

ABSTRACT
Mycobacteriophages are viruses that infect mycobacterial hosts and are prevalent in the environment. Nearly 700 mycobacteriophage genomes have been completely sequenced, revealing considerable diversity and genetic novelty. Here, we have determined the protein complement of mycobacteriophage Giles by mass spectrometry and mapped its genome-wide protein interactome to help elucidate the roles of its 77 predicted proteins, 50% of which have no known function. About 22,000 individual yeast two-hybrid (Y2H) tests with four different Y2H vectors, followed by filtering and retest screens, resulted in 324 reproducible protein-protein interactions, including 171 (136 nonredundant) high-confidence interactions. The complete set of high-confidence interactions among Giles proteins reveals new mechanistic details and predicts functions for unknown proteins. The Giles interactome is the first for any mycobacteriophage and one of just five known phage interactomes so far. Our results will help in understanding mycobacteriophage biology and aid in development of new genetic and therapeutic tools to understand Mycobacterium tuberculosis.
IMPORTANCE Mycobacterium tuberculosis causes over 9 million new cases of tuberculosis each year. Mycobacteriophages, viruses of mycobacterial hosts, hold considerable potential to understand phage diversity, evolution, and mycobacterial biology, aiding in the development of therapeutic tools to control mycobacterial infections. The mycobacteriophage Giles protein-protein interaction network allows us to predict functions for unknown proteins and shed light on major biological processes in phage biology. For example, Giles gp76, a protein of unknown function, is found to associate with phage packaging and maturation. The functions of mycobacteriophage-derived proteins may suggest novel therapeutic approaches for tuberculosis. Our ORFeome clone set of Giles proteins and the interactome data will be useful resources for phage interactomics.
INTRODUCTION
The continuous emergence of bacterial pathogens resistant to antibiotics is an increasing medical problem (1). Mycobacterium tuberculosis is prominent among these pathogens, with over 9 million new cases of tuberculosis reported each year. There is an urgent need for alternate ways to control M. tuberculosis infections, and one potential strategy involves using mycobacteriophages for prophylaxis or therapy (2). The emergence of extensively drug-resistant (XDR) and totally drug-resistant (TDR) strains of M. tuberculosis, both of which are especially difficult to control (3), has spurred renewed interest in the therapeutic use of bacteriophages.
Mycobacteriophages are known to infect many different species of both fast- and slow-growing mycobacteria, including M. tuberculosis and Mycobacterium smegmatis (4, 5). Over the past decade, thousands of mycobacteriophages have been isolated, and hundreds have been completely sequenced (http://phagesdb.org/). Mycobacteriophage genomes are highly mosaic due to horizontal genetic exchange (6–8) but can be grouped into 20 clusters and eight singletons, i.e., phages for which no close relatives have yet been identified (9, 10). Genomic characterization of nearly 700 of these phages (http://phagesdb.org/) reveals a staggeringly large number of genes (>40,000) coding for products of unknown function.
Giles, a temperate mycobacteriophage, has a 53,746-bp genome coding for 77 proteins (11, 12). Giles belongs to the Q cluster of mycobacteriophages, which includes four other phages with very similar genome sequences. More than half of the proteins encoded by Giles are functionally uncharacterized, and most of its unknown proteins do not have close homologs or orthologs in other mycobacteriophages outside the Q cluster (12). Thus, no clues are available for these proteins of unknown function. One key to understanding Giles biology will rely upon elucidating uncharacterized proteins through their protein-protein interactions (PPIs). The full set of PPIs for this virus (that is, its interactome) should provide functional clues unattainable by studying proteins individually (13).
PPIs are essential to understanding how different proteins in phages perform their functions in vivo, either alone or in a well-coordinated, cross-regulated interaction network. Understanding the interactome of a given organism provides new insights into the key steps of major biological pathways. A better comprehension of these pathways, especially in the context of bacterial host pathogenicity, will assist us in treating diseases. Mapping PPIs is specifically essential to unraveling the biology of uncharacterized proteins in Giles. An established strategy to investigate protein function, especially among bacteriophage proteins, is to identify PPIs that may link uncharacterized proteins to well-studied proteins (14). A protein interaction network for any mycobacteriophage may also shed light on the function, of many proteins in hundreds of related phages. Exploring the activities of mycobacteriophage-derived proteins provides an arsenal of potential therapies for M. tuberculosis and other mycobacterial infections.
In the present study, we have comprehensively and systematically analyzed the proteome of mycobacteriophage Giles for binary protein-protein interactions using a combination of four yeast two-hybrid (Y2H) vectors to maximize coverage and reliability of the protein-protein interaction data set. This study provides the first interactome of any mycobacteriophage and one of only 5 published phage interactomes to date (15–18). A comparison of bacteriophage interactomes is shown in Table 1.
TABLE 1.
Comparison of bacteriophage interactomes
| Phage | Host | Genome size (bp) | No. of ORFs | No. of ORFs tested in Y2H | Final no. of PPIs | Reference |
|---|---|---|---|---|---|---|
| Giles (mycobacteriophage) | M. smegmatis | 53,746 | 77 | 75 | 136 | This study |
| Lambda | E. coli | 48,490 | 73 | 68 | 93 | 16 |
| T7 | E. coli | 39,937 | 55 | 55a | 25 | 17 |
| Dp-1 | S. pneumoniae | 56,506 | 72 | 72 | 156 | 15 |
| Cp-1 | S. pneumoniae | 19,345 | 28 | 28 | 15 | 18 |
This study used libraries of random phage genome fragments rather than full-length ORFs.
MATERIALS AND METHODS
Bait-and-prey array construction.
All Giles open reading frames (ORFs) were cloned into Gateway-compatible bait (pGBGT7g and pGBKCg) and prey (pGADT7g and pGADCg) vectors as previously described (19). Bait and prey plasmids were isolated from Escherichia coli, and the yeast strains AH109 (MATa) and Y187 (MATα) were transformed with both bait and prey plasmids, respectively (20, 21), using a slightly modified lithium acetate (LiAc) method (22). Briefly, the log-phase cells were washed and suspended in 1 ml of 0.1 M LiAc–Tris-EDTA (TE) buffer for 30 min before transformation. The cells were then suspended in a reaction mix of 40% polyethylene glycol (PEG) and 2.5 μl of 10-mg/ml boiled carrier DNA (Ambion sheared salmon sperm DNA; Life Technologies). At least 100 ng of plasmid DNA was added to each well/tube. After gentle shaking, tubes/plates were placed at 30°C for 45 min and then transferred to 42°C for 15 to 30 min. Plates were centrifuged, and cells were resuspended in 50 to 100 μl of sterile distilled water (dH2O) and spread on selective medium plates (e.g., without Leu [−Leu] or −Trp). All the bait and prey clones were grown in yeast extract-peptone-dextrose (YPD) supplemented with adenine in a 96-well plate overnight and were pinned onto selective medium plates (−Leu and −Trp, respectively) to verify transformation and growth. All bait-and-prey arrays were constructed and stored on selective (−Trp or −Leu) media. Working arrays were kept on YPD plates with 0.54 mM adenine to increase the mating efficiency.
A combination of N-terminal (pGBGT7g and pGADT7g) and C-terminal (pGBKCg and pGADCg) protein fusions (19) was used to enhance coverage and produce a reliable and credible interactome data set. Here, an N-terminal or a C-terminal protein fusion refers to the bait or prey containing the DNA binding domain or activation domain at its N terminus or C terminus, respectively. Each protein was tested both as both bait and as prey, with the exception of gp20 (the tape measure protein) and gp22 (a predicted minor tail subunit), two constructs we were unable to produce in any vector. The constructs of gp38 (a protein of unknown function) in pGADCg and gp50 (also a protein of unknown function) in pGBKCg could also not be produced but were each used with the other three members of the vector set. With the missing preys, 75 Giles preys (73 of which were used as both N- and C-terminal protein fusions) were available out of 77 protein-coding genes. In total, the use of two different protein fusion variants and two different bait-versus-prey arrangements yielded eight different configurations for each protein pair.
Autoactivation tests.
To ensure that our bait collections (in pGBGT7g and pGBKCg) were not acting as autoactivators, we mated them with yeast carrying empty prey vectors (pGADT7g and pGADCg). Giles baits gp11, gp28, and gp62 in pGBGT7g and gp11, gp25, and gp77 in pGBKCg showed weak to strong autoactivation (see Table S1B in the supplemental material). Interaction strength was titrated using concentrations of 3-aminotriazole (3-AT) between 1 and 100 mM; 3-AT is used as a competitive inhibitor of the HIS3 enzyme in yeast two-hybrid screens, allowing titration of HIS3 expression levels and growth resulting from positive results (20, 23). The baits gp11, gp25, and gp77 showed background growth with empty prey even at 100 mM 3-AT and were screened only in the pGBGT7g Y2H vector system. The remaining autoactivator baits were screened with the concentration of 3-AT found to minimize background growth.
Once the autoactivator baits were identified, we conducted our bait-versus-prey Y2H screens.
Array-based high-throughput multivector (MV)-Y2H screening.
Yeast two-hybrid (Y2H) array screens were performed as previously described (16). A flow chart of the procedure is shown in Fig. 1. Briefly, each bait (DBD-X) was mated with each prey (AD-Y) on rich medium (YPD plus adenine) in a 384-colony format for 36 to 48 h at 30°C. Diploid cells were selected for by pinning cultures from mating plates onto selective agar plates (−Leu −Trp) and growing them for 2 to 3 days. The diploids were then screened for interacting pairs by pinning them onto selective screening medium (−Leu −Trp −His) and incubating at 30°C for another 4 to 7 days. All baits (including self-activating baits) were screened on −Leu −Trp −His plates containing 3-AT to suppress nonspecific background; at least two different 3-AT concentrations between 1 and 100 mM were used for each screen to avoid elimination of true positives. The plates were monitored each day, and positive colonies were evaluated with respect to the background growth on each plate. A representative screen is shown in Fig. S1 in the supplemental material.
FIG 1.

(A) Overview of the bait-and-prey array construction for Y2H screens. This method includes use of a C-terminal and a N-terminal protein fusion for each bait-and-prey construct. (B) Size and coverage of the Giles interactome. A total of 91.12% of all possible bait-prey combinations (100%) were screened in this study.
Completeness of the interactome space was calculated as follows: the tested space is 74 × 73/2 = 2,701 pairs, the search space is 77 × 77/2 = 2,964 pairs, and percent completeness is (tested space/entire space) × 100 or 2,701/2,964 × 100 = 91.12%.
Filtering and retesting of raw interaction data.
Array-based Y2H screens can reduce the number of false positives (which are the nonreproducible signals that arise by self-activation or because of the “sticky” nature of some preys). We filtered out nonspecific raw Y2H data on the basis of prey count, with a few exceptions. Prey count is defined as the number of times a defined prey protein is found to be an interacting partner for a bait. The preys found to interact with 12 or more baits (an arbitrarily defined value specific to our raw data set only) were predicted to be the result of nonspecific interactions and were, with some exceptions, not included in the retest Y2H data set. A sticky prey was included in the retest data set if it was found to interact specifically and strongly at a 3-AT concentration with no background growth visible on the same plate.
We used the filtered set of raw protein-protein interactions to form a retest set. These interactions were tested as described above in a 384-colony format in quadruplicate (each colony was plated four times on each plate) for each bait-and-prey combination in all different vector configurations. Fresh bait-and-prey arrays were prepared specifically for these retests. All protein-protein interactions were quantitatively titrated against background using a series of different concentrations of 3-AT between 0 and 50 mM.
Quantitative assessment of results (% IScore).
A score, % 3-ATS, was calculated for each interacting bait-prey pair using the formula % 3-ATS = (CPPI − CB/CPPI) × 100, where % 3-ATS is the % 3-AT score calculated for each PPI, CPPI is the highest concentration of 3-AT at which a PPI was scored, and CB is the concentration of 3-AT at which background was observed. Thus, each interacting pair was assessed quantitatively and assigned a % 3-ATS which was used to calculate an overall interaction score (% IScore). Once we retested all PPIs, the % IScore was used to select high-confidence PPIs. The % IScore was calculated as IScore = 3-ATS+ ∑wk, where 3-ATS is the 3-AT score assigned to each PPI as described above and ∑wk = w1 + w2 + w3, where w1 is the weight value for PPIs detected in multiple vectors, directly proportional to the IScore (w1 = 0 if a PPI was detected by only a single vector or 33 if detected by at least 2 vectors), w2 is the weight value for reciprocal interactions, also directly proportional to the IScore (w2 = 0 if not found in a reciprocal set of interactions [e.g., A-B and B-A] or 50 if it is a reciprocal interaction), and w3 is the weight value for the prey count, inversely proportional to the IScore (w3 = 0, −5, −10, −15, −20, −25, or −30 for prey counts of 1, 2 to 5, 6 to 10, 11 to 15, 16 to 20, 21 to 25, or 26 to 30, respectively. Then, % IScore = (actual IScore for a given interacting pair/highest IScore observed for any interacting pair) × 100. See Fig. S5 in the supplemental material for a flowchart of this calculation and Table S2C in the supplemental material for a list of interactions with corresponding IScores.
Giles essential and nonessential proteins.
Each Giles protein was assigned an essentiality value based on that determined by Dedrick et al. (12). All proteins determined to be likely essential for the phage lytic cycle, whether by experimental observation or by their role as phage structural components, were designated “essential.” All other gene products were designated “nonessential.”
ER.
Excess retention (ER) values were calculated as described by Wuchty and Almaas (24). These values, when used in a comparison of essential versus nonessential nodes in the protein-protein interaction network, correspond to the degree to which essential proteins are over- or underrepresented relative to the full network (see Fig. S5 in the supplemental material). Values are provided for each k-core, that is, the subnetwork in which all nodes have a degree of at least k. In short, excess retention is defined for a particular k-core as ERKA = ekA/EA, where ekA is the fraction of proteins with property A for a k-core of Nk nodes and EA is the fraction of proteins with property A for the whole network. Because essentiality of each node in the network has been defined in a binary fashion such that each node is either essential or nonessential, a lower value in one category results in higher values for the other.
Protein interaction networks and bioinformatics analysis.
The PPI networks were constructed and analyzed using Cytoscape 3.1 (25), http://www.cytoscape.org/. Giles protein sequences were analyzed further using HHpred secondary structure prediction software (26) and the STRING protein association network tool (27).
Phamerator software (28) was used to create a map for illustration of protein-protein interactions. Twenty-two Giles proteins have homologs present in phages outside cluster Q (nondraft genomes in the database at http://phagesdb.org/).
Mass spectrometry of Giles particles and infected Mycobacterium smegmatis.
Wild-type Mycobacterium smegmatis mc2155 was infected with mycobacteriophage Giles at a multiplicity of infection (MOI) of 3. At 30 min and 2.5 h postinfection, a 1-ml aliquot was centrifuged, the supernatant removed, and the cell pellet immediately frozen. A high-titer Giles lysate was cesium chloride band purified twice and then submitted for mass spectrometry (MS) analysis along with the samples from the 30-min and 2.5-h postinfection time points. The mass spectrometry was performed by the University of California at Davis Proteomics Core on an LC-MS/MS Q-Exactive as described by Pope et al. (13). This study refers to three MS fractions: an early fraction (30 min postinfection), a late fraction (2.5 h postinfection), and the phage particle (whole virion only). Individual proteins may be present in more than one MS fraction.
RESULTS
Mapping the Giles interactome with a high-throughput multivector yeast two-hybrid approach.
We successfully screened a total of 2 × 74 and 2 × 73 bait strains (using all vectors) corresponding to 100% of all the available ORFs or ∼95% of ORFs in the mycobacteriophage Giles genome (Fig. 1A; see Table S1A in the supplemental material for the full list). Our Y2H screens covered 91.12% of all 2.96 × 103 possible bait-prey combinations, corresponding to 2.7 × 103 possible interactions. The first set of Y2H screens yielded 2,662 raw binary interactions. This set was filtered to produce a set of retesting candidates (∼1,000); retesting these baits and preys yielded 324 reproducible interactions, of which 171 PPIs (∼53% of the reproducible results, ∼17% of the filtered set, and ∼6% of the raw data) were deemed “high confidence.” Representative screens are shown in Fig. S1 to S3 in the supplemental material.
In this study, we used four different Gateway-compatible vectors (pGBGT7g, pGADT7g, pGBKCg, and pGADCg) to test for interactions among nearly all protein-protein pairs in the mycobacteriophage Giles proteome. We constructed bait-and-prey arrays by transferring Gateway-compatible Giles entry clones into the Y2H expression vectors, followed by transformation into mating-competent yeast strains. Our final arrays contained 74 and 73 baits (in pGBGT7g and pGBKCg as fusions to the Gal4 DNA binding domain, respectively) and 73 preys (in both pGADT7g and pGADCg as fusions to the Gal4 activation domain) (Fig. 1A). Clones corresponding to Giles proteins gp20 and gp22 were not available and were not included in the final arrays (see Table S1A in the supplemental material).
After constructing the arrays, the self-activating baits were identified within each vector combination (pGBKCg versus pGADCg, pGBKCg versus pGADT7g, pGBGT7g versus pGADT7g, and pGBGT7g versus pGADCg). The self-activating baits (a full list is available in Table S1B in the supplemental material) allowed for background growth signal when mated with an empty prey vector (that is, in the absence of any interacting protein partner). Hence self-activating baits are not ignored but are screened and included in the data set.
Special precautions were taken to ensure that bait self-activation did not lead to false-positive results. A series of 3-AT concentrations between 0 and 100 mM was used to suppress the background from self-activating baits (see Materials and Methods for details regarding the use of 3-AT). A few baits (gp11 and gp25), when expressed from pGBKCg, show background growth even at 100 mM 3-AT. The bait gp25 was an autoactivator in pGBKCg but not in other vector combinations.
In an effort to maximize assay sensitivity, we screened all the baits in binary pairs with preys. All interactions discussed in this study were collected from binary screens, followed by retesting. Images of each screen plate were produced (raw result images can be provided on request). All screens were performed in a 384-colony format, with each plate including ∼73 preys screened against each bait, performed in quadruplicate. An overview of the process is presented in Fig. 1A.
A series of 3-AT concentrations (0, 1, 3, 10, 25, and 50 mM) was used in Y2H retest screens to quantitatively assess PPIs. The retesting of filtered PPIs resulted in a data set of 324 reproducible PPIs. This data set includes 75 out of the 77 proteins (∼97%) in the mycobacteriophage Giles proteome (Fig. 1B).
Coverage and completeness of the Giles interactome.
Each protein was tested in 8 different configurations, either as bait or prey, using four sets of Y2H expression vectors (pGBKCg/pGADCg, pGBKCg/pGADT7g, pGBGT7g/pGADCg, and pGBGT7g/pGADT7g) (Fig. 1A). Out of the 77 predicted Giles proteins (or 75 proteins tested as either bait or prey) available in the Giles proteome, 75 proteins were found to be involved in protein-protein interactions (Fig. 1B). Out of 74 baits, 71 (95.9%) were observed in the interactome. In contrast, out of 73 tested preys, just 58 (79.4%) contributed to the interactome, though all 75 proteins contributed interactions across all bait-versus-prey combinations. Thus, the Giles interactome covers between 95% and 100% of its available, predicted proteome. The interactome size was calculated only for reproducible PPIs in our retest data set (see Table S2B in the supplemental material). For the set of high-confidence PPIs (IScore of >0; 171 PPIs), 70 Giles proteins were found to have at least one interacting partner, representing about 90% of the Giles predicted proteome. Ten Giles proteins (including scaffold protein gp8 and tail protein gp16, among others) were involved in only one interacting pair each (excluding self-interactions), and 6 proteins (gp24, gp31, gp39, gp60, gp62, and gp65) were found to have more than 10 interacting partners. The completeness of the Giles interactome space (that is, the percentage of tested pairs out of all possible protein pairs) was 91.12%. Full lists of protein-protein interactions are available in Tables S2A, S2B, and S2C in the supplemental material.
Each vector combination (2 N-terminal and 2 C-terminal fusions) (Fig. 2A) produced different, nonoverlapping, PPI sets. For example, the vector combination “CC” produced a set of 79 total PPIs, including 14 overlapping interactions with other vector combinations (e.g., 1 with NN and 13 with NC) (Fig. 2B). None of the high-confidence interactions were visible in more than two expression vector combinations. For example, the bait gp62, a putative DNA methylase protein, produced a total of 9 PPIs with CC and CN combinations, yielding 1 and 3 interactions, respectively. The N-terminal fusions of gp62 (NC and NN) yielded 3 and 2 interactions, respectively. No gp62 PPI was observed in more than one vector pair, confirming that different protein fusion result in different sets of PPIs. Numerous other examples are present across the interactome data set. Only 20 redundant PPIs (that is, those detected by more than one expression vector combination) were observed, comprising 6% of the data set. No interactions were detected in three or four vector combinations. The results clearly indicate that different vector combinations have different potencies to dissect the Giles interactome by exploring interactions resulting from different regions of each protein. Furthermore, the vector combinations produced a varied number of total PPIs at different 3-AT concentrations (Fig. 2A). The data shown here were reproducible in retests and were obtained at 3-AT concentrations which clearly differentiate background signal from true positive interactions.
FIG 2.
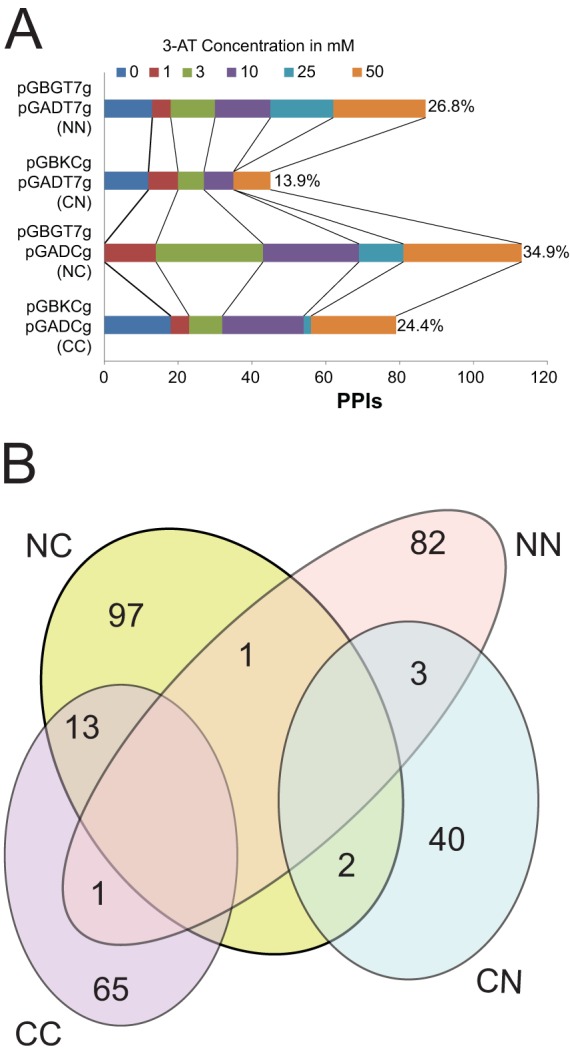
(A) Differences between Y2H vectors in dissecting the Giles interactome. The total number of PPIs detected by each vector pair at different 3-AT concentrations is shown. Percentages represent the contribution of each vector pair to the Giles interactome. (B) Reproducible PPIs detected by different Y2H vector combinations (CC, CN, NN, and NC). Note that 20 interactions are redundant in this Venn diagram.
Identification of high-confidence PPIs.
The filtering and retesting of the raw Y2H data resulted in a reproducible data set of 324 protein-protein interactions (PPIs). To further select the highest-confidence PPIs, an IScore was calculated and assigned to each reproducible interacting pair (see Materials and Methods and Fig. S4 and S8 in the supplemental material). The IScore for all PPIs ranges from −20 to 100. A PPI with an IScore of 100 was classified as the most reliable, and one with an IScore of −20 was the least reliable. All PPIs with an IScore of >0 were classified as high-confidence PPIs. Further scoring of PPIs resulted in a final set of 136 high-confidence (excluding redundant) PPIs (see Table S2C in the supplemental material).
Structure of the Giles interactome.
The Giles protein-protein interactome appears to be tightly connected and intricately cross-regulated. Few portions of the phage proteome appear to be enriched for interactions with neighboring proteins or with proteins in distinct regions (Fig. 3A). Rather, some segments of the proteome do not appear to interact with other segments: gp3 through gp12 produced very few interactions with each other, while proteins gp42 through gp57 produced few interactions among themselves or with gp1 through gp12. Most other regions of the interactome space yielded a pattern of interactions scattered across the other regions. The Giles lytic-phase transcripts (described in further detail in reference 12) place the interactome in the context of gene expression (Fig. 3A). This context reveals that protein interactions appear to cross transcript boundaries, a finding suggesting intricate cross-regulation.
FIG 3.
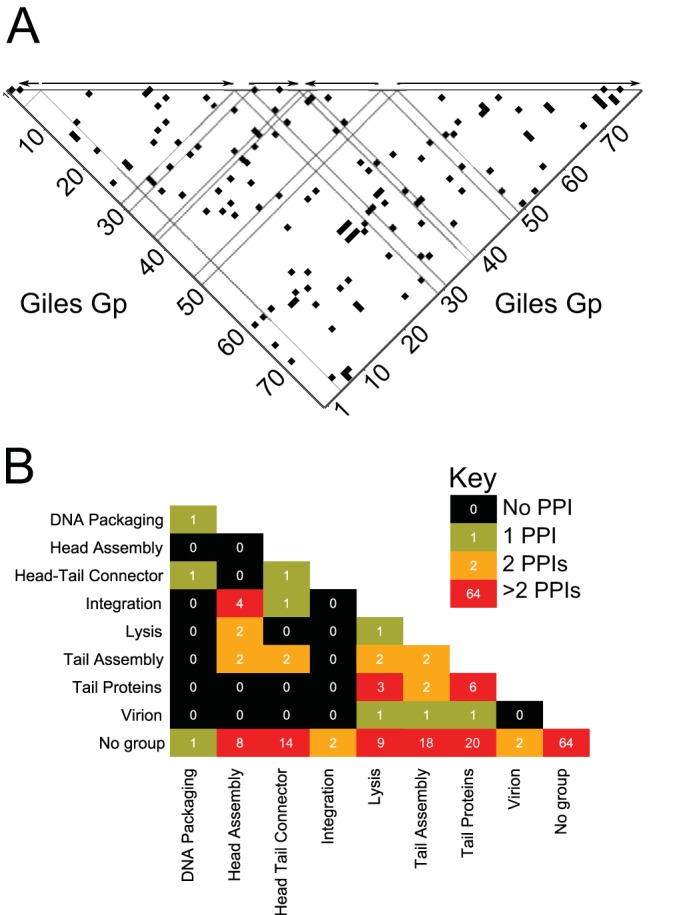
(A) All binary (including redundant) PPIs in the high-confidence set in the context of transcription. Each black square in the heat map represents an interaction between two proteins, regardless of their role as bait or prey. Dashed lines indicate borders of putative transcriptional units as described by Dedrick et al. (12). (B) Number of identified PPIs within and between functional groups.
The predicted proteome of Giles can be organized into functional groups, including structural (head and tail assembly), recombination (integration and excision), etc. We found many interactions within groups, such as those between phage structural proteins (∼11% of the total PPIs) (Fig. 3B; see Table S2F in the supplemental material). For example, gp6 (portal) was found to interact with gp24 and gp25, two tail assembly proteins.
There are 131 cross-functional interactions (∼40.5%) between structural proteins and other functional/regulatory proteins, including proteins of unknown function. For instance, gp26 (a tail assembly protein) and gp15 (a minor tail protein) interact with gp31 (LysA), thus connecting two different biological processes.
Another 157 PPIs (∼48.6%) were detected between nonstructural proteins, which include all 45 proteins of unknown function. For example, gp62, a DNA methylase, interacts with gp56 and gp57, two uncharacterized, potential regulatory proteins. Out of all PPIs, 136 are detected between two proteins of unknown function, accounting for about 86% of nonstructural PPIs or 42% of total PPIs. Thus, a major part of the Giles interactome is between functionally uncharacterized proteins (Fig. 3B), emphasizing the need for further protein characterization. Some proteins, including the RecE- and RecT-like gp52 and gp53, respectively, have predicted functions based on their homology with other protein sequences but are considered uncharacterized in this context due to their low sequence similarity with their closest potential homologues.
Topology of the Giles protein interaction network.
Figure 4A presents the Giles protein interaction network: each edge indicates a pair of nodes that interact at least once in the high-confidence data. The full high-confidence interaction set contains 171 pairs, though these include reciprocal interactions and make distinctions between different expression vector pairs. The compressed network of high-confidence interactions among Giles proteins, as seen in Fig. 4A, is a set of 70 nodes and 136 edges, where each node is a unique protein and each edge is an interaction between two proteins. The average number of neighbors within this network is 3.69; all nodes except gp1 are connected in at least one path. The network demonstrates a loose fit to the generally expected power law distribution (r2 = 0.615), though this may be the result of a small data set and the filters used to render it more biologically relevant. Despite its small size, this network has a clustering coefficient of 0.068, a value similar to those observed for yeast and Caenorhabditis elegans PPI networks (29). Even after filtering, the Giles interactome demonstrates high interconnectivity, with the majority of shortest paths between nodes including 4 edges at most.
FIG 4.

(A) The protein-protein interaction network of bacteriophage Giles. Proteins are represented as nodes; the node color corresponds to the general functional role as noted in the key. Proteins found or predicted to be essential to the phage lytic cycle are denoted by node shape. Proteins found in any MS fraction of those obtained are denoted by black outlines; those not found are outlined in red. Interactions are represented as edges between nodes. All edges are representative of high-confidence Y2H results; brighter green edges are those with % IScores approaching a maximum confidence value of 100, while those approaching zero are gray. Red edges are those with % IScores of zero. Solid edges are those found both in Y2H results and between proteins found to be present in the same mass spectrometry (MS) fraction (out of three possible fractions; see Materials and Methods for more details). Dashed edges represent predicted interactions found in Y2H results between proteins not found to be present in the same MS fraction. (B) Protein interactions related to Giles virion structure. The node color corresponds to the general functional role as noted in the key; these groups differ from those in panel A. Shapes represent the presence of a particular protein at specific infection stages as shown by MS data.
The Giles interactome network can be separated into subgraphs on the basis of gene essentiality, such that each subgraph contains only interactions between proteins essential or nonessential to the lytic cycle. These subgraphs contain all nodes with a specified property (essential or nonessential), with the exception of those which do not interact with any proteins with the same property. Examining interactions between essential, or likely essential (12), proteins (Fig. 4A), we see that this set of 32 nodes contains predominantly structural and assembly proteins (18 in total). Out of these 32 proteins, 9 have no known or predicted function. Conversely, the set of interactions between nonessential proteins (Fig. 4A) is enriched for uncharacterized proteins. This 25-node set contains just 5 nodes with any predicted function.
Benchmarking and validating the Giles interactome using mass spectrometry.
Neither the genome nor the interactome provides any information about the abundance of proteins or whether they are expressed. One method of benchmarking detected protein-protein interactions is to measure coexpression of proteins. If two interacting proteins are coexpressed at the same time interval during the bacteriophage life cycle, they are more likely to interact than two proteins present during different times of the cycle. Thus, to provide further support for our data, we did mass spectrometry analysis of phage particles and infected host cells (see Materials and Methods and Table S1A in the supplemental material). Interestingly, we were able to detect 46 of the 77 Giles proteins by MS (Fig. 4; see Table S1A in the supplemental material), and this subset was enriched for essential proteins (29 in total, versus 12 among undetected proteins) and structural/assembly proteins (19 in total, versus 2 among undetected proteins). A set of 42 proteins was detected in the Giles particle alone, while early (30-min postinfection) and late (2.5-h postinfection) lysate samples contained 16 and 42 unique proteins, respectively. When arranged with the interaction network, these results also provide evidence for interactions among more than two proteins at a time. For instance, the minor tail protein gp21 may participate in interactions with both gp36 (a predicted virion protein without a known function) and the repressor protein gp47.
About one-quarter of interacting pairs are coexpressed and detected in the same sample, validating the PPI data set. Thirty-one Giles proteins, however, including both essential and structural proteins, were not captured in any MS sample (Fig. 4).
Predicting functions for unknown Giles proteins.
Connections between proteins of known and unknown function can provide evidence to aid in function prediction. For example, gp59, a protein of unknown function that is nonessential for lytic growth, interacts with almost all of the tail assembly proteins (including gp24, gp25, and gp26). The gp59 deletion mutant has an increased rate of lysogeny and reduced fecundity (12). Due to its interactions with tail assembly proteins, we hypothesize that gp59 is also required for tail assembly and thus that deletion of the protein may lead to structural defects that cause failure in assembly and prevent completion of the lytic cycle.
gp50 (a protein of unknown function) appears to interact with gp61 (DnaQ), a replication-associated protein. Thus, gp50 may have a shared functional role in DNA replication. Additionally, the results of fecundity and lysogeny assays with gp50 deletion mutants (12) suggest that gp50 could be a protein that affects a major biological process common to both lytic and lysogenic phases. Dedrick et al. (12) also predicted that gp50 is a DNA replication-associated protein, further supporting our data.
Protein-protein interactions involving structural proteins.
Proteins in the virion are expected to be among the most abundant phage proteins; hence, they are detected relatively easily by MS (see Table S1A in the supplemental material). For instance, the head-tail connector structure is well-represented: gp12 and 14 both interact with tail assembly proteins. The structural role of head-tail connector proteins gp10 and 11 is less clear, as they interact with each other but not with any other head-tail connector components. Out of 9 tail proteins confirmed to be present in the virion (12), 6 yielded direct interactions with other structural components (see Table S2D in the supplemental material), and 7 were found in interactions with other nonstructural proteins (see Table S2E in the supplemental material), though just 5 interactions are between proteins found cooccurring in the MS results (Fig. 4). This suggests that many more proteins are part of the virion (or involved in its assembly) than are detected by MS.
Topological coherence of the Giles interactome.
To estimate the degree to which essential and nonessential proteins are over- or underrepresented within the network (see Fig. S5 in the supplemental material), we investigated the excess retention among essential proteins in the network (24). Essential proteins are all those identified by Dedrick et al. (12) to be essential for the phage's lytic cycle, whether by experimental observation or inferred from predicted structural roles. Essential and nonessential Giles proteins appear to retain similar topological characteristics for k values of <10. For k values of >10, essential proteins appear to be overrepresented. This trend decreases for k values of >17, potentially due to the limited number of nodes present in these k-cores. It is likely that many of the connections seen for very highly connected nodes (i.e., those with k values of >15) are false positives, though these results show that the most highly connected nodes are generally essential nodes (see Materials and Methods for more details).
Conserved Giles interactome.
Giles is an unusual mycobacteriophage; it has only a few relatives, and they are nearly identical. As a consequence, only 19 Giles proteins have homologs outside its cluster (Q). We have summarized homologies of Giles (see Fig. S6 in the supplemental material) alongside its high-confidence interactions (score of >30) (see Fig. S7 in the supplemental material). Surprisingly, only two interaction pairs are conserved in other phages outside the Q cluster: gp5 and gp76 were found to interact in Giles, and homologs of both proteins are found in cluster P phages (e.g., Fishburne). Similarly, homologs of the Giles interaction pair gp26 and gp31 are found in the cluster F phage Brocalys.
DISCUSSION
More than half of the mycobacteriophage Giles proteome is without functional annotation, including many essential and nonessential proteins of unknown function. Thus, dissecting the Giles proteome can provide hints about putative protein functions and how these proteins form functional or structural associations in a tightly connected Giles PPI network. We initially attempted a pooled screening approach by screening pools of 5 to 7 non-self-activating baits at a time. Pooling baits appeared to decrease assay sensitivity and reduced the number of observed interactions per bait. We suspect that diminished sensitivity is the result of a reduced number of yeast cells per bait available for mating with prey-containing yeast cells. Thus, an array-based Y2H system was used to map the protein interactome of mycobacteriophage Giles.
More than 45% of the reproducible PPIs in our screens are of high confidence (see Table S2C in the supplemental material) and are shown in the final PPI network (Fig. 4A). The essential proteins in the Giles PPI network are more tightly connected than nonessential proteins. Thus, essential proteins are overrepresented in the Giles interactome. About 86% of the nonstructural PPIs or 42% of the total PPIs in this study involve proteins of unknown functions. A major part of the Giles interactome is between functionally unknown proteins, emphasizing the need to characterize and understand their role in phage biology. Multiple vectors in a Y2H screen mimic the PPI data detected by different methods (30). We used an MV-Y2H system to test each protein as N- and C-terminal bait and prey fusions. Thus, each protein was tested in 8 different configurations, which increases the chances of detecting an interaction 8-fold. The use of multiple vectors also minimizes the impact of self-activating baits. The baits found as self-activating in one vector combination did not show self-activation in others. Thus, the multivector Y2H can balance the quality and size of the interactome by reducing both false positives and false negatives. In this study, we took additional care to minimize false negatives and false positives by considering multiple parameters of each interaction across all four vector pairs.
The Giles gp76 protein interacts strongly with gp5, a large subunit of terminase enzyme required for DNA packaging and maturation. gp76 has similarity to HNH endonucleases. HNH motif-containing endonuclease proteins may interact with phage terminase proteins to promote phage DNA packaging and maturation (31). For example, a recent report showed that gp74 (an HNH endonuclease) of phage HK97 interacts with its terminase protein (32) and that the HNH motif of endonuclease is essential for interacting with terminase and completion of DNA packaging. Also, lambda gpFI, an endonuclease, interacts with lambda terminase (16). Many phages encode an HNH protein (endonuclease) located adjacent to the phage DNA-packaging enzyme terminase, suggesting roles in phage DNA packaging and maturation (33). It is important and interesting that all of the phages, including mycobacteriophage Giles, harboring hnh genes are cos phages with linear DNA containing cohesive ends. Also, the phages ϕ12 and the ϕSLT require HNH nuclease as well as TerL (terminase large subunit) for cos site cleavage and ultimately for DNA packaging (34). A hypothetical protein of Mycobacterium avium 104 (MAV_0815), found to be similar to Giles gp76, interacts with the large subunit of phage terminase (MAV_0813, STRING [27]). Thus, the existing evidence and the nature of the Giles gp76-gp5 interaction observed in this study suggest that Giles gp76 may be essential for DNA packaging and maturation of the phage. Interestingly, both gp5 and gp76 appear to be conserved in some mycobacteriophages from the P cluster, suggesting that this functional pairing is maintained/conserved in more distantly related phages.
Somewhat unexpectedly, no interaction could be found with the capsid protein gp9, not even with itself, possibly because the capsid proteins have not been processed or because no scaffold proteins were present in our screens. The gp6 (portal) and gp5 (terminase) proteins, though expected to interact, did not produce reproducible interactions due to high background signal. This PPI was also not observed between portal and terminase in the Streptococcus phage Dp-1 interactome (15). Interactions between gp20 (tape measure protein) and other proteins were expected but were not tested in this study, though the observed interaction between tail protein gp15 and gp31 (LysA) may present a similar type of interaction.
Harnessing phage activities and phage-derived proteins may offer new venues for phage therapy with mycobacterial infections. Recent research into mycobacteriophage therapy for Mycobacterium ulcerans (35) has shown promise. Similar studies with other mycobacterial infections and phages could prove similarly fruitful. Mycobacteriophages, with their mosaic genomes and great genetic diversity, offer a multitude of options for manipulating or controlling mycobacteria.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the U.S. National Institutes of Health grants R01GM109895 and 8U54HD080784.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00164-15.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.Hatfull GF. 2014. Mycobacteriophages: windows into tuberculosis. PLoS Pathog 10:e1003953. doi: 10.1371/journal.ppat.1003953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang KC, Yew WW. 2013. Management of difficult multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: update 2012. Respirology 18:8–21. doi: 10.1111/j.1440-1843.2012.02257.x. [DOI] [PubMed] [Google Scholar]
- 4.Rybniker J, Kramme S, Small PL. 2006. Host range of 14 mycobacteriophages in Mycobacterium ulcerans and seven other mycobacteria including Mycobacterium tuberculosis—application for identification and susceptibility testing. J Med Microbiol 55:37–42. doi: 10.1099/jmm.0.46238-0. [DOI] [PubMed] [Google Scholar]
- 5.Jones WD., Jr 1975. Phage typing report of 125 strains of “Mycobacterium tuberculosis”. Ann Sclavo 17:599–604. [PubMed] [Google Scholar]
- 6.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182. doi: 10.1016/S0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 7.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, Hoyte NN, Bowman CA, Tantoco AT, Paladin EC, Myers MS, Smith AL, Grace MS, Pham TT, O'Brien MB, Vogelsberger AM, Hryckowian AJ, Wynalek JL, Donis-Keller H, Bogel MW, Peebles CL, Cresawn SG, Hendrix RW. 2010. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J Mol Biol 397:119–143. doi: 10.1016/j.jmb.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, Alcoser TA, Alexander LM, Alfano MB, Alford ST, Amy NE, Anderson MD, Anderson AG, Ang AAS, Ares M Jr, Barber AJ, Barker LP, Barrett JM, Barshop WD, Bauerle CM, Bayles IM, Belfield KL, Best AA, Borjon A Jr, Bowman CA, Boyer CA, Bradley KW, Bradley VA, Broadway LN, Budwal K, Busby KN, Campbell IW, Campbell AM, Carey A, Caruso SM, Chew RD, Cockburn CL, Cohen LB, Corajod JM, Cresawn SG, Davis KR, Deng L, Denver DR, Dixon BR, Ekram S, Elgin SCR, Engelsen AE, English BEV, Erb ML, Estrada C, Filliger LZ, et al. . 2011. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One 6:e16329. doi: 10.1371/journal.pone.0016329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatfull GF, Pedulla ML, Jacobs-Sera D, Cichon PM, Foley A, Ford ME, Gonda RM, Houtz JM, Hryckowian AJ, Kelchner VA, Namburi S, Pajcini KV, Popovich MG, Schleicher DT, Simanek BZ, Smith AL, Zdanowicz GM, Kumar V, Peebles CL, Jacobs WR Jr, Lawrence JG, Hendrix RW. 2006. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet 2:e92. doi: 10.1371/journal.pgen.0020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatfull GF. 2014. Molecular genetics of mycobacteriophages. Microbiol Spectrum 2(2):MGM2-0032-2013. doi: 10.1128/microbiolspec.MGM2-0032-2013. [DOI] [PubMed] [Google Scholar]
- 11.Morris P, Marinelli LJ, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2008. Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J Bacteriol 190:2172–2182. doi: 10.1128/JB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. 2013. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol Microbiol 88:577–589. doi: 10.1111/mmi.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pope WH, Jacobs-sera D, Russell DA, Rubin DHF, Kajee A, Msibi ZNP, Larsen MH, Jacobs WR, Lawrence JG, Hendrix RW, Hatfull F. 2014. Genomics and proteomics of mycobacteriophage Patience, an accidental tourist in the mycobacterium neighborhood mBio 5(6):e02145-11. doi: 10.1128/mBio.02145-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauser R, Blasche S, Dokland T, Haggard-Ljungquist E, von Brunn A, Salas M, Casjens S, Molineux I, Uetz P. 2012. Bacteriophage protein-protein interactions. Adv Virus Res 83:219–298. doi: 10.1016/B978-0-12-394438-2.00006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabri M, Hauser R, Ouellette M, Liu J, Dehbi M, Moeck G, Garcia E, Titz B, Uetz P, Moineau S. 2011. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J Bacteriol 193:551–562. doi: 10.1128/JB.01117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajagopala SV, Casjens S, Uetz P. 2011. The protein interaction map of bacteriophage lambda. BMC Microbiol 11:213. doi: 10.1186/1471-2180-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartel PL, Roecklein JA, SenGupta D, Fields S. 1996. A protein linkage map of Escherichia coli bacteriophage T7. Nat Genet 12:72–77. doi: 10.1038/ng0196-72. [DOI] [PubMed] [Google Scholar]
- 18.Häuser R, Sabri M, Moineau S, Uetz P. 2011. The proteome and interactome of Streptococcus pneumoniae phage Cp-1. J Bacteriol 193:3135–3138. doi: 10.1128/JB.01481-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stellberger T, Hauser R, Baiker A, Pothineni VR, Haas J, Uetz P. 2010. Improving the yeast two-hybrid system with permutated fusions proteins: the varicella zoster virus interactome. Proteome Sci 8:8. doi: 10.1186/1477-5956-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816. doi: 10.1016/0092-8674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- 22.Gietz RD, Woods RA. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96. [DOI] [PubMed] [Google Scholar]
- 23.Cagney G, Uetz P, Fields S. 2000. High-throughput screening for protein-protein interactions using two-hybrid assay. Methods Enzymol, 328:3–14. [DOI] [PubMed] [Google Scholar]
- 24.Wuchty S, Almaas E. 2005. Peeling the yeast protein network. Proteomics 5:444–449. doi: 10.1002/pmic.200400962. [DOI] [PubMed] [Google Scholar]
- 25.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang PL, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, Bader GD. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biegert A, Mayer C, Remmert M, Soding J, Lupas AN. 2006. The MPI bioinformatics toolkit for protein sequence analysis. Nucleic Acids Res 34:W335–W339. doi: 10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. 2011. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395. doi: 10.1186/1471-2105-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedel CC, Zimmer R. 2006. Inferring topology from clustering coefficients in protein-protein interaction networks. BMC Bioinformatics 7:519. doi: 10.1186/1471-2105-7-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YC, Rajagopala SV, Stellberger T, Uetz P. 2010. Exhaustive benchmarking of the yeast two-hybrid system. Nat Methods 7:667–668. doi: 10.1038/nmeth0910-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moodley S, Maxwell KL, Kanelis V. 2012. The protein gp74 from the bacteriophage HK97 functions as a HNH endonuclease. Protein Sci 21:809–818. doi: 10.1002/pro.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kala S, Cumby N, Sadowski PD, Hyder BZ, Kanelis V, Davidson AR, Maxwell KL. 2014. HNH proteins are a widespread component of phage DNA packaging machines. Proc Natl Acad Sci U S A 111:6022–6027. doi: 10.1073/pnas.1320952111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu SY, Gupta YK. 2013. Natural zinc ribbon HNH endonucleases and engineered zinc finger nicking endonuclease. Nucleic Acids Res 41:378–390. doi: 10.1093/nar/gks1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quiles-Puchalt N, Carpena N, Alonso JC, Novick RP, Marina A, Penades JR. 2014. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc Natl Acad Sci U S A 111:6016–6021. doi: 10.1073/pnas.1320538111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trigo G, Martins TG, Fraga AG, Longatto-Filho A, Castro AG, Azeredo J, Pedrosa J. 2013. Phage therapy is effective against infection by Mycobacterium ulcerans in a murine footpad model. PLoS Negl Trop Dis 7:e2183. doi: 10.1371/journal.pntd.0002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.