

ABSTRACT
Clostridium difficile is a major nosocomial pathogen and the principal causative agent of antibiotic-associated diarrhea. The toxigenic C. difficile strains that cause disease secrete virulence factors, toxin A and toxin B, that cause colonic injury and inflammation. C. difficile toxins have no export signature and are secreted by an unusual mechanism that involves TcdE, a holin-like protein. We isolated a TcdE mutant of the epidemic R20291 strain with impaired toxin secretion, which was restored by complementation with functional TcdE. In the TcdE open reading frame (ORF), we identified three possible translation start sites; each translated isoform may play a specific role in TcdE-controlled toxin release. We created plasmid constructs that express only one of the three TcdE isoforms and complemented the TcdE mutant with these isoforms. Western blot analysis of the complemented strains demonstrated that TcdE is translated efficiently from the start codon at the 25th and 27th positions in the predicted ORF, producing proteins with 142 amino acids (TcdE142) and 140 amino acids (TcdE140), respectively. TcdE166 was not detected when expressed from its own ribosomal binding site (RBS). The effects of all three TcdE isoforms on C. difficile cell viability and toxin release were determined. Among the three isoforms, overexpression of TcdE166 and TcdE142 had a profound effect on cell viability compared to the TcdE140 isoform. Similarly, TcdE166 and TcdE142 facilitated toxin release more efficiently than did TcdE140. The importance of these variations among TcdE isoforms and their role in toxin release are discussed.
IMPORTANCE C. difficile is a nosocomial pathogen that has become the most prevalent cause of antibiotic-associated diarrhea in North America and in several countries in Europe. Most strains of C. difficile produce two high-molecular-weight toxins that are regarded as the primary virulence factors. The mechanism by which these large toxins are secreted from bacterial cells is not yet clear but involves TcdE, a holin-like protein. In this work, we show that TcdE could be translated from three different start codons, resulting in the production of three TcdE isoforms. Furthermore, we investigated the role of these isoforms in toxin release and cell lysis in C. difficile. An understanding of TcdE-dependent toxin secretion may be helpful for the development of strategies for preventing and treating C. difficile infections.
INTRODUCTION
Clostridium difficile is a major nosocomial pathogen and the principal causative agent of antibiotic-associated diarrhea. The economic impact of C. difficile infection (CDI) is profound and represents a major financial drain on the health care system (1). Pathogenic C. difficile strains produce two toxins, toxin A (enterotoxin) and toxin B (cytotoxin), which belong to the family of large clostridial glucosylating toxins (LCGTs) (2). Other members of this family include Clostridium sordellii lethal (TcsL) and hemorrhagic (TcsH) toxins, Clostridium novyi alpha-toxin (TcnA), and Clostridium perfringens TpeL toxin. These toxins act as glucosyl transferases and inactivate small GTPases, such as Rho, Rac, and Cdc42, in target cells by glucosylation, which leads to the disruption of vital signaling pathways and cell death (3–5). LCGTs are secreted by bacteria during infection and directly influence disease severity. Most extracellular proteins carry an N-terminal or C-terminal signal peptide, such as the Tat signal peptide or another clearly definable signal that directs the secretion process (6). Interestingly, LGCTs are secreted without any recognizable secretion signals and without lysing the bacterial cell (7), suggesting that they are secreted by an unconventional mechanism.
In C. difficile, the toxin genes lie within the pathogenicity locus (PaLoc) (8). In addition to the toxin-encoding genes, the locus also contains the tcdR, tcdC, and tcdE genes (see Fig. S1A in the supplemental material). TcdR is an alternative sigma factor that specifically directs transcription from the toxin promoters as well as its own promoter, and TcdC is an antagonist of TcdR (9–14). We previously demonstrated that TcdE is necessary for toxin secretion (15).
TcdE is predicted to contain three transmembrane domains (TMDs), a short hydrophilic stretch at the N terminus, and a series of charged residues at the C terminus (see Fig. S1B in the supplemental material) (16). The primary sequence and structural features and primary sequence similarities strongly suggest that TcdE is a member of the class I family of holins (17). This family of holins includes the lambda phage S protein, and we previously demonstrated that TcdE could complement a lambda phage with a defective S holin (15). Holins are small membrane proteins encoded by double-stranded DNA phages that are required for programmed host cell lysis after intracellular phage development (17–21). Lambda phage S is produced in two forms, S105 and S107, which are translated from a single open reading frame (ORF) by an alternate translation start mechanism. The ribosome initiates translation from the annotated start site to form a 107-amino-acid S protein (S107) and from the nearest downstream AUG codon to translate a protein with 105 amino acid residues (S105) (22–24). The S105 protein has holin activity, whereas S107, which has two additional amino acid residues at the N terminus, retards lysis by interfering with the activity of S105 and thus possesses antiholin activity (25). By maintaining a defined proportion of holin (S105) and antiholin (S107), lambda phage controls the timing of lysis and maximizes progeny production before release (26). In the tcdE ORF, there are three possible translational start sites that may result in three different TcdE isoforms of different sizes (15, 27). The presence of different TcdE isoforms in certain proportions may be necessary for efficient toxin release in C. difficile.
In this study, we isolated a tcdE mutant from the high-toxin-producing R20291 strain and confirmed the role of TcdE in toxin release in this strain. We complemented the R20291::tcdE mutant with various mutant tcdE constructs to identify possible translation start sites in the tcdE ORF. Our results indicate that tcdE is efficiently translated from the 25th and/or 27th AUG codon in the predicted ORF, resulting in two major TcdE isoforms. The effects of all three TcdE isoforms on C. difficile cell viability and toxin release were determined and are discussed.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. difficile strains R20291 (28) and CD646 (29) and tcdE mutants were grown anaerobically (10% H2, 10% CO2, and 80% N2) in TY (tryptose-yeast extract) broth or TY agar, as described previously (11). C. difficile strains carrying pRPF185-derived plasmids were grown in TY medium supplemented with 15 μg/ml thiamphenicol. Escherichia coli strain S17 (30), which was used for conjugation, was cultured aerobically in LB medium. E. coli cultures were supplemented with 30 μg/ml chloramphenicol or 100 μg/ml ampicillin as necessary. Plasmids were constructed by using standard procedures.
C. difficile toxin assay.
Culture supernatants were collected and filtered, and the cell pellets were resuspended in 10 mM Tris buffer (pH 8.0) containing a protease inhibitor cocktail (Roche, Mannheim, Germany). The cytosolic contents were obtained by sonication of the cells, followed by brief centrifugation to remove unbroken cells and cell debris. The total protein concentration was determined by using the Bio-Rad protein assay reagent. Equal amounts of cytosolic and supernatant proteins were assayed for their relative toxin contents by using an enzyme-linked immunosorbent assay (ELISA) kit (Alere Wampole C. difficile Tox A/B) according to the manufacturer's directions.
Generation of TcdE-specific antibodies.
A female New Zealand rabbit was used to raise TcdE-specific antibodies. A peptide with the TcdE C-terminal sequence, GLPVPKRLKEKIAILLDAMTDEMNAKDEK, was synthesized by Selleck Chemicals Services (Houston, TX). One milligram of this synthetic peptide conjugated with keyhole limpet hemocyanin (KLH) was used for the first immunization on day 1, followed by two boosters on days 30 and 45. Immunizations and blood collections were performed by Lampire Biologicals, PA, in their animal care facilities. Two weeks after the second booster, blood was collected from the immunized animal. TcdE-specific antibodies were enriched after preadsorbing the collected serum against total proteins from the R20129::tcdE mutant strain.
Complementation of C. difficile TcdE mutants.
The predicted tcdE gene was amplified by using primers ORG102 and ORG103 to introduce BamHI and SacI restriction sites and a C-terminal 6×His tag (see Table S2 in the supplemental material). The PCR product was cloned into the pGEM-T Easy vector, and the resulting plasmid was used as a template for site-directed mutagenesis to introduce mutations in the TcdE coding region. After confirmation by sequencing, the mutated tcdE ORFs were excised from pGEM-T Easy and cloned into pRPF185 under a tetracycline-inducible promoter. The various mutations introduced are presented in Fig. 2A and 3A, while the primers and resulting plasmids are presented in Tables S1 and S2 in the supplemental material. To express TcdE with 166 amino acids (TcdE166) from an altered ribosomal binding site (RBS), tcdE166 was PCR amplified from plasmid pRG126 by using primers ORG107 and ORG102. Forward primer ORG107 introduces the gus (beta-glucuronidase) RBS upstream of the tcdE166 ORF. The amplified product was cloned under the tetracycline-inducible promoter in the pRPF185 vector to create pRGL267.
FIG 2.

Expression of TcdE isoforms in C. difficile. (A) TcdE sequence. All possible translation start sites are indicated as Met1, Met25, and Met27; the potential Shine-Dalgarno (SD) sequences are underlined, and mutated nucleotides in the specified constructs are in boldface type. (B) Cytoplasmic membrane protein analysis of C. difficile strains expressing various TcdE isoforms. Western blots were probed with anti-TcdE, anti-6×His, or anti-ATPase beta-subunit antibodies.
FIG 3.

FACS analysis of C. difficile cells expressing TcdE isoforms for membrane damage by propidium iodide and SYTO9 staining. C. difficile R20291:: tcdE mutants carrying plasmids expressing either the TcdE WT or different TcdE isoforms were induced with 50 ng/ml ATc for 3 h and subjected to FACS analysis following PI and SYTO9 staining. Shown are data from a representative experiment of four independent experiments.
Plasmids carrying the wild-type (WT) or the mutated tcdE ORF under control of the tetracycline-inducible promoter were introduced into the R20291::tcdE mutant strain by transconjugation. As controls, the pRPF185 vector alone was introduced into the R20291 parent strain and the R20291::tcdE mutant strain. Transconjugants carrying either the tcdE-expressing plasmids or pRPF185 alone were grown overnight in TY medium supplemented with thiamphenicol. Fresh 100-ml cultures were initiated by using 1 ml of cultures grown overnight and were grown for 4 h in TY medium with thiamphenicol before induction with anhydrous tetracycline (ATc). Culture supernatants were harvested after induction to detect released toxins by an ELISA. ATc-induced cultures were harvested after induction to detect TcdE-6×His by using a 6×His antibody and a TcdE antibody, as described below.
Creation of C. difficile strains that express toxins at the early exponential stage.
The alternate sigma factor TcdR, which is essential for the transcription of toxin genes, was expressed from the tetracycline-inducible promoter. The tcdR ORF was PCR amplified from the JIR8094 strain by using primers ORG341 and ORG359 and cloned into pRPF185 by digestion with BamHI and SacI. The resulting plasmid, pRGL209, was introduced into R20291 and the R20291::tcdE mutant by conjugation to produce the transconjugant strains RGL100 and RGL101, respectively.
Western blot analysis.
Soluble membrane proteins were harvested from bacterial cultures as described previously (31), separated by SDS-PAGE, and immobilized onto a polyvinylidene difluoride (PVDF) membrane by using the semidry blot technique. Membranes were probed with antibodies specific for TcdE (1:500) followed by horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibodies. Anti-6×His-HRP-conjugated antibodies were used at a dilution of 1:5,000. The membrane with test samples was also probed with anti-ATP synthetase β-subunit antibody (32) at a dilution of 1:5,000 when necessary.
Fluorescence-activated cell sorter (FACS) analysis.
C. difficile cells were washed with equal proportions of 0.9% NaCl and stained with SYTO9 and propidium iodide (PI) for 5 min. Flow cytometry was performed immediately after staining, using a FACSCalibur instrument (Becton Dickinson, San Jose, CA) equipped with an air-cooled 15-mW argon ion laser operating at 488 nm. The green fluorescence of the SYTO9 dye (FL1) was collected by using a 530- to 630-nm-band-pass filter; the red fluorescence emitted from PI (FL3) was collected by using a 630- to 610-nm-band-pass filter. The data were analyzed by using FCS Express software from De Nova Software.
RESULTS
TcdE is required for efficient secretion of toxins A and B in C. difficile.
We recently reported that TcdE is required for the secretion of toxins in C. difficile strain JIR8094, an erythromycin-sensitive derivative of the 630 strain (15). We measured secreted and cytosol-accumulated toxins in the parent and tcdE mutant strains and demonstrated that TcdE mediates toxin secretion in C. difficile without inducing cell lysis (15). In contrast, Olling et al. demonstrated that a tcdE mutant of C. difficile strain 630E was not defective in toxin secretion and exhibited cell lysis-mediated toxin release (27). This discrepancy may be attributable to differences in the parental strains used in these studies. The rate of toxin production in C. difficile varies widely among strains. For example, ribotype 027 strains produce significantly more toxins than do strains of other ribotypes (33). We hypothesized that the effect of a tcdE mutation on toxin secretion would be clearly observed in a high-toxin-producing strain. To test this hypothesis, we created tcdE mutants of R20291 (ribotype 027) and CD646 (ribotype 078) using the ClosTron system developed by Heap and coworkers (see Fig. S2A in the supplemental material) (34, 35). We measured the secreted toxins in the supernatants of bacterial cultures at different time points using a toxin-specific ELISA (Fig. 1). Culture supernatants were also probed for cytoplasmic markers (ribosomal protein L7/L12) to detect cell lysis. The toxin secretion defect was not observed in the tcdE mutant of CD646, consistent with the results reported previously for the 630E strain (27). A rapid decrease in the optical density (OD) at 600 nm (Fig. 1) and detection of the cytoplasmic ribosomal L7/L12 protein in the culture supernatants confirmed cell lysis of the CD646 strain (see Fig. S2B in the supplemental material). Due to this rapid cell lysis and low level of toxin production, we could not measure the true effect of the TcdE mutation on the CD646 strain. However, for R20291, a clear difference in toxin secretion between the R20291::tcdE mutant and the parent strain was observed (Fig. 1). The absence of the ribosomal protein L7/L12 in the culture supernatant confirmed the absence of cell lysis in R20291 cultures at 8 and 12 h (see Fig. S2B in the supplemental material). The clear difference in the secreted toxin titers of the tcdE mutant and parent R20291 strains at the late exponential growth phases (Fig. 1) indicated that TcdE is important for toxin secretion in C. difficile, at least in the R20291 strain. We did not detect a significant difference in total toxin contents (i.e., from culture supernatants and cytosolic proteins) between the mutant and parent strains, confirming that the defect is mostly in the secretion of the toxins (see Fig. S3 in the supplemental material). As we clearly observed the toxin secretion defect in the R20291 background, we used the R20291::tcdE mutant strain for the remainder of our studies.
FIG 1.
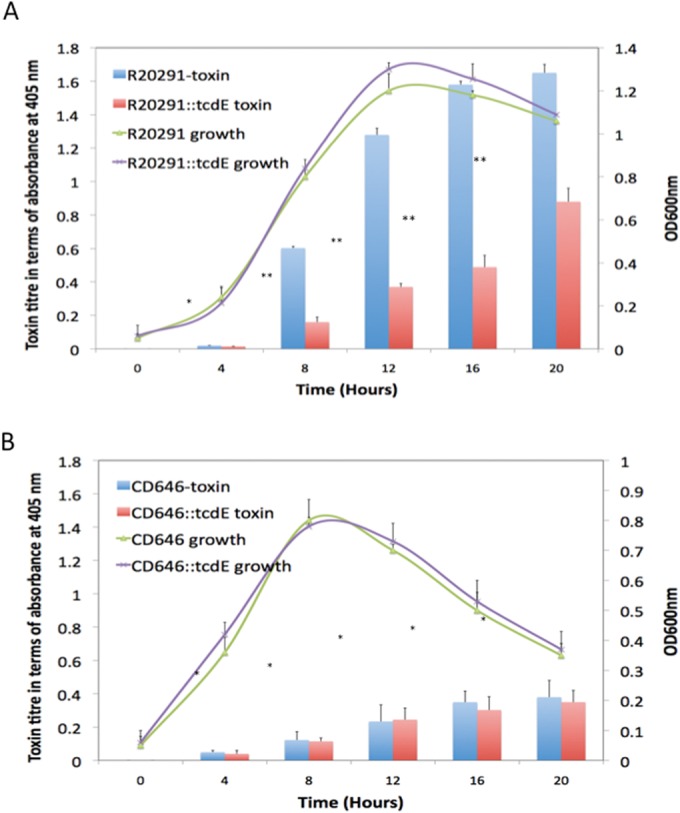
Characterization of tcdE mutants of C. difficile strains R20291 and CD646. Shown are growth curves and toxin titers of the parent strains and their corresponding tcdE mutants. Bacterial cultures were harvested at different time points, and the toxins in the supernatants were quantified by an ELISA. The signal from the test was recorded as the absorbance at 405 nm. Data are expressed as the means ± standard errors from three replicative samples. Student's t test was used for statistical analysis to compare toxin titers. **, P < 0.05; *, P > 0.5.
Translation of the tcdE-predicted ORF starts only at Met25 and/or Met27.
Structural and sequence analyses suggested that TcdE is similar to class I holins (15). A well-characterized class I holin is lambda phage S. The S holin ORF can be translated from two different start codons to produce the S105 and S107 proteins, which comprise 105 and 107 amino acid residues, respectively (23). S105 is a holin, and S107 is an antiholin; together, they control the timing of lysis during the lambda phage lytic cycle (25). As with lambda S, more than one potential ATG start codon is present in the tcdE ORF (Fig. 2A). Three potential initiator methionines are present at positions 1 (Met1), 25 (Met25), and 27 (Met27) of the putative full-length TcdE protein. The specific mutations indicated in Fig. 2A were introduced in the tcdE ORF to express the TcdE166, TcdE142, or TcdE140 isoform. Translation starting at Met1 would be expected to result in the production of the TcdE166 protein, while translation starting at Met25 or Met27 would result in the production of the TcdE142 or TcdE140 protein, respectively. TcdE WT refers to the expression of the tcdE ORF without any mutations, in which more than one TcdE isoform may be expressed. Different tcdE ORFs with six histidine amino acid residues at the C terminus were cloned under the control of the tetracycline-inducible promoter in the pRPF185 vector (36). The resulting plasmid constructs were introduced into the R20291::tcdE mutant through conjugation. Membrane proteins were harvested from cultures of selected transconjugants and probed with anti-6×His- or TcdE-specific antibodies to detect different TcdE isoforms. ATPase was detected in the same blots as a loading control. Western blotting revealed that the tcdE ORF is translated from the Met25 and Met27 codons in C. difficile to produce TcdE142 and TcdE140, respectively (Fig. 2B). Both TcdE isoforms were detectable when expressed from the tetracycline-inducible promoter. Surprisingly, we did not detect TcdE166, the isoform resulting from translation initiating from the Met1 codon. This result suggests that the TcdE166 isoform either is not translated or is translated in C. difficile at levels below the detectable threshold. In the TcdE WT construct, the tcdE ORF was expressed without any mutation, and we expected to detect more than one TcdE isoform. By comparing the sizes of TcdE142, TcdE140, and the TcdE WT, it is clear that TcdE166 is absent or present at undetectable levels and that the major expressed TcdE isoform is either TcdE140 or a mixture of both TcdE140 and TcdE142, which are hard to separate if coexpressed (Fig. 2B).
In tcdE, a potential RBS (GGTGG) is present 10 nucleotides (nt) upstream of the Met1 codon (see Fig. S4A in the supplemental material). Further analysis revealed potential start codons 3 nt and 6 nt downstream of the RBS (underlined in red in Fig. S4A in the supplemental material). However, translation from these codons would result in a frameshift, with the production of a small peptide with 4 or 5 amino acids instead of TcdE166. We hypothesized that bacteria use this alternate translation strategy to limit the production of TcdE166. If TcdE166 is a potent holin protein, limited production may be important to minimize its lethal effect. To test this hypothesis, we introduced a mutation in the ATG sequences (3 nt and 6 nt downstream of the RBS) and replaced them with TTA in the TcdE166 construct (see Fig. S4A in the supplemental material). The single adenine 9 bases downstream of the GGTGG sequence (indicated by a box in Fig. S4A in the supplemental material) was also deleted to prevent a potential frameshift. Western blotting demonstrated that TcdE166 is not produced, even after removal of the alternate translational start codons near Met1. Removal of the adenine that could result in a frameshift also did not result in translation of the higher-molecular-weight TcdE166 protein. From these results, we conclude that the tcdE ORF in C. difficile is not translated from the Met1 position or is translated at undetectable levels and that the GGTGG sequence upstream of the tcdE166 start codon is a weak RBS. The tcdE transcript is efficiently translated from the Met25 and/or Met27 codon of the predicted ORF, probably because of the stronger RBS (GGAGG) upstream of the tcdE140 and tcdE142 start codons.
Effect of TcdE isoforms on toxin secretion and cell viability in C. difficile.
In our previous work (15), we showed that complementation of a tcdE mutant using a multicopy plasmid (with the tcdE ORF from its own promoter) was unsuccessful. However, we successfully complemented the tcdE mutant by expressing tcdE from the tetracycline-inducible promoter (15). These results demonstrated that TcdE is lethal to C. difficile, if expressed above a certain threshold level. In the present study, we wanted to determine which TcdE isoform is responsible for inducing cell death in C. difficile. Plasmids expressing TcdE WT, TcdE166, TcdE142, and TcdE140 were introduced into the R20291::tcdE mutant by conjugation, and the transconjugants were grown to an OD at 600 nm of 0.5 before being induced with 10 ng or 50 ng/ml ATc. Three hours after induction, bacterial cultures were collected and serially diluted to enumerate the population of living cells (see Fig. S5A in the supplemental material). Induction of TcdE isoforms at a lower level (with 10 ng/ml ATc) did not result in cell death (see Fig. S5A in the supplemental material), but overexpression of the TcdE isoforms and the TcdE WT induced a certain level of cell death in C. difficile. However, their levels of lethality were significantly different from one another. Overexpression of TcdE166 and TcdE142 in C. difficile resulted in cell death, while TcdE140 had little effect on cell viability. Even though the serial dilution method was efficient, to precisely quantify the extent of cell death in TcdE-expressing C. difficile cultures, we performed FACS analysis after staining the cells with the fluorescent nucleotide-binding dyes SYTO9 and PI. SYTO9 stains live cells, whereas PI is excluded by an intact cell membrane and stains only dead cells with a compromised membrane. Cells stained with both dyes represent an injured (or dying) population (Fig. 3). Even though TcdE166 was undetectable by Western blot analysis (Fig. 2B), induction of TcdE166 resulted in cell death in nearly 15% (dead and injured) of the population (see Fig. S5B in the supplemental material). This result indicated that even a very small amount of TcdE166 is enough to trigger membrane damage in C. difficile. For TcdE140 and TcdE142, induction of TcdE142 resulted in rapid cell death compared to TcdE140. Nearly 40% of the population was found to have membrane damage due to TcdE142, whereas only 13% of the population was affected when TcdE140 was expressed in C. difficile (Fig. 3). The lethal effects due to TcdE166 and TcdE142 were similar when they were expressed in E. coli as well (15). The TcdE WT (which may express more than one TcdE isoform) exhibited a low level of cell death, where 90% of the population was alive, with an intact cell membrane. This result suggests that coexpression of the less lethal isoform TcdE140 along with the lethal isoforms TcdE142 and TcdE166 somehow minimized membrane damage and subsequent cell death. Lower-level expression of TcdE166, TcdE140, TcdE142, and the TcdE WT by induction with 10 ng/ml ATc did not affect bacterial viability (see Fig. S4A in the supplemental material). Toxin titers in the supernatant were determined by using these cultures. The TcdE142-complemented mutant secreted more toxin than did the TcdE140-complemented mutant (Fig. 4). We detected more toxins in the TcdE166-expressing cultures than in the vector-alone control cultures. This result, combined with the results of the TcdE166 expression analysis (Fig. 2B and 3), suggests that expression of TcdE166, even at undetectable levels, can trigger toxin release. Expression of the TcdE WT completely complemented the toxin secretion defect of the R20291::tcdE mutant. None of the TcdE isoforms were as efficient as the TcdE WT in complementing the toxin secretion defect of the tcdE mutant. When just one TcdE isoform was expressed in the R20291::tcdE mutant strain, it only partially regained the toxin secretion ability, suggesting that the expression of TcdE166, TcdE142, and TcdE140 together is necessary for optimal toxin secretion in C. difficile.
FIG 4.
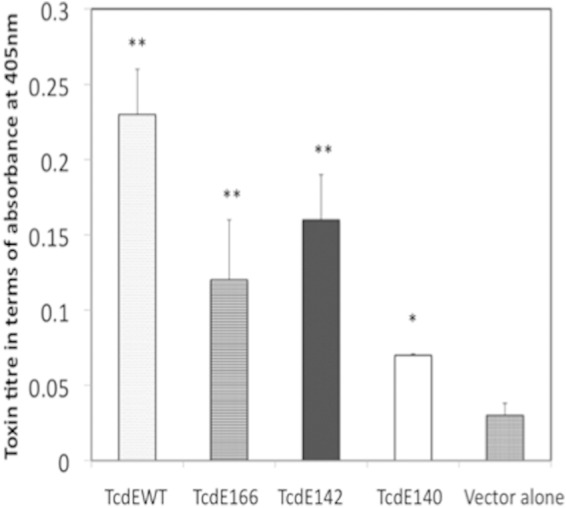
Effect of TcdE isoforms on toxin secretion. C. difficile R20291::tcdE mutants carrying plasmids expressing either the TcdE WT or different TcdE isoforms were induced with 10 ng/ml ATc for 3 h, the toxin titers in the culture supernatants were determined by an ELISA, and the signal from the test was recorded as the absorbance at 405 nm. Data are expressed as the means ± standard errors from three replicate samples. Student's t test was used for statistical analysis. *, P < 0.05; **, P < 0.01 (versus the vector control).
An asparagine residue is important for the holin activity of TcdE142.
The lambda phage holin S105 and antiholin S107 are identical in sequence, with the exception of two extra amino acids, a methionine and a lysine, at the N terminus of S107. The TcdE142 isoform differs from TcdE140 only by the presence of a methionine and an asparagine (Asn) at its N terminus. In lambda phage, the shorter polypeptide S105 acts as a holin and induces cell death, whereas in TcdE, the longer form, TcdE142, is more lethal than the shorter form, TcdE140. To determine if the presence of the extra Asn in the N terminus of TcdE142 is important for this phenotype, we replaced this residue in TcdE142 with a valine (TcdE142N26V), alanine (TcdE142N26A), or glutamine (TcdE142N26Q) by site-directed mutagenesis. We then overexpressed these mutated TcdE142 isoforms by inducing their expression with 50 ng/ml of ATc. The lethal effects of TcdE142N26V, TcdE142N26V, TcdE142N26V, and TcdE142 were studied by enumerating the number of live cells by spotting serial dilutions (Fig. 5A). Membrane proteins prepared from the induced bacterial cultures were probed with anti-TcdE antibodies to confirm the expression of various TcdE142 forms (Fig. 5B). TcdE proteins carrying a valine, glutamine, or alanine at position 26 instead of Asn were nearly 2- to 3-fold less lethal than TcdE142. These results suggest that the N-terminal Asn residue is required for the holin activity of TcdE142. Supernatants were collected from cultures induced with 10 ng/ml of ATc, and toxin levels were measured by using an ELISA. The results showed that TcdE142N26V, TcdE142N26V, and TcdE142N26V were less efficient in releasing the toxin than TcdE142 and were similar to the TcdE140 isoform. These results suggest that the holin activity of TcdE is necessary for aiding in toxin release from C. difficile cells.
FIG 5.

Importance of Asn26 for the holin function of TcdE142. (A) The expression of mutant forms of TcdE142 was induced with 50 ng/ml ATc for 3 h. After induction, 10-fold serial dilutions were prepared, and 5 μl of the dilutions was spotted for overnight growth at 37°C. (B) Membrane proteins prepared from C. difficile strains expressing various TcdE142 constructs were separated by SDS-PAGE and probed with anti-TcdE antibodies and anti-ATPase antibodies. The mutant forms of TcdE142 and TcdE WT along with TcdE140 were induced with 10 ng/ml ATc for 3 h, the culture supernatants were collected, and the toxin titers were determined by an ELISA. Data are expressed as the means ± standard errors from three replicate samples. Student's t test was used for statistical analysis for comparing the effects of TcdE142N26A, TcdE142N26V, and TcdE142N26Q to those of TcdE142. Results of statistical analyses performed to compare the effects of all test samples to the effect of the vector alone were significant (P < 0.001).
Overexpression of TcdE166 is lethal to C. difficile.
To identify the effect of TcdE166 on toxin release, we expressed TcdE166 from an altered RBS. Interestingly, TcdE166 overexpression in C. difficile resulted in cell lysis, which was detected by a rapid reduction in the optical density (Fig. 6A). Lysis was evident even in cultures induced with a lower concentration of ATc (Fig. 6B). Western blot analysis of membrane proteins from the cultures induced with anti-6×His antibodies confirmed the expression of TcdE166 (Fig. 6C). This result suggests that TcdE166 is a potent holin and that its expression is tightly controlled in C. difficile. A weak RBS and the pair of alternate start codons (see Fig. S4A in the supplemental material) upstream of the tcdE166 start codon may be important in regulating the expression of TcdE166 in C. difficile to limit its lethal effect.
FIG 6.

Expression of TcdE166 from a modified RBS. (A) Growth curve of the R20291::tcdE mutant complemented with TcdE166. The strain with the vector alone was used as a control. The arrow indicates the time point at which ATc (10 ng/ml) was added to induce protein expression. (B) Clearing of the culture was observed ∼4 h after induction. (C) Membrane proteins from R20291::tcdE mutants carrying pRGL60 (TcdE WT) and pRGL297 (TcdE166) were probed with anti-6×His antibodies to detect the TcdE WT and TcdE166, respectively.
TcdE-mediated toxin release occurs without cell lysis in C. difficile.
Overexpression of the TcdE isoforms TcdE142 and TcdE166 in C. difficile resulted in cell death and cell lysis, respectively, raising the question of whether toxins in C. difficile are released by TcdE-mediated cell lysis. TcdE-mediated toxin release occurs during the late exponential and stationary phases of bacterial culture growth. Certain levels of autolysis occur during these growth stages, particularly as the bacteria initiate the sporulation pathway, which complicates the detection of the true effect of TcdE on toxin release or its effect on cell lysis in C. difficile. To overcome this difficulty, we expressed the toxin regulator tcdR from the tetracycline-inducible promoter in WT strain R20291 and the R20291::tcdE mutant to generate strains RGL100 and RGL101, respectively. The creation of these strains enabled toxin expression in the early exponential stage, when autolysis is absent. Toxins could be detected in the cytosol of these cultures even in the absence of ATc induction. These results demonstrate that even a very low level of TcdR during this growth stage is sufficient to induce high levels of toxin production. Toxins were detected the cytosol even in 1-h-old bacterial cultures (data not shown). The production and release of toxins in the early exponential stage did not affect bacterial growth (Fig. 7A). The effect of TcdE on toxin release is clearly evident in these strains. When TcdR was expressed at the exponential phase, toxins were observed only in the cytoplasm in RGL101 and were not released from the cells (Fig. 7B). Bacterial cells were collected at this earlier growth stage (4 h old) and analyzed for cell lysis. Twofold dilutions of these bacterial cultures were prepared and spotted to enumerate live bacteria during toxin release. No difference in cell counts was observed between the toxin-releasing strain RGL100 (R20291 background) and strain RGL101 (R20291::tcdE mutant background), which did not release toxins (data not shown). Furthermore, we examined the effects of TcdE and toxin release on C. difficile membrane integrity by FACS analysis of cells exposed to the fluorescent nucleotide-binding dyes SYTO9 and PI. R20291 and the R20291::tcdE mutant carrying the vector alone were used as controls (see Fig. S6 in the supplemental material). Analysis of the tcdE mutant and parental strains harvested at 4 h of growth revealed no difference in the proportions of intact and membrane-permeable cell populations. The membrane-intact live-cell fraction was >97% in all four strains tested, indicating the absence of cell lysis during TcdE-mediated toxin release (Fig. 7C; see also Fig. S6 in the supplemental material).
FIG 7.

Effect of TcdE on toxin secretion in C. difficile strains that produce toxins in the early exponential growth phase. (A) Growth curves of C. difficile strains RGL100 and RGL101. The arrows indicate the time points at which the bacterial cultures were harvested for evaluation of toxin production and secretion and for FACS analysis. (B) Bacterial cultures were harvested 4 h after inoculation, and the toxins in the cytoplasm and supernatant were quantified by an ELISA. Fifty microliters of the culture supernatants and 25 μg of cytosolic proteins harvested from RGL100 and RGL101 were used in ELISAs to determine toxin titers. The signal from the test was recorded as the absorbance at 405 nm. Data are expressed as the means ± standard errors from three replicate samples. (C) FACS analysis of the membrane permeability of C. difficile cells by using PI and SYTO staining. RGL100 and RGL101 cells were harvested at 4 h and subjected to FACS analysis following PI and SYTO staining. Shown here are representative results from four independent experiments.
DISCUSSION
In this study, we provide additional genetic evidence that TcdE, an apparent holin, is required for efficient toxin secretion by C. difficile. We performed this study based on the hypothesis that the effect of TcdE on toxin release would be more obvious in high-toxin-producing C. difficile strains than in low-toxin-producing strains. We created tcdE mutants of the high-toxin-producing R20291 strain and demonstrated that the mutants are defective in toxin release into the culture medium. However, we did not detect a significant change in toxin secretion between the CD646::tcdE mutant and its parent strain. The CD646 strain produced very low levels of toxin and was lysed more rapidly as it entered the stationary growth phase (Fig. 1B). Hence, toxins from this strain are released into the culture medium mostly due to cell lysis independent of TcdE. We tried to introduce the plasmid expressing tcdR (pRGL209) into CD646 and the CD646::tcdE mutant to overexpress toxins in these strains. However, our repeated attempts were unsuccessful for unknown reasons. Our observations of the CD646::tcdE mutant strain are consistent with those of Olling et al., who did not detect an effect of TcdE on toxin release when they used the low-toxin-producing 630E strain (27). Taken together, these results suggest that the discrepancy in the apparent role of TcdE in toxin secretion may be due mainly to differences in the parental strains used in these studies. Our results with the R20291 strain validate the role of TcdE in toxin secretion in C. difficile.
Holins are grouped into three main classes based on the number of predicted TMDs. We predicted three TMDs in TcdE (http://www.cbs.dtu.dk/services/TMHMM) with characteristics of class I holins (37). Similar to many class I holins, TcdE expressed in E. coli and C. difficile was localized to the cytoplasmic membrane in an oligomeric form (15). We previously provided the first experimental evidence for the holin function of TcdE by expressing TcdE downstream of the lambda late promoter in E. coli MC1063 λ (cI857Sam7), which resulted in cell lysis upon phage induction (15). Holin activity and holin-encoding genes are regulated at various levels. A unique holin regulatory mechanism is the common occurrence of two potential translation start sites separated by only a few codons (22, 24). The TcdE coding sequence includes three potential start codons, at positions 1, 25, and 27. We tested all three potential TcdE isoforms (TcdE166, TcdE142, and TcdE140) for their ability to complement the R20291::tcdE mutant strain. Initial expression analysis revealed that only TcdE142 and TcdE140 were translated in C. difficile, while the TcdE166 isoform was undetectable. Upon induction, TcdE142 induced some level of cell death, whereas the cell death induced by TcdE140 was less obvious. TcdE WT constructs, which presumably express all isoforms simultaneously, exhibited reduced cell death. The toxin secretion profiles of different complemented R20291::tcdE mutant strains demonstrated that the TcdE WT facilitated efficient toxin release, suggesting that combinations of different TcdE isoforms may be necessary for this process.
In the lambda phage S107 antiholin, the presence of a highly charged lysine reside in its N terminus hinders the penetration of the first TMD (38). Thus, with only two TMDs, S107 is less efficient in inducing cell lysis than S105, which contains three TMDs (38). In contrast to lambda, the longer form of TcdE (TcdE142) induced more cell death than did the shorter form (TcdE140). TcdE142 differs from TcdE140 by two additional amino acids, Met and Asn, at the N terminus. Site-directed mutagenesis revealed that this Asn residue is important for the holin function of TcdE142. Previous studies demonstrated that Asn side chains in membrane proteins can form a strong site for transmembrane helix stabilization and oligomerization (39). Thus, the extra Asn residue in TcdE142 may help the protein form a stable first TMD and/or facilitate its oligomerization. These hypotheses are under investigation.
Overexpression of TcdE166 from a modified RBS was lethal in C. difficile. Interestingly, expression of TcdE166 resulted in a rapid decrease in the turbidity of the bacterial cultures, mimicking host cell lysis during phage release, which involves both the holin and an endolysin. The proteins released from the TcdE166-overexpressing cultures were concentrated and analyzed in zymograms (using Micrococcus cells and C. difficile cells) to detect endolysin activity. No endolysins were detected, suggesting that the rapid lysis was due mainly to the expression of TcdE166 (data not shown). It is unclear how the first 25 amino acid residues of TcdE166 trigger such a dramatic cell lysis effect in bacterial cells. We detected toxin release by an ELISA and cell death by FACS analysis in the strain that expresses TcdE166 from its native RBS, suggesting that TcdE166 is translated albeit at a very low level, below the detection threshold. Although TcdE166 is lethal to C. difficile, its presence along with TcdE142 and TcdE140 may be necessary for the efficient release of toxins in C. difficile. Stem and loop structures are predicted for the tcdE transcript RNA (see Fig. S7 in the supplemental material), and these structures may play a role in controlling the translation rate of TcdE140 and TcdE142. A less efficient RBS and alternate translation start codons that can lead to a frameshift (see Fig. S4A in the supplemental material) may stringently control the translation of TcdE166 production. Under natural conditions, C. difficile presumably translates different amounts of all three TcdE isoforms to release toxins with minimal or no cell lysis.
To exclude a role of TcdE-mediated cell lysis during toxin release, we altered the timing of toxin expression to the early exponential stage of bacterial growth. When TcdR was expressed from the tetracycline-inducible promoter, toxins were produced as early as 1 h after inoculation, and no cell lysis was detected. The tet promoter is leaky in nature, and toxin production was observed even in the absence of any induction with ATc. This result suggested that even a very small amount of the TcdR sigma factor is enough to induce high levels of toxin production in C. difficile. Analysis of the C. difficile tcdR mutant in our laboratory suggests that TcdR is necessary for tcdE expression (our unpublished data). Hence, when TcdR was expressed at an earlier time point in the wild-type strain (RGL100), it must have been inducing the production of both toxins and TcdE, enabling the efficient release of the toxins from bacterial cells. However, in the absence of TcdE, a defect in toxin release in the tcdE mutant strain (RGL101) was clearly evident. At the 12-h time point, the toxin levels in the supernatants of RGL100 and RGL101 were very similar to the toxin levels observed in the supernatants of 12-h-old R20291 and R20291::tcdE mutant cultures (data not shown). This suggests that the smaller amount of toxins detected in the R20291::tcdE strain supernatant during this late exponential growth stage is due to a lower level of TcdE-independent cell lysis in C. difficile.
How TcdR induces the transcription of tcdE is still under investigation. It has been proposed that tcdE may carry its own promoter or could be transcribed as a polycistronic message from the upstream tcdB promoter. In our previous study, we failed to complement the tcdE mutant by cloning tcdE from its own promoter in a multicopy plasmid (15). This result suggested that tcdE could be transcribed from its own upstream region and that its overexpression from a multicopy plasmid could be lethal to C. difficile cells. Complementation of the tcdE mutant became possible when TcdE was expressed at an optimal level from an inducible promoter (15). In this study, when TcdR was expressed during the early exponential growth stage (in RG100), tcdE expression was driven by the chromosome and the TcdE protein. In fact, quantitative reverse transcriptase PCR (qRT-PCR) analysis showed that induction of tcdE from PtetR using 10 ng/ml of ATc produces nearly 30-fold more tcdE transcripts than those present in the early exponential culture of RG100 (data not shown). Thus, induction of chromosomal tcdE by TcdR enabled efficient toxin release from this strain. These toxin-overexpressing strains provide new tools to analyze TcdE-mediated toxin release in C. difficile. How does TcdE mediate toxin release? Why are there three TcdE isoforms? Does TcdE act with other proteins to aid toxin release? These questions regarding the toxin secretion mechanism in C. difficile remain to be answered.
Supplementary Material
ACKNOWLEDGMENTS
We thank Linc Sonenshein (Tufts University), Joeseph Sorg (Texas A&M), and Craig Ellermeier (University of Iowa) for their advice on the manuscript; Nigel Minton, University of Nottingham, for plasmid pMTL007C-E5; Robert Fagan for plasmid pRPF185; and Brintha Parasumanna Girinathan for her assistance throughout the study.
This work was supported by funds from the Johnson Cancer Center, KSU, and startup grants to R.G. from KINBRE, supported by the National Center for Research Resources (P20RR016475) and the National Institute of General Medical Sciences (P20GM103418), and from a pilot project to R.G. supported through COBRE grant P30 496 GM103326 to Joe Lutkenhaus, University of Kansas.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00224-15.
REFERENCES
- 1.McGlone SM, Bailey RR, Zimmer SM, Popovich MJ, Tian Y, Ufberg P, Muder RR, Lee BY. 2012. The economic burden of Clostridium difficile. Clin Microbiol Infect 18:282–289. doi: 10.1111/j.1469-0691.2011.03571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popoff MR, Bouvet P. 2009. Clostridial toxins. Future Microbiol 4:1021–1064. doi: 10.2217/fmb.09.72. [DOI] [PubMed] [Google Scholar]
- 3.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. 1995. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 4.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B Nature 375:500–503. [DOI] [PubMed] [Google Scholar]
- 5.Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95:1026–1031. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Natale P, Bruser T, Driessen AJ. 2008. Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—distinct translocases and mechanisms. Biochim Biophys Acta 1778:1735–1756. doi: 10.1016/j.bbamem.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee K, Karlsson S, Burman LG, Akerlund T. 2002. Proteins released during high toxin production in Clostridium difficile. Microbiology 148:2245–2253. [DOI] [PubMed] [Google Scholar]
- 8.Braun V, Hundsberger T, Leukel P, Sauerborn M, von Eichel-Streiber C. 1996. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 181:29–38. doi: 10.1016/S0378-1119(96)00398-8. [DOI] [PubMed] [Google Scholar]
- 9.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, Buckley AM, Antunes A, Kotsanas D, Jenkin GA, Dupuy B, Rood JI, Lyras D. 2011. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7:e1002317. doi: 10.1371/journal.ppat.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27:107–120. [DOI] [PubMed] [Google Scholar]
- 12.Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98:5844–5849. doi: 10.1073/pnas.101126598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mani N, Lyras D, Barroso L, Howarth P, Wilkins T, Rood JI, Sonenshein AL, Dupuy B. 2002. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J Bacteriol 184:5971–5978. doi: 10.1128/JB.184.21.5971-5978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matamouros S, England P, Dupuy B. 2007. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol 64:1274–1288. doi: 10.1111/j.1365-2958.2007.05739.x. [DOI] [PubMed] [Google Scholar]
- 15.Govind R, Dupuy B. 2012. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 8:e1002727. doi: 10.1371/journal.ppat.1002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan KS, Wee BY, Song KP. 2001. Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J Med Microbiol 50:613–619. [DOI] [PubMed] [Google Scholar]
- 17.Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol 54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- 18.Young I, Wang I, Roof WD. 2000. Phages will out: strategies of host cell lysis. Trends Microbiol 8:120–128. doi: 10.1016/S0966-842X(00)01705-4. [DOI] [PubMed] [Google Scholar]
- 19.Catalao MJ, Gil F, Moniz-Pereira J, Sao-Jose C, Pimentel M. 2013. Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev 37:554–571. doi: 10.1111/1574-6976.12006. [DOI] [PubMed] [Google Scholar]
- 20.Young R. 2013. Phage lysis: do we have the hole story yet? Curr Opin Microbiol 16:790–797. doi: 10.1016/j.mib.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young R. 2014. Phage lysis: three steps, three choices, one outcome. J Microbiol 52:243–258. doi: 10.1007/s12275-014-4087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasi U, Nam K, Hartz D, Gold L, Young R. 1989. Dual translational initiation sites control function of the lambda S gene. EMBO J 8:3501–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CY, Nam K, Blasi U, Young R. 1993. Synthesis of two bacteriophage lambda S proteins in an in vivo system. Gene 133:9–16. doi: 10.1016/0378-1119(93)90218-R. [DOI] [PubMed] [Google Scholar]
- 24.Blasi U, Young R. 1996. Two beginnings for a single purpose: the dual-start holins in the regulation of phage lysis. Mol Microbiol 21:675–682. doi: 10.1046/j.1365-2958.1996.331395.x. [DOI] [PubMed] [Google Scholar]
- 25.Grundling A, Smith DL, Blasi U, Young R. 2000. Dimerization between the holin and holin inhibitor of phage lambda. J Bacteriol 182:6075–6081. doi: 10.1128/JB.182.21.6075-6081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CY, Nam K, Young R. 1995. S gene expression and the timing of lysis by bacteriophage lambda. J Bacteriol 177:3283–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olling A, Seehase S, Minton NP, Tatge H, Schroter S, Kohlscheen S, Pich A, Just I, Gerhard R. 2012. Release of TcdA and TcdB from Clostridium difficile cdi 630 is not affected by functional inactivation of the tcdE gene. Microb Pathog 52:92–100. doi: 10.1016/j.micpath.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keel K, Brazier JS, Post KW, Weese S, Songer JG. 2007. Prevalence of PCR ribotypes among Clostridium difficile isolates from pigs, calves, and other species. J Clin Microbiol 45:1963–1964. doi: 10.1128/JCM.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng F, Murray BE, Weinstock GM. 1998. Conjugal transfer of plasmid DNA from Escherichia coli to enterococci: a method to make insertion mutations. Plasmid 39:182–186. doi: 10.1006/plas.1998.1336. [DOI] [PubMed] [Google Scholar]
- 31.Govind R, Vediyappan G, Rolfe RD, Fralick JA. 2006. Evidence that Clostridium difficile TcdC is a membrane-associated protein. J Bacteriol 188:3716–3720. doi: 10.1128/JB.188.10.3716-3720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deckers-Hebestreit G, Altendorf K. 1986. Accessibility of F0 subunits from Escherichia coli ATP synthase. A study with subunit specific antisera. Eur J Biochem 161:225–231. [DOI] [PubMed] [Google Scholar]
- 33.Vohra P, Poxton IR. 2011. Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology 157:1343–1353. doi: 10.1099/mic.0.046243-0. [DOI] [PubMed] [Google Scholar]
- 34.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods 80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493. doi: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young R. 2002. Bacteriophage holins: deadly diversity. J Mol Microbiol Biotechnol 4:21–36. [PubMed] [Google Scholar]
- 38.White R, Tran TA, Dankenbring CA, Deaton J, Young R. 2010. The N-terminal transmembrane domain of lambda S is required for holin but not antiholin function. J Bacteriol 192:725–733. doi: 10.1128/JB.01263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choma C, Gratkowski H, Lear JD, DeGrado WF. 2000. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol 7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.