

ABSTRACT
Mycobacterium smegmatis encodes several DNA repair polymerases that are adept at incorporating ribonucleotides, which raises questions about how ribonucleotides in DNA are sensed and removed. RNase H enzymes, of which M. smegmatis encodes four, are strong candidates for a surveillance role. Here, we interrogate the biochemical activity and nucleic acid substrate specificity of M. smegmatis RnhC, a bifunctional RNase H and acid phosphatase. We report that (i) the RnhC nuclease is stringently specific for RNA:DNA hybrid duplexes; (ii) RnhC does not selectively recognize and cleave DNA-RNA or RNA-DNA junctions in duplex nucleic acid; (iii) RnhC cannot incise an embedded monoribonucleotide or diribonucleotide in duplex DNA; (iv) RnhC can incise tracts of 4 or more ribonucleotides embedded in duplex DNA, leaving two or more residual ribonucleotides at the cleaved 3′-OH end and at least one or two ribonucleotides on the 5′-PO4 end; (v) the RNase H activity is inherent in an autonomous 140-amino-acid (aa) N-terminal domain of RnhC; and (vi) the C-terminal 211-aa domain of RnhC is an autonomous acid phosphatase. The cleavage specificity of RnhC is clearly distinct from that of Escherichia coli RNase H2, which selectively incises at an RNA-DNA junction. Thus, we classify RnhC as a type I RNase H. The properties of RnhC are consistent with a role in Okazaki fragment RNA primer removal or in surveillance of oligoribonucleotide tracts embedded in DNA but not in excision repair of single misincorporated ribonucleotides.
IMPORTANCE RNase H enzymes help cleanse the genome of ribonucleotides that are present either as ribotracts (e.g., RNA primers) or as single ribonucleotides embedded in duplex DNA. Mycobacterium smegmatis encodes four RNase H proteins, including RnhC, which is characterized in this study. The nucleic acid substrate and cleavage site specificities of RnhC are consistent with a role in initiating the removal of ribotracts but not in single-ribonucleotide surveillance. RnhC has a C-terminal acid phosphatase domain that is functionally autonomous of its N-terminal RNase H catalytic domain. RnhC homologs are prevalent in Actinobacteria.
INTRODUCTION
The human pathogen Mycobacterium tuberculosis and its avirulent relative Mycobacterium smegmatis have a large roster of DNA repair enzymes, including a shared set of eight DNA polymerases (1–5), four of which—LigD-POL, PolD1, PolD2, and DinB2—have the distinctive properties of low fidelity and readily incorporating ribonucleotides in lieu of deoxyribonucleotides during primer extension and gap repair in vitro (4–10). LigD-POL, PolD1, and PolD2 are paralogous members of the AEP (archaeal/eukaryal polymerase/primase) polymerase family (4, 10, 11). They incorporate between one and four sequential ribonucleoside monophosphates (rNMPs) at a DNA primer terminus; after four rNMPs, they cease to elongate (4, 8). This effect is attributed to their inability to extend an RNA:DNA hybrid primer terminus with a fully A-form helical conformation (8). DinB2 is a Y family polymerase, and its natural ribonucleotide preference reflects the absence of an aromatic steric gate that confers sugar selectivity (5). DinB2 is distinguished from LigD-POL, PolD1, and PolD2 by its ability to synthesize long RNA tracts on a DNA template strand (5).
There has been a surge of interest in the biological impact of ribonucleotides embedded in DNA in the wake of many studies, primarily in eukaryal systems, showing that persistent ribonucleotides in genomic DNA are promutagenic and that there are distinct pathways of ribonucleotide surveillance and ribonucleotide excision repair (RER) that deal with these potentially harmful “lesions” (reviewed in reference 12). We are intrigued by the potential connections in mycobacteria between ribonucleotide utilization and replicative quiescence, which is central to the long-term carriage of M. tuberculosis in a clinically dormant state. Quiescent cells that are not replicating their DNA are generally thought to have reduced deoxynucleoside triphosphate (dNTP) pools compared to actively dividing cells, resulting in a high ribonucleoside triphosphate (rNTP)/dNTP ratio in the polymerase substrate pool. Although, to our knowledge, the intracellular concentrations of dNTPs and rNTPs in mycobacteria have not been reported for any growth conditions, we assume that rNTPs prevail in nonreplicating mycobacteria. It is attractive to think that that DNA repair with a “ribopatch” by polymerase utilization of available rNTPs provides an intelligent strategy for quiescent cells to avoid otherwise deadly chromosome damage.
The in vivo impact of ribonucleotide incorporation by mycobacterial polymerases is undoubtedly blunted, if not obscured, by the presence in the M. smegmatis proteome of four different RNase H enzymes: MSMEG_5562/RnhA (13), MSMEG_4305 (14), MSMEG_2442/RnhB, and MSMEG_5849 (15). RNase H enzymes incise the RNA strand of RNA:DNA hybrid duplexes; they are classified as type I (H1) or type II (H2 and H3) (16–19). RNase H1 requires an oligoribonucleotide tract and is unable to incise a single ribonucleotide embedded in duplex DNA. RNase H2 is uniquely capable of incising a single embedded rNMP. None of the four mycobacterial RNase H enzymes has been rigorously characterized with respect to its RNA requirements, which is a relevant issue, given that the rNTP-utilizing mycobacterial polymerases differ with respect to how many sequential ribonucleotides they can embed.
In the present study, we purify and characterize the MSMEG_4305 protein. The 365-amino-acid (aa) MSMEG_4305 polypeptide consists of two putative domains: an N-terminal RNase H1 module and a C-terminal acid phosphatase module (Fig. 1A). It is homologous to Streptomyces coelicolor SCO2299, a bifunctional nuclease-phosphatase enzyme with bona fide RNase H activity in vitro that is inherent to an autonomous N-terminal domain, which is as active in this regard as full-length SCO2299 (20). The C-terminal domain of SCO2299 has a generic phosphatase activity that hydrolyzes p-nitrophenyl phosphate (20). The M. tuberculosis homolog Rv2228c was characterized by Watkins and Baker (14), who reported that (i) the full-length Rv2228c was equally adept at cleaving RNA:DNA and RNA:RNA duplexes and (ii) the isolated N-terminal RNase H domain was 100-fold less active than the full-length Rv2228c.
FIG 1.

Recombinant RnhC. (A) Primary structure. The amino acid sequence of M. smegmatis (Msm) MSMEG_4305/RnhC is aligned with that of M. tuberculosis (Mtu) Rv2228c. The positions of side chain identity are denoted by dots above the residues. Gaps in the alignments are denoted by dashes. The Glu49 and Asp73 RNase H active-site residues and the His173 phosphatase active-site residue that were mutated are shaded in black. The margins of the autonomous N-terminal RNase H and C-terminal acid phosphatase domains, as defined by deletion analysis, are indicated by reverse and forward arrows above the RnhC sequence. (B) Purification. Aliquots (4 μg) of recombinant wild-type RnhC (WT) and E49Q and D73N mutants were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (in kilodaltons) of marker polypeptides are indicated on the left.
We find here that MSMEG_4305 is a vigorous magnesium- or manganese-dependent RNase H with stringent specificity for incision of an RNA:DNA hybrid duplex. It does not cleave an RNA:RNA duplex with otherwise identical primary structure. MSMEG_4305 can recognize and cleave a 4-nucleotide (nt) ribotract embedded in duplex DNA but not an embedded diribonucleotide or monoribonucleotide. Therefore, we name this type I RNase H enzyme RnhC. We show that the N-terminal 140-aa segment of RnhC suffices for RNase H activity, whereas the C-terminal segment (aa 155 to 365) suffices for acid phosphatase activity.
MATERIALS AND METHODS
Recombinant RnhC proteins.
A 1,098-nt DNA fragment comprising the MSMEG_4305 (rnhC) open reading frame (ORF) was amplified from M. smegmatis genomic DNA by PCR with primers that introduced a BglII site at the start codon and an XhoI site immediately after the stop codon. The PCR product was digested with BglII and XhoI and inserted between the BamHI and XhoI sites of pET28b-His10Smt3 to generate the expression plasmid pET-His10Smt3·RnhC. The missense mutations E49Q, D73N, and H173A were introduced into the rnhC ORF by PCR with mutagenic primers. Truncated ORFs encoding RnhC-(1-140) and RnhC-(1-149) were generated by PCR amplification with antisense-strand primers that introduced a stop codon and an XhoI site after codons for Asp140 and Pro149, respectively. Truncated ORFs encoding RnhC-(141-365) and RnhC-(155-365) were synthesized by PCR amplification with sense strand primers that introduced a BglII site overlying the codons for Lys141 and Val156, respectively. The truncated PCR products were digested with BglII and XhoI and inserted between the BamHI and XhoI sites of pET28b-His10Smt3. The inserts in each plasmid were sequenced to verify the fusion junctions and to ensure that no unwanted coding changes were introduced during amplification and cloning.
The RnhC expression plasmids were transfected into Escherichia coli BL21(DE3) RIPL cells. Cultures (1 liter) amplified from single transformants were grown at 37°C in Terrific broth containing 50 μg/ml kanamycin, 17.5 μg/ml chloramphenicol, 12.5 μg/ml streptomycin, and 0.4% (vol/vol) glycerol until the A600 reached 0.6. The cultures were chilled on ice for 1 h, adjusted to 2% (vol/vol) ethanol and 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and then incubated for 20 h at 17°C with constant shaking. All subsequent steps of purification were performed at 4°C. The cells were harvested by centrifugation and resuspended in 35 ml of buffer A (50 mM Tris-HCl, pH 7.5, 500 mM KCl, 15 mM imidazole, 10% glycerol) containing one protease inhibitor cocktail tablet (Roche). The cells were lysed by sonication, and the insoluble material was removed by centrifugation at 38,000 × g for 30 min. The supernatants were mixed for 1 h with 2 ml of His60-Ni Superflow Resin (Clontech) that had been equilibrated with buffer A. The resins were poured into gravity flow columns and then washed with 25 ml of buffer B (50 mM Tris-HCl, pH 7.5, 2 M KCl, 15 mM imidazole, 10% glycerol), followed by 10 ml of buffer A. The adsorbed proteins were step eluted with 300 mM imidazole in buffer A. The polypeptide compositions of the eluate fractions were monitored by SDS-PAGE, and the peak fractions containing each recombinant protein were pooled. The His10-Smt3 tags were cleaved by supplementing the protein solutions with the Smt3-specific protease Ulp1 and dialyzing them overnight against 1,000 ml of buffer C (20 mM Tris-HCl, pH 7.5, 5% glycerol) containing either 125 mM KCl [for wild-type (WT) RnhC, RnhC-H173A, RnhC-(1-140), RnhC-(1-149), RnhC-(141-365), and RnhC-(155-365)] or 200 mM KCl (for RnhC-E49Q and RnhC-D73N). The tag-free RnhC proteins were separated from His10-Smt3 by applying the dialysates to 2-ml Ni-nitrilotriacetic acid (NTA)–agarose columns that had been equilibrated with buffer C. The flowthrough fractions containing the RnhC proteins were mixed with an equal volume of buffer C and then applied to prepacked 5-ml HP Q-Sepharose columns (GE Healthcare) equilibrated in buffer D (50 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol [DTT], 1 mM EDTA, 2.5% glycerol) containing either 125 mM KCl [for wild-type RnhC, RnhC-E49Q, RnhC-D73N, RnhC-H173A, RnhC-(1-140), and RnhC-(1-149)] or 60 mM KCl [for RnhC-(141-365) and RnhC-(155-365)]. The absorbed proteins were eluted with a 75-ml linear gradient of 125 mM to 1,000 mM KCl in buffer D [for wild-type RnhC, RnhC-E49Q, RnhC-D73N, RnhC-H173A, RnhC-(1-140), and RnhC-(1-149)] or 60 mM to 1,000 mM KCl in buffer D [for RnhC-(141-365) and RnhC-(155-365)]. Peak fractions containing RnhC proteins were pooled and subjected to gel filtration through a Superdex-200 column (GE Healthcare) equilibrated in 10 mM Tris-HCl, pH 7.5, 3 mM DTT, 150 mM KCl. Peak fractions were pooled; concentrated by centrifugal ultrafiltration to 1.9 mg/ml (wild-type RnhC), 0.5 mg/ml (RnhC-E49Q), 4.0 mg/ml (RnhC-D73N), 2.6 mg/ml (RnhC-H173A), 15.2 mg/ml [RnhC-(1-140)], 32.5 mg/ml [RnhC-(1-149)], 1.9 mg/ml [RnhC-(141-365)], and 2.9 mg/ml [RnhC-(155-365)]; and then stored at −80°C. Protein concentrations were determined from the A280 measured with a Nanodrop spectrophotometer (Thermo Scientific), applying extinction coefficients calculated using Protparam.
RNase H substrates.
RNA and DNA oligonucleotides were purchased from Dharmacon and Thermo Fisher Scientific, respectively. Oligonucleotides were 5′ 32P labeled by reaction with T4 polynucleotide kinase and [γ32P]ATP and then gel purified. The labeled oligonucleotides were annealed to a 4- to 8-fold molar excess of the unlabeled complementary strands in buffer containing 0.2 M NaCl to form the various duplex substrates shown in the figures. Partial alkaline hydrolysates of the 5′ 32P-labeled strands were prepared by incubation in a solution of 40 mM NaHCO3 (pH 8.0), 60 mM Na2CO3 (pH 10.5) for 12 min at 95°C.
RESULTS
Recombinant MSMEG_4305 has RNase H activity.
Alignment of the primary structure of M. smegmatis MSMEG_4305 (365 aa) to that of the M. tuberculosis Rv2228c homolog (364 aa) highlights 265 positions of amino acid identity (indicated by dots in Fig. 1A) and 26 positions of side chain similarity. Two short gaps in the alignment flank the putative linker between an N-terminal RNase H domain and a C-terminal acid phosphatase domain. Four conserved acidic residues, Asp8, Glu49, Asp73, and Asp123, are predicted to coordinate two catalytic metal ions in the RNase H active site (14). We produced full-length wild-type MSMEG_4305 in E. coli as a His10-Smt3 fusion and isolated the protein from a soluble extract by nickel-agarose chromatography. The His10-Smt3 tag was removed with the Smt3-specific protease Ulp1. Native MSMEG_4305 was separated from the tag by Ni-agarose chromatography. Subsequent anion-exchange chromatography and gel filtration through Superdex 200 yielded a discrete monomeric component comprising a single predominant MSMEG_4305 polypeptide, as gauged by SDS-PAGE (Fig. 1B, WT). In parallel, we produced and purified two mutants, E49Q and D73N, that have conservative substitutions at the metal binding site (Fig. 1B).
In order to assay RNase H activity, we annealed a 5′ 32P-labeled 24-mer RNA oligonucleotide to an unlabeled complementary 24-mer DNA strand and reacted the RNA:DNA hybrid (at 20 nM concentration) with 0.4 nM MSMEG_4305 and 5 mM divalent cation for 20 min at 37°C. The reactions were quenched with EDTA, after which the products were analyzed by urea-PAGE and visualized by autoradiography. In the presence of magnesium, MSMEG_4305 incised the RNA:DNA hybrid to yield shorter end-labeled RNA fragments (Fig. 2A, Mg). No RNase activity was detected when the divalent cation was omitted. Whereas manganese could replace magnesium as the requisite metal, calcium, copper, cadmium, cobalt, nickel, and zinc could not (Fig. 2A). Mutations E49Q and D73N abolished the magnesium-dependent nuclease activity (Fig. 2B) and the manganese-dependent nuclease activity (not shown). We conclude that the observed RNase H activity is inherent to the recombinant MSMEG_4305 protein, which we henceforth rename RnhC.
FIG 2.
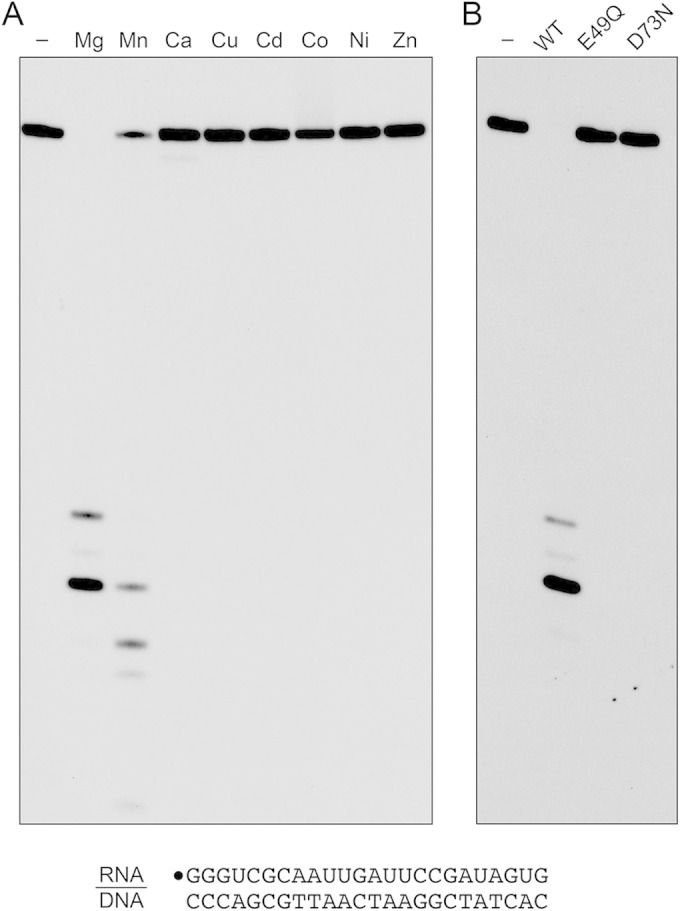
Metal-dependent RNase H activity. (A) Reaction mixtures (10 μl) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 20 nM (200 fmol) 32P-RNA:DNA hybrid duplex (depicted at the bottom), 0.4 nM (4 fmol) wild-type RnhC, and 5 mM the indicated divalent cation (as the chloride salt) were incubated at 37°C for 20 min. Divalent cation was omitted from a control reaction in lane −. (B) Reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 7.5); 50 mM NaCl; 10 mM MgCl2; 1 mM DTT; 20 nM (200 fmol) RNA:DNA hybrid duplex; and 0.4 nM (4 fmol) wild-type RnhC, RnhC-E49Q, or RnhC-D73N were incubated at 37°C for 20 min. RnhC was omitted from a control reaction in lane −. The reactions were quenched with an equal volume of 90% formamide, 50 mM EDTA, 0.3% bromophenol blue. The reaction products were analyzed by electrophoresis through a 40-cm 18% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The products were visualized by autoradiography.
Nucleic acid substrate specificity of the RnhC nuclease.
To probe substrate specificity, we prepared a series of 5′ 32P-labeled 24-bp duplex nucleic acids in which the labeled strand was RNA or DNA and the complementary unlabeled strand was either RNA or DNA (Fig. 3A). When RnhC was reacted with the RNA:DNA hybrid duplex in the presence of magnesium, all of the labeled RNA strand was cleaved to generate two predominant 32P-labeled species. A partial alkaline hydrolysis ladder was analyzed in parallel to provide a rough indication of the product size. (Note that partial alkaline hydrolysis generates a mixture of 2′-phosphate, 3′-phosphate, and 2′,3′-cyclic phosphate ends; the monophosphate and cyclic phosphate species are resolved as doublets only for the shortest fragments in the ladder. The RNase H cleavage products, which have 3′-OH termini, migrate more slowly during PAGE than end-labeled oligonucleotides of the same chain length in the alkaline ladder.) In order to accurately assign the sites of RNA cleavage, we analyzed the RnhC reaction products in parallel with a partial digest of the 24-mer 32P-RNA strand with purified M. smegmatis polynucleotide phosphorylase, which produces a ladder of 5′ 32P-labeled RNAs with 3′-OH termini (21). We thereby determined that the predominant 32P-labeled RnhC reaction products derived from incisions at the sixth and eighth internucleotide phosphodiesters, the sites indicated by arrowheads in Fig. 3A.
FIG 3.

Substrate specificity and kinetics. (A and B) Reaction mixtures (10 μl) containing 25 mM Tris-HCl (pH 7.5); 50 mM NaCl; either 10 mM MgCl2 (A) or 10 mM MnCl2 (B); 1 mM DTT; either 200 fmol 32P-RNA:DNA, 32P-DNA:DNA, or 32P-RNA:RNA duplexes or 200 fmol 32P-RNA single strand (as depicted at the bottom, with the 5′ radiolabel denoted by a dot); and RnhC as specified were incubated at 37°C for 20 min. The products were analyzed by urea-PAGE and visualized by autoradiography. An alkaline hydrolysis ladder of the 32P-labeled 24-mer RNA strand was analyzed in parallel in lanes −OH. (C) Time course of RNA:DNA cleavage. Reaction mixtures (110 μl) containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 20 nM 32P-RNA:DNA hybrid duplex (depicted at the bottom), and 0.4 nM RnhC were incubated at 37°C. Aliquots (10 μl) were withdrawn at the times specified and quenched with formamide-EDTA. The reaction products were analyzed by urea-PAGE and visualized by autoradiography. The principal sites of RNase H incision are indicated by arrowheads above the 32P-labeled RNA strands shown at the bottom.
In contrast, we detected no cleavage of a DNA:DNA duplex and no cleavage of an RNA:RNA duplex (Fig. 3A). Moreover, RnhC did not incise the 32P-labeled RNA single strand (Fig. 3A). We verified by native gel electrophoresis that the radiolabeled RNA:RNA substrate was indeed a duplex by virtue of its retarded mobility vis à vis the labeled RNA single strand.
Ohtani et al. (22) have reported that the RNase H1 from the archaeon Sulfolobus tokodaii is able to cleave RNA:RNA duplexes when manganese replaces magnesium as the metal cofactor, although the specific activity of the enzyme on an RNA:RNA substrate in manganese was several orders of magnitude less than the activity on an RNA:DNA hybrid in magnesium. Watkins and Baker reported that M. tuberculosis Rv2228c cleaved an RNA:RNA duplex in the presence of manganese and that the Km and Vmax for double-stranded RNase (dsRNase) activity were identical to the Km and Vmax for RNase H activity (14). In light of their results, we surveyed the substrate specificity of RnhC in the presence of manganese as the metal cofactor. RnhC was less active with manganese than with magnesium, requiring severalfold more enzyme (50 fmol) to achieve quantitative cleavage of the RNA strand of the RNA:DNA hybrid duplex (Fig. 3B). Also, the sites of incision in the presence of manganese (indicated by arrowheads in Fig. 3B) were shifted toward the 5′ end of the labeled strand. The salient finding was that 50 fmol of RnhC failed to cleave the RNA:RNA duplex. We conclude that M. smegmatis RnhC is stringently specific for cleaving the RNA strand of an RNA:DNA hybrid, whether the metal cofactor is magnesium or manganese.
Further characterization of the RnhC RNase.
The kinetic profile of reaction of RnhC with the 24-bp RNA:DNA hybrid under conditions of substrate excess showed that, at early times (0.5, 1, and 1.5 min), when not all of the input 24-mer RNA strand had been incised, cleavage generated a mixture of 32P-labeled 7-mer, 9-mer, 11-mer, and 12-mer strands, reflecting initial incision at four different internucleotide phosphodiesters (Fig. 3C, sites denoted by arrowheads). As the input substrate was consumed, the product distribution shifted over time so that the shortest 7-mer end-labeled cleavage product predominated (Fig. 3C). By plotting the rate of consumption of the input 24-mer RNA strand, we estimated a steady-state turnover number of 27 min−1.
The effect of the enzyme concentration on the product profile of a 20-min reaction is shown in Fig. 4A. At limiting enzyme level, when not all of the input 24-mer was consumed, we again detected a mixture of 32P-labeled 7-mer, 9-mer, 11-mer, and 12-mer products. At saturating enzyme level, when all of the substrate was cleaved, the 7-mer product predominated. By plotting the extent of 24-mer consumption as a function of RnhC, we estimated a specific activity of 440 fmol of RNA cleaved per fmol of enzyme, which corresponded to a turnover number of 22 min−1. The effect of the pH on the RNase H activity is shown in Fig. 4B. Activity was optimal at pH 8.0 to 9.0 and was curtailed sharply at acidic pH (≤5.0).
FIG 4.

Enzyme dependence and effect of pH. (A) RnhC titration. Reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 20 nM (200 fmol) 32P-RNA:DNA hybrid duplex, and increasing amounts of RnhC (0.02, 0.04, 0.1, 0.2, 0.4, 1, 2, and 4 fmol, from left to right) were incubated at 37°C for 20 min. RnhC was omitted from a control reaction in lane −. (B) pH profile. Reaction mixtures (10 μl) containing 50 mM buffer {either Tris-acetate, pH 4.5, 5.0, 5.5, 6.0, or 6.5; Tris-HCl, pH 7.0, 7.5, 8.0, 8.5, or 9.0; or CHES [2-(cyclohexylamino)ethanesulfonic acid], pH 9.5}, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 20 nM (200 fmol) 32P-RNA:DNA duplex, and 0.4 nM (4 fmol) RnhC were incubated at 37°C for 5 min. The reaction products were analyzed by urea-PAGE and visualized by autoradiography.
Cleavage of chimeric RNA-DNA strands.
Chimeric duplex substrates were prepared in which the 32P-labeled scissile strand consisted of a 5′ segment of 12 ribonucleotides and a 3′ segment of 12 deoxynucleotides (R12D12) or a 5′ segment of 12 deoxynucleotides and a 3′ segment of 12 ribonucleotides (D12R12) (Fig. 5). The R12D12 duplex is analogous to an Okazaki fragment; the D12R12 duplex is analogous to the fill-in repair reaction product synthesized by M. smegmatis DinB2 in the presence of rNTPs. RnhC was reacted with the chimeric substrates in parallel with an RNA:DNA hybrid with an all-RNA strand (R24) with identical primary structure. Partial alkaline hydrolysis confirmed the chimeric nature of the R12D12 and D12R12 strands (Fig. 5).
FIG 5.
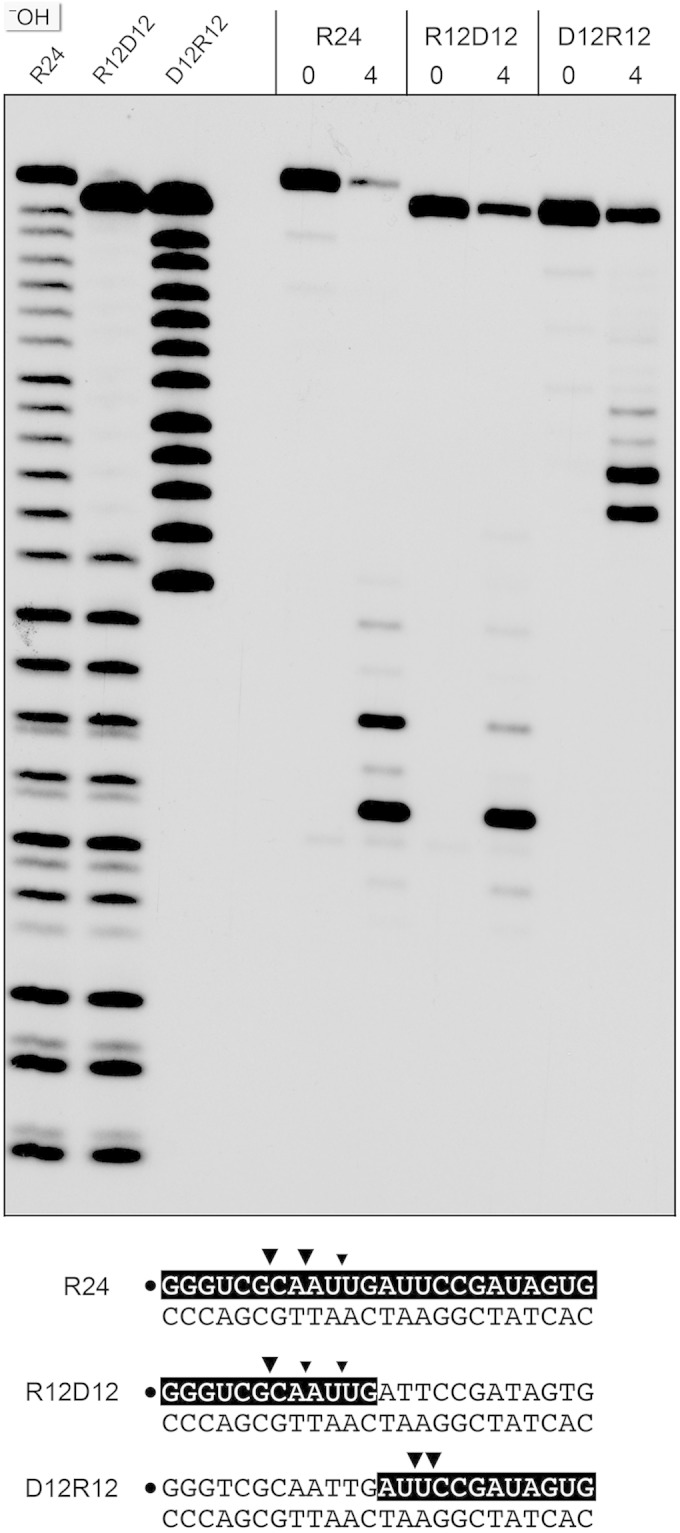
Cleavage of chimeric RNA-DNA junction substrates. Reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 8.0); 50 mM NaCl; 10 mM MgCl2; 1 mM DTT; 200 fmol 32P-labeled 24-mer duplex R24, R12D12, or D12R12 (shown at the bottom, with the 32P label denoted by a dot and the ribonucleotides shaded black); and 0 or 4 fmol RnhC was incubated for 20 min at 37°C. The products were resolved by urea-PAGE and visualized by autoradiography. Alkaline hydrolysis ladders of the 32P-labeled R24, R12D12, and D12R12 strands were analyzed in parallel in the three lanes on the left (−OH). Major and minor cleavage sites are denoted by large and small arrowheads, respectively.
Reaction of 1 nM RnhC with 20 nM R12D12 duplex resulted in incision of the RNA segment of the chimeric strand at the same sites that were cleaved in the R24 control duplex (Fig. 5). RnhC cleaved the RNA segment of the D12R12 duplex to yield two major radiolabeled products, which consisted of the proximal DNA segment plus two or three 3′-terminal ribonucleotides (Fig. 5). We surmise that (i) RnhC does not preferentially cleave the junctions of RNA and DNA segments and (ii) RnhC does not completely remove a ribonucleotide tract installed 3′ of DNA to yield a “clean” DNA 3′-OH end.
Minimal RNA requirement.
Further insights into substrate specificity were gleaned by comparing the reactions of M. smegmatis RnhC and E. coli RNase H2 (a prototypical type II RNase H) with a series of chimeric 24-bp substrates, of otherwise identical primary structure, in which one, two, four, or six ribonucleotides were embedded between 5′- and 3′-flanking DNA segments (Fig. 6A). Reactions with the all-RNA:DNA hybrid duplex (R24) were included as positive controls. Partial alkaline hydrolysis of the 32P-labeled strands verified the lengths and positions of the ribonucleotide tracts (Fig. 6B). As expected, E. coli RNase H2 efficiently incised the R1 duplex 5′ of the embedded ribonucleotide to yield a single 32P-labeled 7-mer species with a clean DNA 3′-OH end (Fig. 6B). (The reaction also yields an unlabeled distal fragment with a 5′-PO4 monoribonucleotide terminus.) The key findings in regard to RNase H2 were that it consistently and specifically incised the R2, R4, and R6 duplexes at the phosphodiester immediately 5′ of the ribonucleotide at the RNA-DNA junction (Fig. 6B). The RNase H2 incision sites are indicated by open arrowheads in Fig. 6A. However, E. coli RNase H2 cleaved the R24 substrate at eight different sites in the all-RNA strand (Fig. 6A and B). These results underscore the selectivity of RNase H2 for an RNA-DNA junction.
FIG 6.

Minimum RNA requirement. (A) Substrates and cleavage sites. The 32P label is denoted by a dot. Ribonucleotides are shaded in black. Sites of incision by E. coli RNase H2 and M. smegmatis RnhC are indicated by open and solid arrowheads, respectively. (B) Reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton X-100, 20 nM (200 fmol) duplex substrate as specified and either no enzyme (lanes −) or 5 U E. coli RNase H2 (lanes +) were incubated at 37°C for 20 min. (C) Reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 20 nM (200 fmol) duplex substrate as specified, and either no enzyme (lanes −) or 5 nM (50 fmol) RnhC were incubated at 37°C for 20 min. The products were analyzed by urea-PAGE and visualized by autoradiography. Alkaline hydrolysis ladders of the 32P-labeled strands were analyzed in parallel in lanes OH.
In contrast, M. smegmatis RnhC was unable to incise the R1 and R2 duplexes, though it did cleave the R4 and R6 substrates (Fig. 6C). Unlike RNase H2, RnhC incised the R4 and R6 duplexes at two sites and three sites, respectively (Fig. 6C), at the positions denoted by open arrowheads in Fig. 6A. The RnhC cleavage patterns on the R4 and R6 strands suggest that the enzyme requires at least two ribonucleotides (rN) on the “upstream” side of the scissile phosphodiester (↓) and at least one ribonucleotide on the “downstream” side: p(rN)p(rN)↓p(rN). (The cleavage pattern on the R4 strand suggests that a three-ribonucleotide tract might suffice for incision by RnhC, but we did not test this directly.) We conclude from this analysis that RnhC is a type I RNase H enzyme.
The N-terminal domain of RnhC suffices for RNaseH activity.
To query the activity and functional autonomy of the predicted N-terminal RNase H domain and the C-terminal domain of RnhC, we produced and purified the recombinant tag-free truncated proteins RnhC-(1-140), RnhC-(1-149), RnhC-(141-365), and RnhC-(155-365) in parallel with full-length wild-type RnhC (Fig. 7A). The two isolated N-terminal domains displayed vigorous RNase H activity on the 32P-RNA:DNA hybrid substrate (Fig. 7B). Whereas the two predominant sites of incision by wild-type RnhC were also seen with the isolated N-terminal domains, the latter enzymes yielded a population of minor products cleaved further from the 5′ ends of the labeled RNA strand. The isolated C-terminal domains had no RNase H activity (Fig. 7B). We conclude that the C-terminal domain is dispensable for the RnhC nuclease.
FIG 7.

RNase H activities of the isolated domains. (A) Aliquots (5 μg) of full-length RnhC (WT), RnhC-(1-140), RnhC-(1-149), RnhC-(141-365), and RnhC-(155-365) were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (in kilodaltons) of marker polypeptides are indicated on the left. (B) Reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 8.0); 50 mM NaCl; 10 mM MgCl2; 1 mM DTT; 20 nM (200 fmol) 32P-RNA:DNA hybrid duplex (R24); and 1 nM (10 fmol) full-length wild-type RnhC (WT), full-length RnhC-H173A, or the truncated RnhC proteins as specified were incubated at 37°C for 20 min. The products were analyzed by urea-PAGE and visualized by autoradiography.
Acid phosphatase activity of RnhC is inherent to the C-terminal domain.
Members of the histidine acid phosphatase superfamily catalyze metal-independent phosphoryl transfer via a covalent enzyme-(histidinyl-N)–phosphate intermediate (23). They are characterized by a signature active-site peptide motif, present in RnhC as 168FLLRHGQT176, in which His173 is the putative histidine nucleophile. Recombinant full-length wild-type RnhC readily hydrolyzed p-nitrophenylphosphate to p-nitrophenol at pH 5.5 (the optimum pH reported for the S. coelicolor homolog [20]) without a divalent cation cofactor (Fig. 8A), with an apparent turnover of 4.8 min−1. The D73N mutation that effaced RNase H activity had virtually no effect on the phosphatase activity of RnhC (Fig. 8A). However, the phosphatase activity was abolished when His173 was replaced by alanine (Fig. 8A). In contrast, the H173A mutation did not affect RNase H activity (Fig. 7B). As one might expect, the isolated N-terminal RNase H domains RnhC-(1-140) and RnhC-(1-149) had no phosphatase activity (Fig. 8B). The instructive finding was that RnhC-(141-365) and RnhC-(155-365) retained phosphatase activity (Fig. 8B), with apparent turnover numbers of 4.3 min−1 and 4.4 min−1, respectively. Further characterization revealed that RnhC phosphatase activity was actually optimal at pH 4.5 (Fig. 8C), with an apparent turnover number of 19.7 min−1. RnhC phosphatase activity was effaced at pHs of ≥7.0 (Fig. 8C).
FIG 8.

Acid phosphatase activity inherent to the C-terminal domain. (A) Aliquots (5 μg) of purified RnhC and RnhC-H173A were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (in kilodaltons) of marker polypeptides are indicated on the left. (B and C) Phosphatase reaction mixtures (100 μl) containing 50 mM Tris-acetate (pH 5.5); 10 mM p-nitrophenylphosphate (Sigma); and either 25 μg RnhC (WT), RnhC-H173A, or RnhC-D73N (B) or 25 μg RnhC-(141-365), RnhC-(1-140), RnhC-(155-365), or RnhC-(1-149) (C) were incubated at 37°C for 30 min. The reactions were quenched by adding 900 μl of 1 M sodium carbonate. Release of p-nitrophenol was measured by A410 and interpolating the value to a p-nitrophenol standard curve. The data are the averages of the results of three separate experiments plus standard errors of the mean (SEM). (D) pH profile. Reaction mixtures (100 μl) containing 50 mM buffer (either Na-actetate, pH 4.0; Tris-acetate, pH 4.5, 5.0, 5.5, 6.0, or 6.5; or Tris-HCl pH 7.0, 7.5, or 8.0) and 19 μg RnhC were incubated at 37°C for 30 min. Release of p-nitrophenol is plotted as a function of the pH. Each datum is the average of three separate experiments ± SEM.
DISCUSSION
Here, we provide new biochemical insights into the substrate specificity of M. smegmatis RnhC nuclease that have implications for its potential function in ribonucleotide surveillance. The chief findings are that (i) the RnhC nuclease is stringently specific for RNA:DNA hybrid duplexes, (ii) RnhC does not selectively recognize and cleave DNA-RNA or RNA-DNA junctions in duplex nucleic acid compared to an all-RNA:DNA hybrid duplex with the same primary structure, (iii) RnhC cannot incise an embedded monoribonucleotide or diribonucleotide in duplex DNA, (iv) RnhC can incise tracts of 4 or more ribonucleotides embedded in duplex DNA but in doing so leaves two or more residual ribonucleotides at the cleaved 3′-OH end and at least one or two ribonucleotides on the 5′-PO4 end, and (v) the RNase H activity is inherent to an autonomous 140-aa N-terminal domain of RnhC. The substrate and cleavage specificities of RnhC are clearly distinct from those of RNase H2, which selectively incises 5′ of the last ribonucleotide at an RNA-DNA junction. Thus, RnhC can be classified with confidence as a type I RNase H.
The extent of the RNA requirement for incision by RnhC is broadly consonant with the crystal structure of Bacillus halodurans RNase H1 bound to an RNA:DNA hybrid, which showed that the enzyme contacts five sequential ribonucleotides: three “upstream” of the scissile phosphate and two “downstream” of the scissile phosphate: NpNpN↓pNpN (17). Per our inspection, the two amino acids in B. halodurans RNase H1, Ser74 and Gly76, that contact the two downstream ribonucleotides are conserved in M. smegmatis RnhC as Ser11 and Gly13.
These biochemical characteristics of RnhC seem to rule it out as an agent of RER of single ribonucleotides embedded during DNA replication or repair. This RER function, to the extent that it is operative in mycobacteria, would likely default to one or both of the putative type II RNase H enzymes, neither of which has been characterized biochemically. Minias et al. recently reported that deletion of the M. smegmatis rnhB gene encoding an RNase H2 homolog has no impact on bacterial growth in liquid culture, sensitivity to hydroxyurea, or spontaneous mutation rate (24).
On the other hand, the substrate and cleavage specificities of RnhC are consistent with a role in removing the RNA primers at the 5′ ends of Okazaki fragments formed during lagging-strand DNA replication. Because RnhC leaves a residual 5′-PO4 ribonucleotide when it cleaves an RNA-DNA junction, it would likely be assisted in this task by a DNA polymerase and a 5′ nuclease to ultimately generate a ligatable DNA 3′-OH/5′-PO4 nick. As reported by Minias et al. (24), M. smegmatis ΔrnhC and ΔrnhA single-deletion strains are viable, as are ΔrnhC ΔrnhB and ΔrnhA ΔrnhB double-deletion strains. Thus, either one of the two type I RNase H enzymes suffices for M. smegmatis growth. (Additional studies will be required to determine whether RnhA and RnhC are functionally redundant in vivo.)
It is also plausible that RnhC could play a role in initiating the removal of longer RNA tracts laid down during gap repair by ribo-utilizing DNA polymerases (e.g., DinB2). Such a hypothetical pathway would rely on additional activities to clean up the terminal ribonucleotides left by RnhC.
The properties of M. smegmatis RnhC reported here differ from those ascribed to the M. tuberculosis homolog Rv2228c (14). First, whereas Rv2228c displayed identical activity in degrading RNA:DNA and RNA:RNA duplexes, RnhC is exclusively an RNase H enzyme. We detected no dsRNase activity, with either magnesium or manganese as the metal cofactor, under reaction conditions that supported complete cleavage of an RNA:DNA hybrid with identical primary structure. Second, whereas the N-terminal 140-aa domain of Rv2228c displayed extremely feeble RNase activity compared to the full-length protein, the N-terminal 140-aa segment of RnhC comprises an autonomous catalytically active RNase H enzyme.
We show that the isolated C-terminal domain of RnhC is an autonomous acid phosphatase enzyme reliant on His173, which is the putative nucleophile for phosphoryl transfer via a covalent phosphoenzyme intermediate. The physiological substrate(s) for the RnhC phosphatase and the biological rationale for fusion of an acid phosphatase to an RNase H module are presently obscure, yet this functional and physical arrangement is likely meaningful, as bifunctional RnhC homologs are distributed widely among mycobacterial species (including Mycobacterium leprae) and in many other genera of Actinobacteria.
ACKNOWLEDGMENT
This work was supported by U.S. National Institutes of Health grant AI64693.
REFERENCES
- 1.Huberts P, Mizrahi V. 1995. Cloning and sequence analysis of the gene encoding the DNA polymerase I from Mycobacterium tuberculosis. Gene 164:133–136. doi: 10.1016/0378-1119(95)00453-D. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff HI, Reed MB, Barry CE, Mizrahi V. 2003. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113:183–193. doi: 10.1016/S0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 3.Gong C, Bongiorno P, Martins A, Stephanou NC, Zhu H, Shuman S, Glickman MS. 2005. Mechanism of non-homologous end joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat Struct Mol Biol 12:304–312. doi: 10.1038/nsmb915. [DOI] [PubMed] [Google Scholar]
- 4.Zhu H, Bhattarai H, Yan H, Shuman S, Glickman M. 2012. Characterization of Mycobacterium smegmatis PolD2 and PolD1 as RNA/DNA polymerases homologous to the POL domain of bacterial DNA ligase D. Biochemistry 51:10147–10158. doi: 10.1021/bi301202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ordonez H, Uson ML, Shuman S. 2014. Characterization of three Mycobacterial DinB (DNA polymerase IV) paralogs highlights DinB2 as naturally adept at ribonucleotide incorporation. Nucleic Acids Res 42:11056–11070. doi: 10.1093/nar/gku752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ordonez H, Shuman S. 2014. Mycobacterium smegmatis DinB2 misincorporates deoxyribonucleotides and ribonucleotides during templated synthesis and lesion bypass. Nucleic Acids Res 42:12722–12734. doi: 10.1093/nar/gku1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma A, Nair DT. 2012. MsDpo4—a DinB homolog from Mycobacterium smegmatis—is an error-prone DNA polymerase than can promote G:T and T:G mismatches. J Nucleic Acids 2012:285481. doi: 10.1155/2012/285481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu H, Shuman S. 2005. Novel 3′-ribonuclease and 3′-phosphatase activities of the bacterial non-homologous end-joining protein, DNA ligase D. J Biol Chem 280:25973–25981. doi: 10.1074/jbc.M504002200. [DOI] [PubMed] [Google Scholar]
- 9.Yakovleva L, Shuman S. 2006. Nucleotide misincorporation, 3′-mismatch extension, and responses to abasic sites and DNA adducts by the polymerase component of bacterial DNA ligase D. J Biol Chem 281:25026–25040. doi: 10.1074/jbc.M603302200. [DOI] [PubMed] [Google Scholar]
- 10.Pitcher RS, Brissett NC, Picher AJ, Andrade P, Juarez R, Thompson D, Fox GC, Blanco L, Doherty AJ. 2007. Structure and function of a mycobacterial NHEJ DNA repair polymerase. J Mol Biol 366:391–405. doi: 10.1016/j.jmb.2006.10.046. [DOI] [PubMed] [Google Scholar]
- 11.Zhu H, Nandakumar J, Aniukwu J, Wang LK, Glickman MS, Lima CD, Shuman S. 2006. Atomic structure and NHEJ function of the polymerase component of bacterial DNA ligase D. Proc Natl Acad Sci U S A 103:1711–1716. doi: 10.1073/pnas.0509083103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Potenski CJ, Klein HL. 2014. How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res 42:10226–10234. doi: 10.1093/nar/gku773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawes SS, Crouch RJ, Morris SL, Mizrahi V. 1995. Cloning, sequence analysis, overproduction in Escherichia coli and enzymatic characterization of the RNase HI from Mycobacterium smegmatis. Gene 165:71–75. doi: 10.1016/0378-1119(95)00523-9. [DOI] [PubMed] [Google Scholar]
- 14.Watkins HA, Baker EN. 2010. Structural and functional characterization of an RNase HI domain from the bifunctional protein Rv2228c from Mycobacterium tuberculosis. J Bacteriol 192:2878–2886. doi: 10.1128/JB.01615-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murdeshwar MS, Chatterji D. 2012. MS_RHII-RSD, a dual-function RNase HII-(p) ppGpp synthetase from Mycobacterium smegmatis. J Bacteriol 194:4003–4014. doi: 10.1128/JB.00258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tadokoro T, Kanaya S. 2009. Ribonuclease H: molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J 276:1482–1493. doi: 10.1111/j.1742-4658.2009.06907.x. [DOI] [PubMed] [Google Scholar]
- 17.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. 2005. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 18.Rychlik MP, Chon H, Cerritelli SM, Klimek P, Crouch RJ, Nowotny M. 2010. Crystal structures of RNase H2 in complex with nucleic acid reveal the mechanism of RNA-DNA junction recognition and cleavage. Mol Cell 40:658–670. doi: 10.1016/j.molcel.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Figiel M, Nowotny M. 2014. Crystal structure of RNase H3-substrate complex reveals parallel evolution of RNA/DNA hybrid recognition. Nucleic Acids Res 42:9285–9294. doi: 10.1093/nar/gku615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohtani N, Saito N, Tomita M, Itaya M, Itoh A. 2005. The SCO2299 gene from Streptomyces coelicolor A3(2) encodes a bifunctional enzyme consisting of an RNase H domains and an acid phosphatase domain. FEBS J 272:2828–2837. doi: 10.1111/j.1742-4658.2005.04704.x. [DOI] [PubMed] [Google Scholar]
- 21.Unciuleac M, Shuman S. 2013. Discrimination of RNA versus DNA by polynucleotide phosphorylase. Biochemistry 52:6702–6711. doi: 10.1021/bi401041v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtani N, Yanagawa H, Tomita M, Itaya M. 2004. Cleavage of double-stranded RNA by RNase HI from a thermoacidophilic archaeon, Sulfolobus tokodaii 7. Nucleic Acids Res 32:5809–5819. doi: 10.1093/nar/gkh917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigden DJ. 2008. The histidine phosphatase superfamily: structure and function. Biochem J 409:333–348. doi: 10.1042/BJ20071097. [DOI] [PubMed] [Google Scholar]
- 24.Minias AE, Brzostek AM, Minias P, Dziadek J. 2015. The deletion of rnhB in Mycobacterium smegmatis does not affect the level of RNase HII substrates or influence genome stability. PLoS One 10:e0115521. doi: 10.1371/journal.pone.0115521. [DOI] [PMC free article] [PubMed] [Google Scholar]