):ii116–ii123. doi: 10.1136/annrheumdis-2012-202371. [DOI] [PubMed] [Google Scholar]
- 39.Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–859. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyara M, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005;175(12):8392–8400. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- 41.Kukreja A, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. 2002;109(1):131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol. 2011;131(9):1853–1860. doi: 10.1038/jid.2011.139. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez Rodriguez R, et al. Memory regulatory T cells reside in human skin. J Clin Invest. 2014;124(3):1027–1036. doi: 10.1172/JCI72932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Q, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28(5):687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16(2):249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 46.Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480(7378):538–542. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saadoun D, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365(22):2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- 48.Castela E, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol. 2014;150(7):748–751. doi: 10.1001/jamadermatol.2014.504. [DOI] [PubMed] [Google Scholar]
- 49.Koreth J, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Ianni M, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 51.Smigiel KS, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211(1):121–136. doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol Rev. 2014;259(1):40–59. doi: 10.1111/imr.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med. 2005;202(10):1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–1627. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jovanovic V, Dai X, Lim YT, Kemeny DM, MacAry PA. Fcγ receptor biology and systemic lupus erythematosus. Int J Rheum Dis. 2009;12(4):293–298. doi: 10.1111/j.1756-185X.2009.01426.x. [DOI] [PubMed] [Google Scholar]
- 57.Dörner T, Shock A, Smith KGC. CD22 and autoimmune disease. Int Rev Immunol. 2012;31(5):363–378. doi: 10.3109/08830185.2012.709890. [DOI] [PubMed] [Google Scholar]