

ABSTRACT
A key regulator of swarming in Proteus mirabilis is the Rcs phosphorelay, which represses flhDC, encoding the master flagellar regulator FlhD4C2. Mutants in rcsB, the response regulator in the Rcs phosphorelay, hyperswarm on solid agar and differentiate into swarmer cells in liquid, demonstrating that this system also influences the expression of genes central to differentiation. To gain a further understanding of RcsB-regulated genes involved in swarmer cell differentiation, transcriptome sequencing (RNA-Seq) was used to examine the RcsB regulon. Among the 133 genes identified, minC and minD, encoding cell division inhibitors, were identified as RcsB-activated genes. A third gene, minE, was shown to be part of an operon with minCD. To examine minCDE regulation, the min promoter was identified by 5′ rapid amplification of cDNA ends (5′-RACE), and both transcriptional lacZ fusions and quantitative real-time reverse transcriptase (qRT) PCR were used to confirm that the minCDE operon was RcsB activated. Purified RcsB was capable of directly binding the minC promoter region. To determine the role of RcsB-mediated activation of minCDE in swarmer cell differentiation, a polar minC mutation was constructed. This mutant formed minicells during growth in liquid, produced shortened swarmer cells during differentiation, and exhibited decreased swarming motility.
IMPORTANCE This work describes the regulation and role of the MinCDE cell division system in P. mirabilis swarming and swarmer cell elongation. Prior to this study, the mechanisms that inhibit cell division and allow swarmer cell elongation were unknown. In addition, this work outlines for the first time the RcsB regulon in P. mirabilis. Taken together, the data presented in this study begin to address how P. mirabilis elongates upon contact with a solid surface.
INTRODUCTION
Proteus mirabilis, a Gram-negative member of the family Enterobacteriaceae, is a bacterium well known for its ability to swarm. Swarming is a specialized form of motility displayed by multicellular groups of flagellated bacteria across a solid or semisolid surface. In liquid culture, P. mirabilis exists as a peritrichously flagellated, rod-shaped cell. However, after coming into contact with a solid surface, the cells undergo differentiation into elongated, highly flagellated, multinucleate swarmer cells. Swarmer cells are 20- to 50-fold longer than vegetative cells and express thousands of flagella (1). Together, these swarmer cells form multicellular rafts, which they utilize to move across a solid surface (2). After a period of migration, the swarmer cells undergo consolidation (or dedifferentiation) and revert to vegetative rods. The repeated interchange from differentiation to consolidation is responsible for the characteristic bull's eye pattern that P. mirabilis forms on an agar plate (3, 4). Reviews on P. mirabilis swarming provide additional details on this process (5, 6).
The switch from a rod-shaped cell to a swarmer cell is a complex process involving several global regulatory factors. The regulator of colonic acid capsule synthesis (Rcs) phosphorelay is one of these important regulators. The Rcs phosphorelay consists of a sensor kinase (RcsC), a response regulator (RcsB), and a phosphotransferase (RcsD), which mediates the transfer of the phosphate from RcsC to RcsB (7, 8). An additional protein, RcsF, is an outer membrane lipoprotein that increases the levels of RcsC phosphorylation by some unknown mechanism (9). Once the system is activated, it results in phosphorylated RcsB, which represses flhDC (10). flhDC encodes the master regulator for flagellar synthesis, FlhD4C2, which controls genes central to flagellin production (11). The levels of flhDC increase in swarming cells (10, 12), and flhDC mutants do not swarm (10, 11). Factors that influence flhDC expression, such as activated RcsB, can have dramatic effects on the ability of P. mirabilis to swarm. When the Rcs system is active, for example, the cells exist as vegetative rods due to repression of flhDC; however, in rcs mutants, P. mirabilis cells hyperswarm due, in part, to increased flhDC expression (10, 13, 14). Another interesting phenotype of rcs mutants in P. mirabilis is the ability of cells to differentiate into swarmer cells in liquid; this phenomenon does not occur in wild-type cells or in cells overexpressing flhDC (10, 13, 14), suggesting that other genes within the Rcs regulon are involved in swarmer cell elongation. The external factors that influence the expression of the Rcs phosphorelay and FlhD4C2, and subsequently the cycles of differentiation and consolidation, are unknown. Cell-to-cell contact (4, 15) and extracellular signaling (16) are among the hypothesized factors that could play a role in these genetic and morphological cycles.
In Escherichia coli and other members of the Enterobacteriaceae, RcsB activates genes involved in cell division (17, 18), cell wall synthesis (17, 18), and virulence (19) while repressing genes involved in motility (10, 20). The rationale for investigating genes regulated by the Rcs phosphorelay in P. mirabilis lies in the observation that rcs mutants hyperswarm on solid agar and differentiate into swarmer cells in liquid culture. Therefore, it is inferred that the Rcs phosphorelay regulates the expression of genes important for swarmer cell formation, including elongation. One subset of genes activated by RcsB in other bacteria is those involved in cell division (17, 18). However, the role of the cell division machinery in P. mirabilis swarmer cell formation has not been investigated.
Cell division in many prokaryotes is dictated by the placement of the FtsZ-mediated Z-ring (21), whose positioning is determined by a group of negative regulators known as the Min system. The Min system is comprised of three proteins, MinC, MinD, and MinE, whose oscillation prevents the formation of the Z-ring at the poles of a rod-shaped cell (22, 23). MinC acts as the effector of this system by preventing FtsZ polymerization (23, 24). MinD binds the cell membrane in an ATP-dependent manner (25), where it recruits MinC and activates it 25- to 50-fold (24). The ATPase activity of MinD, which causes it to disassociate with the cell membrane, is induced by MinE (26). This trait of MinE, along with its ability to suppress the activity of MinCD (27), restricts cell division inhibition to one pole at a time and is responsible for the oscillating nature of the complex. When MinE stimulates disassociation of the complex at one pole, MinD-ADP moves to the opposite pole, where it recruits MinC, and the process begins again. A min mutant produces anucleate minicells from the pole of a mother cell, which, as a result of minicell production, is slightly enlarged (28). Though cells elongate during cell division inhibition, the potential roles these three proteins play in swarmer cell formation and motility have not yet been investigated.
In this study, we elucidate the regulation and role of the Min system in P. mirabilis swarming. We found that a minC::Kanr mutation resulted in aberrant cell division in both vegetative and swarmer cells and decreased swarming motility. Additionally, we identified the transcriptional start site of the minCDE operon and demonstrated that the region encompassing this start site is bound by RcsB, the response regulator in the Rcs phosphorelay. Overall, our data demonstrate that the cell division inhibition machinery contributes to swarmer cell formation and identify an important regulator of the Min system.
MATERIALS AND METHODS
Bacterial growth conditions.
Except where indicated, P. mirabilis and E. coli strains (Table 1) were grown in modified Luria-Bertani (LB) broth (10 g tryptone, 5 g yeast extract, 5 g NaCl per liter) at 37°C with shaking at 250 rpm. All the strains were incubated at 37°C. E. coli strains were grown on 1.5% LB agar, and P. mirabilis strains were grown on 3.0% LB agar to prevent swarming. Antibiotic selection for E. coli contained the following concentrations: 100 μg/ml ampicillin, 20 μg/ml kanamycin, and 25 μg/ml streptomycin. P. mirabilis antibiotic selection used 300 μg/ml ampicillin, 20 μg/ml kanamycin, 35 μg/ml streptomycin, and 15 μg/ml tetracycline. When appropriate, a concentration of 12 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was used for the selection of blue or white colonies for both E. coli and P. mirabilis. For induction of protein expression, a concentration of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was used.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description and/or genotypea | Source or reference |
|---|---|---|
| E. coli | ||
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Laboratory stock |
| CC118 λpir | araD139 Δ(ara leu)7697 ΔlacZ74 phoAΔ20 galE galK thi rpsE rpoB argE(Am) recA1 | 37 |
| SM10 λpir | thi thr leu tonA supE recA RP4-2Tc::Mu Kanr λpir | 38 |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | Stratagene |
| P. mirabilis | ||
| PM7002 | Wild type; Tcr | ATCC |
| HI4320 | Wild type; Tcr | 39 |
| PMKH1 | PM7002 minC::Kanr | This study |
| PMKM1 | PM7002 rcsB::Strr | 10 |
| PMKH4 | PM7002 minC::Kanr rcsB::Strr | This study |
| PMKH5 | PM7002 minC::Kanr(pMinCDE) | This study |
| PMKH6 | PM7002(pMinClacZ) | This study |
| PMKH7 | PM7002 rcsB::Strr(pMinClacZ) | This study |
| PMKM2 | HI4320 rcsB::Strr | This study |
| Plasmids | ||
| pBluescriptII SK(−) | ColE1 replicon; lacZα Ampr | Stratagene |
| pBC | High copy number; Cmr | Stratagene |
| pKNG101 | R6K replicon; mob+ sacB+R+ Strr | 40 |
| pTrc99a | ColE1 replicon; trcPO lacIq Ampr | 41 |
| pQF50 | pRO1600 replicon; lacZα (promoterless); Ampr | 29 |
| pET21a | T7 expression vector; Ampr | Novagen |
| pRcsB | pKNG101::rcsB (internal fragment); Strr | 10 |
| pMinC | pKNG101 minC::Kanr Strr | This study |
| pBCMinCDE | pBC minCDE Cmr | This study |
| pMinCDE | pKNG101 minCDE Strr | This study |
| pMinClacZ | pQF50 minCp Ampr | This study |
| pETRcsB | pET21a rcsB Ampr | This study |
Kanr, kanamycin resistant; Tcr, tetracycline resistant; Strr, streptomycin resistant; Ampr, ampicillin resistant; Cmr, chloramphenicol resistant.
DNA manipulations and transformations.
All PCR amplifications were done using Phusion Hot Start II high-fidelity DNA polymerase (Thermo Scientific) and verified by DNA sequencing. All ligations were performed using the Fast-Link DNA ligation kit (Epicentre). For electroporation of plasmids into both E. coli and P. mirabilis, 30 ml of cells was grown to an optical density at 600 nm (OD600) of 0.3 to 0.5 in LB broth. The cells were pelleted by centrifugation at 3,000 × g for 5 min at 4°C before being washed twice with 1 ml of ice-cold 10% glycerol and resuspended in 60 μl of ice-cold 10% glycerol per electroporation. Electroporations were done in Bio-Rad Gene Pulser 0.2-cm-diameter cuvettes with a Bio-Rad MicroPulser electroporator. Cells were plated on the appropriate antibiotics after 1.5 h shaking at 250 rpm at 37°C. For P. mirabilis transformations, competent cells were incubated for 30 min with plasmid DNA on ice prior to electroporation. P. mirabilis electroporations were allowed to recover for 3 h.
Construction of strains.
For bacterial conjugations, overnight cultures were washed 3 times in 1 ml of fresh LB broth. One hundred microliters of each strain was combined in a 1.5-ml microcentrifuge tube before being added to a predried 1.5% LB agar plate; 200 μl of each strain alone was added to separate plates to serve as controls. After 7 h of incubation at 37°C, the cells were resuspended in 4 ml LB broth. The mating mixture and the individual control strains were plated on 3% LB agar containing the appropriate antibiotics and incubated overnight at 37°C.
To construct a minC mutant, the minC gene was amplified using minC-F and minC-R primers (Table 2) and ligated into pBluescript SK (Stratagene) digested with XbaI and BamHI (New England BioLabs). The cloned minC gene was then mutagenized using an EZ-Tn5 <Kan-2> TNP Transposome kit (Epicentre). A minC::EZ-Tn5 insertion in the center of the minC gene at a position 387 bp into the 702-bp minC open reading frame (ORF) was identified by restriction enzyme analysis and confirmed by DNA sequencing. This is referred to here as minC::Kanr. This was then subcloned into pKNG101, creating pMinC. pKNG101 is a suicide vector harboring streptomycin resistance, used to select for initial pKNG101 integration, and the sacB gene, which selects for the loss of pKNG101 in the presence of 10% sucrose. pMinC was maintained in E. coli SM10 λpir. This strain was conjugated with wild-type PM7002, and selection for PM7002 harboring the Campbell-type insertion at the minC locus was done on LB plates containing tetracycline (15 μg/ml) and streptomycin (35 μg/ml). An exconjugant was grown in LB broth for only approximately 10 generations and then plated on LSW agar (10 g tryptone, 5 g yeast extract, 5 ml glycerol, and 20 g agar per liter) containing 10% sucrose and kanamycin to select for clones that had lost pMinC but contained the minC locus harboring the EZ-Tn5 Kanr insertion. Clones that were Strs Kanr and sucrose resistant were subjected to Southern blot analysis to confirm they harbored the minC::Kanr mutation. One of these strains, PMKH1 (PM7002 minC::Kanr), was used for subsequent experiments.
TABLE 2.
Primer sequences used in this study
| Primer | Sequence (5′→3′) | Purpose |
|---|---|---|
| minC-F | TTTTTCTAGATTTTTGAGCGAGGCCGGTAG | minC mutation |
| minC-R | TTTTGGATCCAGCGACTTGCCTGAATATTTTAGT | minC mutation |
| PminC-F | TTTTGTCGACGCGCGAGATCTAGCAACATT | minC promoter activity |
| PminC-R | TTTTGGATCCCCTGAGCTGCTGGCTTAGTT | minC promoter activity |
| GSP1 | CCAATATTGACCAGCAATGGAAACT | 5′-RACE |
| GSP2 | ACACCATAAATATGTACATTACCGTCT | 5′-RACE |
| minCSS-F | TTAAGAATTCCGCGAGATCTAGCAACATTGCA | DNA binding studies |
| minCSS2-F | TTAAGAATTCAGCGACTTGCCTGAATATTTTAGT | DNA binding studies |
| minCSS2-R | TTAAGAATTCAATGGGCGTGTTTGACATCT | DNA binding studies |
| abaI-F | AAGTTAATCGCTTCTTGCAG | DNA binding studies |
| abaI-R | AGCTTATGTGGTCGCTCAAG | DNA binding studies |
| minCDE-F | ATAATTAAAACACAGAGCC | Complementing strain |
| minCDE-R | CCAGATTATAGAAAATTTAC | Complementing strain |
| minCqRT-F | CCCAAGCGCCTCAATTTCTG | qRT-PCR studies |
| minCqRT-R | TTCCCACAACACGTAAGCCA | qRT-PCR studies |
| minDqRT-F | CCACCAACCCAGAAGTCTCC | qRT-PCR studies |
| minDqRT-R | CGGCCTGGATTATAGCGTGT | qRT-PCR studies |
| minEqRT-F | CGCGTAAAAAGTCGACAGCA | qRT-PCR studies |
| minEqRT-R | TAAGCTGGCTCAGTATCGCC | qRT-PCR studies |
| 16S-F | GGCTCAGATTGAACGCTGGC | qRT-PCR studies |
| 16S-R | CGAAGAGCCCCTGGTTTGG | qRT-PCR studies |
| flhD-F | TTGGTTAAGTTGGCTGAAAC | Semiquantitative RT-PCR |
| flhD-R | TTTGTAGCAGAGGTATCATG | Semiquantitative RT-PCR |
| minE-R | CTTAGGTGATTCTTCATTTTCAGG | Operon study |
| rcsB-F | ACAGTCTAGAGATCCGACAGTTGCTGTAGC | RcsB protein purification |
| rcsB-R | AGACGTCGACCGGGTTAATGATGATGATGATGATGGCTTTCTCCGTTAATTTCCTTATC | RcsB protein purification |
To construct PMKH4 (PM7002 minC::Kanr rcsB::Strr), SM10 pKNG101::rcsB containing an internal fragment of rcsB (10) was conjugated to PMKH1. Exconjugants that were resistant to tetracycline, kanamycin, and streptomycin were confirmed using Southern blot analysis as having a disruption in the rcsB gene.
To complement the minC mutation, the minCDE region was amplified from PM7002 genomic DNA using minCDE-F and minCDE-R (Table 2). The minCDE-containing fragment was cloned into the SmaI site of pBC, creating pBCMinCDE. minCDE was digested out of pBCMinCDE using BamHI and XhoI and cloned into the BamHI/SalI sites of pKNG101, creating pMinCDE (pKNG101 plus minCDE). pMinCDE was maintained in E. coli SM10 λpir and conjugated to PMKH1. Exconjugants that were resistant to tetracycline, kanamycin, and streptomycin were confirmed using Southern blot analysis, and one of the clones, PMKH5, was used for subsequent experiments.
pMinClacZ was created by PCR amplification of the minC upstream region using PminC-F and PminC-R and ligating it into the BamHI and SalI sites of pQF50, a low-copy-number vector harboring a promoterless lacZ that can be used to quantify promoter activities (29). pMinClacZ was electroporated into E. coli DH5α, and clones were selected on LB agar plates containing X-Gal and ampicillin. One of these confirmed clones was purified using the Qiagen Plasmid Mini-Kit, and 5 μl of this DNA was used to electroporate PM7002 and PM7002 rcsB::Strr, creating PMKH6 and PMKH7, respectively.
5′-RACE.
5′ rapid amplification of cDNA ends (5′-RACE) was performed on 1.5 μg of RNA isolated from PM7002 using the 5′-RACE system (Invitrogen) according to version 2.0 of the manufacturer's protocol. First-strand cDNA synthesis was performed using GSP1 (Table 2). PCR amplification of the dCTP-tailed cDNA was done using GSP2 (Table 2) and the provided abridged anchor primer. Nested-PCR amplification of the diluted PCR product was done with Phusion Hot Start II high-fidelity DNA polymerase (Thermo Scientific) using the provided abridged universal amplification primer (AUAP) and GSP2. The resulting product was ligated into the SmaI site of pBC.SK and transformed into E. coli DH5α. The transformant was plated on ampicillin and X-Gal, and white colonies that were confirmed to have the 5′-RACE-generated product were sequenced to identify the 5′ end of the minC gene.
Phase-contrast microscopy.
All phase-contrast microscopy images were captured with an Infinity 2-1 charge-coupled-device (CCD) camera (Lumenera) using an Olympus BX51 microscope. For imaging liquid cultures, cells were grown to mid-log phase, and 1.5 μl was pipetted onto a glass slide (VWR International). A coverslip (VWR International) was placed on top of the culture, and images were taken at ×1,000 magnification using oil immersion. Swarm fronts were imaged by adding 650 μl of LB agar to CoverWell imaging chambers (1 by 20 mm by 2.3-mm depth) and then adding 4 μl of an overnight culture to the solidified agar. The cells were grown in a humidified environment at 37°C until they began to swarm. A coverslip was placed on top of the imaging chamber containing the swarming cells, and images were taken at ×1,000 magnification using oil immersion. When applicable, antibiotics and IPTG were added to the media.
β-Galactosidase assays.
Overnight cultures were standardized to the same optical density before 300 μl of each culture was spread plated onto 1.5% LB agar containing ampicillin and incubated at 37°C for 2 or 4 h. After the appropriate incubation period, cells were harvested from agar plates using 2 ml of ice-cold LB broth, and 0.9 ml of the harvested culture was pelleted by centrifugation at 13,000 rpm for 4 min. The pelleted cells were placed on ice for 20 min before lysis using chloroform and 0.1% SDS. β-Galactosidase activity was quantified as described by Miller (30).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed on digoxigenin (DIG)-ddUTP-labeled probes created by PCR amplification using Phusion Hot Start II high-fidelity DNA polymerase (Thermo Scientific) and primers containing EcoRI recognition sites at their ends (Table 2). The amplified DNA was digested with EcoRI for 3 h at 37°C, cleaned up using a PCR purification kit (Qiagen), and boiled for 10 min. After boiling, the digested DNA was placed immediately on ice, and 250 ng was incubated with 1 μl DIG-11-dUTP (Roche), 1 mM dATP (Roche), 4 μl buffer B (6 mM Tris-HCl, 6 mM MgCl2, 50 mM NaCl, pH 7.5), and 1 μl Klenow enzyme (Roche). The labeling reaction mixture was incubated at 37°C for 1 h, followed by a 10-min incubation at 65°C to heat inactivate the Klenow enzyme. After labeling, the probe was cleaned up using the PCR purification kit.
Binding reactions were done by incubating 0.5 μl of labeled probe with 10× binding buffer (500 mM Tris-HCl, 1 M NaCl, 1 mM EDTA, 0.5 μg/μl bovine serum albumin [BSA], 100 mM dithiothreitol [DTT]), 1 μg/μl poly(dI-dC), and 0 to 320 pmol of purified His-tagged RcsB (RcsB-His6) brought to a final volume of 20 μl with molecular biology grade water (Fisher). The reaction mixtures were incubated for 30 min at room temperature before being loaded with 5× DNA loading dye on a prerun Mini-Protean 5% Tris-borate-EDTA (TBE) precast gel (Bio-Rad). The gel was run at 100 V for approximately 3 to 4 h at 4°C until the loading dye was no longer visible on the gel. The DNA was transferred onto a 0.22-μm nylon membrane (MagnaGraph) at 15 V with a 0.4-mA limit for 45 min using a Bio-Rad Transblot SD Semi-Dry transfer cell. The DNA was cross-linked to the membrane using a Stratagene cross-linker. Blocking reagent (10×; Roche) was prepared in maleate buffer (100 mM maleic acid [pH 7.5], 150 mM NaCl), and the membrane was blocked for 30 min at room temperature using 1 times blocking solution diluted in maleic acid wash buffer (maleate buffer with 0.3% Tween 20). The membrane was then incubated for 30 min with a 1:1,000 dilution of antidigoxigenin coupled to alkaline phosphatase (Roche) in maleic acid wash buffer. The membrane was washed twice for 15 min each time with maleic acid wash buffer. The membrane was equilibrated in detection buffer (100 mM Tris-HCl [pH 9.5], 100 mM NaCl) for 2 min and then incubated with 1 ml of a 1:10 dilution of CDP-Star (Roche) for 5 min. After briefly blotting the membrane with Whatman paper, it was placed in a sealable plastic bag and exposed to film.
Swarm assays.
Overnight cultures were standardized to the same optical density, and 4 μl of each culture was pipetted onto the same predried 1.5% LB agar plate. The culture was allowed to dry into the LB plate before being incubated at 37°C. Measurements of the swarm diameter in centimeters were taken every 30 min until the cells reached consolidation. Antibiotics were included in the agar plate when appropriate. The values represent the average diameter of duplicate sets (4 in total) with standard deviations.
Transcriptome sequencing (RNA-Seq).
HI4320 and rcsB::Strr (PMKM2) cells were harvested at an OD600 of 1.1 in triplicate. RNAprotect bacterial reagent was used at a 1:1 ratio (Qiagen). Total RNA was isolated using a MasterPure RNA purification kit (Epicentre), and contaminating DNA was removed using a Turbo DNA-Free kit (Ambion). RNA purity was confirmed on an Agilent 2100 bioanalyzer (Agilent Technologies). DNA-free samples were then enriched for mRNA using a MicrobExpress bacterial mRNA enrichment kit (Ambion). Two rounds of rRNA depletion were performed, and RNA purity was confirmed on an Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA libraries were constructed from HI4320 and rcsB::Strr mRNA using the SuperScript double-stranded cDNA synthesis kit (Invitrogen). The cDNA libraries were purified by phenol extraction and ethanol precipitation. Sequences were analyzed on an Illumina HiSeq2000 instrument with 50-bp paired-end reads and matched to the HI4320 genome using CLC Genomics Workbench (CLC Bio-Qiagen, Aarhus, Denmark). The total number of reads mapped per transcript was determined and used to determine the reads per kilobase per million reads (RPKM). Three separate sets of RNA samples were analyzed, and the average of samples from two sets was used (data available on request).
qRT-PCR.
The same RNA used for RNA-Seq was used for quantitative real-time reverse transcriptase (qRT) PCR. Briefly, the RNA was transcribed into cDNA using an iScript Selected cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. qRT-PCR was performed using a 1:10 dilution of the generated cDNA in iQ SYBR green Supermix (Bio-Rad). The reaction was completed in an iCycler iQ Real-Time PCR detection system (Bio-Rad), and the values were normalized to 16S rRNA expression for each sample. The expression of minC, minD, and minE in HI4320 was calculated relative to PMKM2 (HI4320 rcsB::Strr) using the ΔΔCT method (31). The primers used to quantify the expression of each gene can be found in Table 2.
Semiquantitative RT-PCR.
Total RNA was harvested at hourly intervals (T1, T2, T4, and T6) after placing the cells on 1.5% LB agar plates using a MasterPure RNA purification kit (Epicentre). The T0 sample represents cells immediately before being placed on agar surfaces. Contaminating DNA was removed using a Turbo DNA-Free kit (Ambion). The purified RNA was transcribed into cDNA using an iScript Selected cDNA synthesis kit (Bio-Rad). PCR amplification was performed using 16S-F and 16S-R (Table 2), and aliquots from the PCR mixture were taken after 5, 10, and 15 cycles in order to equilibrate cDNA samples relative to 16S rRNA expression. Once equilibrated, minC expression was examined using minC-F and minC-R (Table 2) and flhD expression was examined using flhD-F and flhD-R (Table 2) at cycles 20, 25, 30, and 35.
Western blot analysis.
Overnight cultures grown to stationary phase in LB were equilibrated to the same optical density, and then 300 μl was spread onto 1.5% LB agar before incubation at 37°C for 5 h. The cells were gently resuspended in cold LB broth to the same optical density, and 1.5 ml was pelleted and stored at −80°C. The cell pellets were lysed using 400 μl CellLytic B cell lysis reagent (Sigma). Equivalent protein levels for each sample were confirmed using a Bradford assay. The cell lysates were resuspended at a 1:1 ratio in Laemmli sample buffer. Samples were run on a 12% Mini-Protean TGX (Bio-Rad) gel and transferred to a nitrocellulose membrane. The primary antibody used was rabbit anti-FlaA obtained from Robert Belas at the University of Maryland. The secondary antibody was ECL peroxidase-labeled anti-rabbit antibody (Life Sciences).
DAPI (4′,6-diamidino-2-phenylindole) staining and microscopy.
Swarmer cells were harvested after 5 h of incubation on 1.5% LB agar and resuspended in 1× phosphate-buffered saline (PBS). The cell suspensions were chemically fixed by the addition of formaldehyde (Sigma) to a 3% (vol/vol) final concentration and incubated for 10 min by gentle rocking. The fixed bacteria were attached to a poly-l-lysine (Sigma-Aldrich)-coated coverglass and incubated with DAPI (AnaSpec, Inc.) at 2 μg/ml (final concentration) in 1× PBS for 5 min in the dark. The excess DAPI solution was removed by washing with 1× PBS. The entire staining procedure was performed at room temperature. The DAPI-stained cells were imaged at room temperature using phase-contrast light and wide-field fluorescence microscopy with an Olympus IX83 microscope. An oil immersion 100× objective and a standard DAPI filter were employed for microscopy. Images were acquired using a Neo 5.5 sCMOS camera (Andor) and MetaMorph software (Molecular Devices). The images were prepared for publication using ImageJ software (NIH).
RESULTS
The minCDE operon is positively regulated by RcsB.
Mutations in rcsB result in a hyperswarming phenotype and differentiation into swarmer cells in liquid, indicating that RcsB regulates genes important for swarmer cell differentiation (10). To identify these genes, RNA-Seq was performed on RNA isolated from liquid cultures of P. mirabilis HI3420 and an isogenic rcsB::Strr mutant derivative at an A600 of 1.1. At this optical density, cells of the rcsB mutant were highly elongated, in contrast to the short rods of wild-type cells. When a cutoff of 2-fold or greater was applied, 87 genes were found to be RcsB repressed and 46 were RcsB activated (data available on request). Among the RcsB-regulated genes, we were interested in those that might contribute to cell elongation and initially focused on cell division genes. Expression of the ftsA and ftsZ genes was reduced 2.4- and 3.3-fold, respectively, in the rcsB mutant. However, the essential nature of these genes precluded a direct analysis of their role in cell elongation during swarming. Therefore, we focused on the minC and minD genes encoding components of a cell division inhibition complex, where expression decreased 3.4- and 6.7-fold, respectively, in the rcsB mutant relative to wild-type cells. The minE gene, located immediately downstream of minD, was not identified in the RNA-Seq as RcsB regulated, which was surprising, as minE is part of an operon with minCD in other bacteria (32, 33). To determine if the minCDE genes formed an operon in P. mirabilis, a primer specific to minE was used to prime cDNA synthesis, and the cDNA was used in a PCR to amplify the minC to minE region. The minC to minE region was amplified using the cDNA, but no product was observed with the RNA only, indicating that the minCDE genes form an operon (data not shown).
To confirm the RNA-Seq data, the expression of minC and minD was examined by qRT-PCR using the same RNA used for RNA-Seq. A 7.4-fold decrease in minC expression was observed in the rcsB mutant relative to the wild type, and a 6.3-fold decrease in minD was observed (Fig. 1C). In addition, qRT-PCR analysis indicated that the expression of minE was reduced 2.4-fold in the rcsB mutant (Fig. 1C). These results indicate that RcsB positively regulates the minCDE operon.
FIG 1.
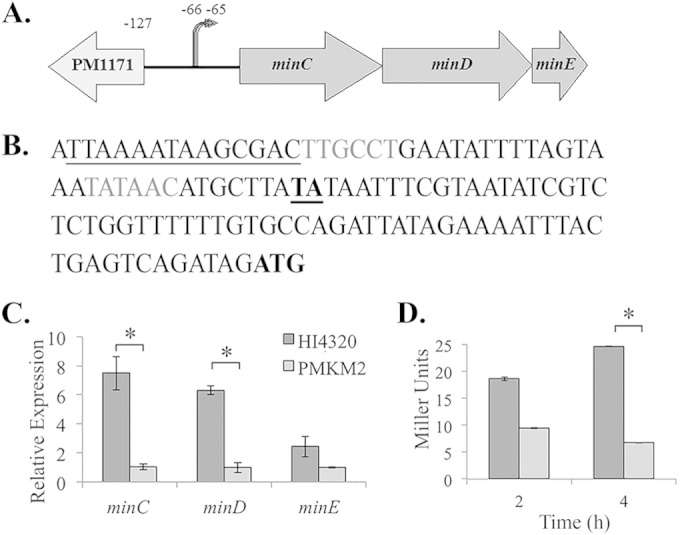
The min operon is positively regulated by RcsB. (A) Organization of the minCDE open reading frame. minC (702 bp), minD (813 bp), and minE (207 bp) are located in the same operon. The noncoding region upstream of minC is 127 bp long. Two transcriptional start sites for minCDE are located at positions −65 and −66 upstream of the minC start codon. (B) Sequence upstream of minCDE. The start codon of minC is in boldface. The two transcriptional start sites identified using 5′-RACE are underlined and in boldface. They are 65 and 66 bp upstream of the start codon. Upstream of the two identified start sites are putative −10 and −35 regions (gray letters). A potential RcsB binding site is underlined based on the E. coli consensus sequence (TAAGAATAATCCTA). (C) qRT-PCR analysis of minC, minD, and minE expression levels in P. mirabilis HI4320 and P. mirabilis HI4320 rcsB::Strr (PMKM2). minC expression is 7.5 times lower in PMKM2 than in the wild type, minD expression is 6.3 times lower in PMKM2 than in the wild type, and minE expression is 2.4 times lower in PMKM2 than in the wild type. (D) β-Galactosidase activity of pMinCLacZ in the PM7002 wild type (PMKH6) (dark-gray bars) and PM7002 rcsB::Strr (PMKH7) (light-gray bars). The activity of the minC promoter is decreased 2-fold in PMKH7 compared to PMKH6 after 2 h on solid agar and is decreased 4-fold in PMKH7 after 4 h on solid agar. The asterisks indicate a P value of <0.05.
Identification of the minC transcriptional start site and analysis of a minC-lacZ transcriptional fusion.
The minC promoter region was identified by first determining the 5′ end of the minC mRNA using 5′-RACE with RNA harvested from wild-type PM7002. Two alternative transcriptional start sites were identified 65 and 66 bp upstream of the minC start codon. Upstream of the identified start sites were putative −10 and −35 regions and a possible RcsB binding site based on the E. coli consensus sequence AAGAATAATCCTA (Fig. 1B). To confirm that this region represented the min promoter, a DNA fragment encompassing from −169 to +371 relative to the transcriptional start site and containing the putative −10 and −35 regions was amplified using PCR. This fragment was ligated into pQF50, a low-copy-number vector harboring a promoterless lacZ gene (29), in order to quantify the minC promoter activity. The construct, pMinClacZ, was transformed into PM7002 and PM7002 rcsB::Strr, and cells were collected after 2 (T2) and 4 (T4) h of incubation on agar plates at 37°C. The expression of β-galactosidase from pMinClacZ was 2-fold lower in the rcsB mutant at T2 than in the wild type and was approximately 4-fold lower at T4 (Fig. 1D).
RcsB directly regulates minCDE expression.
The above-mentioned data indicated that RcsB positively regulated minCDE expression. Next, to determine whether RcsB exerted its effect on minCDE expression directly, EMSAs were performed using purified His-tagged RcsB and a 292-bp region upstream of minCDE. This 292-bp region included the transcriptional start site identified using 5′-RACE and extended from −208 to +82 relative to the transcriptional start site. A partial shift was observed when 75 pmol of RcsB was added to the reaction mixture, and this shift further increased at RcsB concentrations of 170 pmol and above (Fig. 2A). To confirm that the binding of RcsB was specific to the region upstream of minC, a competitive EMSA was performed with the unlabeled 292-bp minC promoter fragment and a 249-bp fragment containing the abaI gene from Acinetobacter nosocomialis strain M2 using the abaI-F and abaI-R primers (Table 2). At a concentration 50 times that of the labeled minC fragment, the unlabeled minC DNA prevented a shift. However, the unlabeled 249-bp abaI fragment did not affect the shift of the labeled minC fragment (Fig. 2B). Together, these data indicate that RcsB binds the region containing the minC promoter and that this binding is specific.
FIG 2.
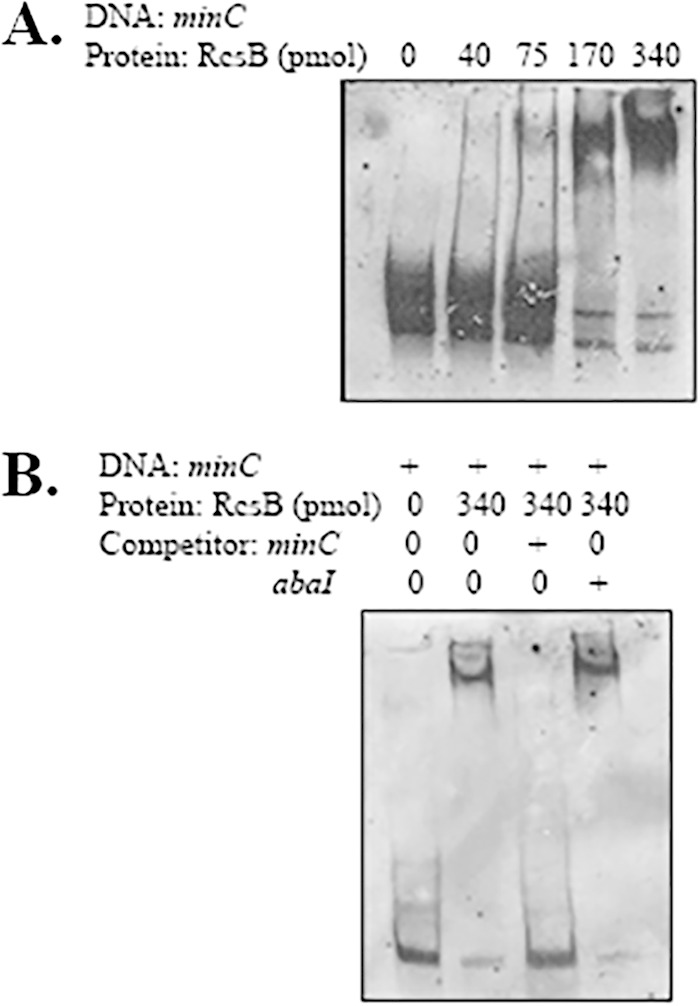
RcsB binds to the minC promoter region. Shown is binding of purified His-tagged RcsB to the minC upstream region. (A) Binding of the minC upstream region (290 bp) with increasing concentrations of RcsB. Lane 1 (from left), free labeled minC probe; lane 2, minC probe with 40 pmol RcsB; lane 3, minC probe with 75 pmol RcsB; lane 4, minC probe with 170 pmol RcsB; lane 5, minC probe with 340 pmol RcsB. (B) The digoxigenin-labeled 290-bp fragment was incubated with 340 pmol His-tagged RcsB. The binding was competed with unlabeled minC DNA (290 bp) and unlabeled abaI DNA (249 bp). Lane 1 (from left), free minC probe without RcsB or competitor; lane 2, minC probe with 340 pmol RcsB; lane 3, minC probe with 340 pmol RcsB and unlabeled minC DNA; lane 4, minC probe with 340 pmol RcsB and unlabeled abaI DNA.
Expression of the minCDE operon during swarmer cell differentiation.
To determine if minCDE expression varied during swarmer cell differentiation, expression was examined by semiquantitative RT-PCR from cells collected at hourly intervals after differentiation was initiated by placing the cells on agar plates. The expression of minC increased sharply by T2 and then decreased to undetectable levels by T4 (Fig. 3). As a marker for swarmer cell differentiation, flhD was also examined, and expression began to increase at T1, peaked at T2 to T4, and then decreased by T6 (Fig. 3).
FIG 3.
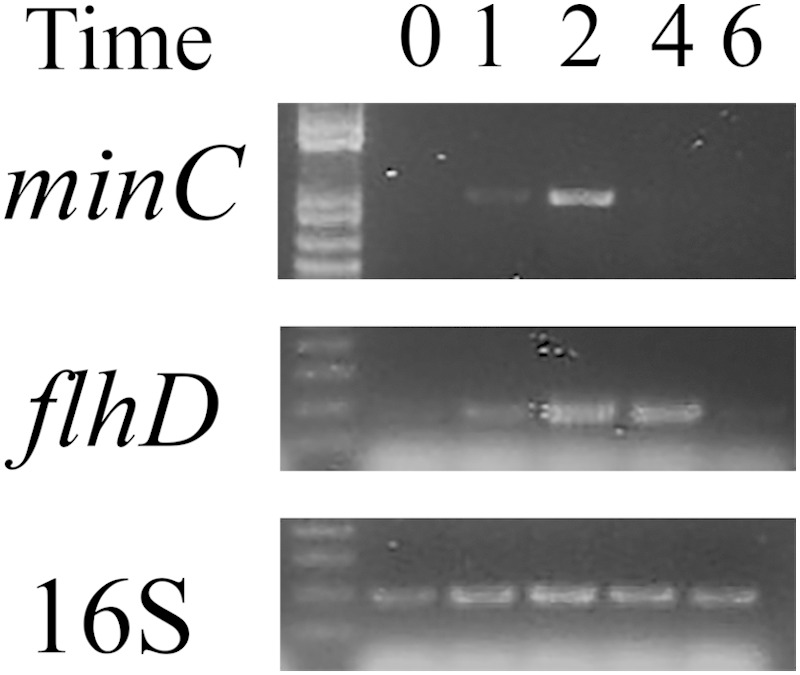
Semiquantitative RT-PCR of minC, flhD, and 16S RNA expression during swarmer cell differentiation. Total RNA was harvested at hourly intervals (T1, T2, T4, and T6) after placing cells on agar plates. The T0 sample represents the cells immediately before being placed on agar surfaces. PCRs on cDNA samples prepared from RNA at each time point were amplified for 10 cycles (16S rRNA), 30 cycles (minC), or 25 cycles (flhD).
A minC mutation results in aberrant cell division and minicell production.
Since RcsB regulates genes important for swarmer cell differentiation, it seemed likely that the identification of the MinCDE cell division inhibitors as RcsB regulated indicated they probably had a role in swarmer cell elongation. To address the effect of the min system on P. mirabilis cell division and swarmer cell formation, a minC::Kanr mutant was constructed. Cell morphologies were first examined in liquid media and imaged by phase-contrast microscopy. PM7002 produced short rod-shaped cells in liquid with an average length of 2.34 ± 0.6 μm (n = 147), while the minC::Kanr mutant produced a typical min mutant phenotype with both enlarged cells and minicells, resulting in an average length of 3.72 ± 1.8 μm (n = 102) (Fig. 4). In contrast, no minicells were observed in wild-type cells. The minC::Kanr mutation was complemented using the minCDE operon in single copy, as PMKH5 (PM7002 minC::Kanr plus pMinCDE) shared the division phenotype of PM7002, with an average cell length of 2.37 ± 0.6 μm (n = 82) (Fig. 4).
FIG 4.
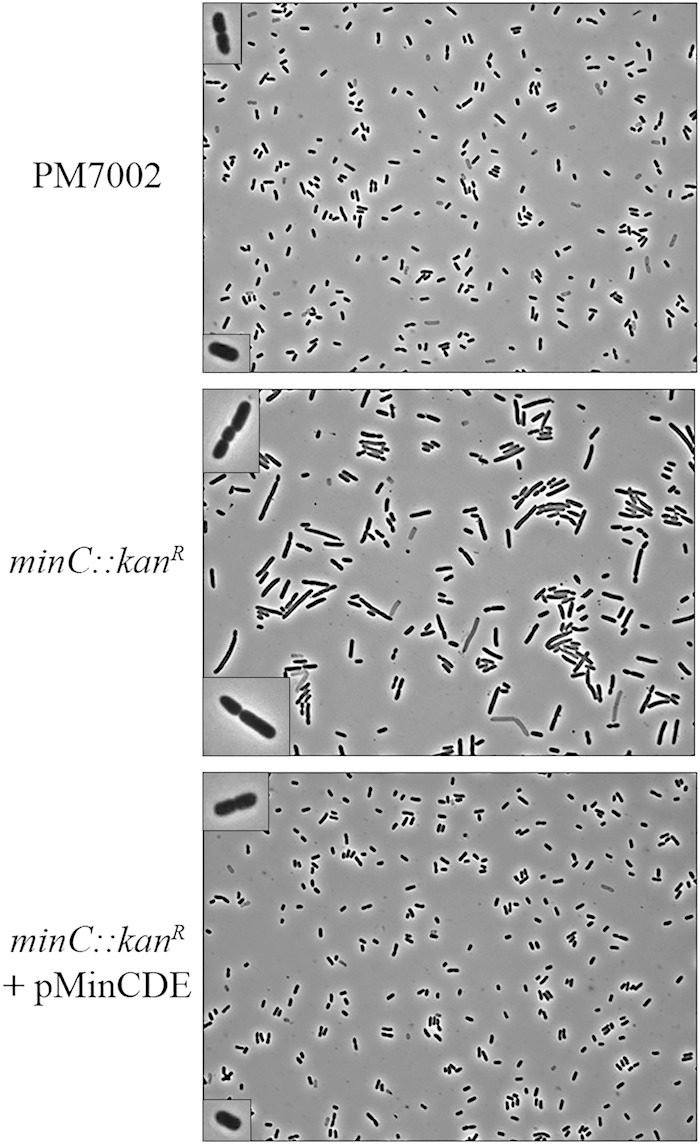
A minC::Kanr mutation results in aberrant cell division in liquid culture. The cell division phenotypes for wild-type strain PM7002 (top), PMKH1 (PM7002 minC::Kanr) (middle), and PMKH5 (PM7002 minC::Kanr plus pMinCDE) (bottom) are shown. Cells were grown to mid-log phase, and 1.5 μl was imaged using phase-contrast microscopy. Liquid cultures of PM7002 exist as rod-shaped cells, often as doublets, with septa positioned at the centers of the cells. The minC::Kanr mutation causes a large percentage of the cells to divide at the poles (middle image, insets). Minicells are often present. The division phenotype can be complemented by the addition of minCDE in single copy (bottom). The images were taken at ×1,000 magnification. All cell sizes in the insets have been increased equally.
The minC::Kanr mutation reduces swarming motility.
To investigate whether the min system was important for swarming in both wild-type and rcsB::Strr mutant backgrounds, wild-type PM7002, a minC::Kanr mutant, a rcsB::Strr mutant, and the minC::Kanr rcsB::Strr double mutant were subjected to swarm assays. Measurements of the swarm diameter were taken every 30 min until the strains reached the first consolidation phase. PM7002 initiated swarming at approximately 4 h (T4) after being spotted onto an agar plate (Fig. 5A). In contrast, the minC::Kanr mutant experienced a half-hour delay in the onset of swarming and did not reach wild-type levels of swarming until the consolidation phase at T6. The swarming phenotype of the minC::Kanr mutant complemented with pMinCDE was not significantly different than that of the minC::Kanr mutant alone (Fig. 5A), and the possible basis for this is discussed below. As expected, the rcsB mutant hyperswarmed and initiated swarming an hour before the wild type (Fig. 5B). The minC::Kanr rcsB::Strr double mutant initiated swarming at the same time as the rcsB::Strr mutant, but migration was 60% lower (Fig. 5B).
FIG 5.

A minC mutation reduces P. mirabilis swarming in wild-type and rcsB::Strr backgrounds. (A) The minC::Kanr mutation was introduced into PM7002 (wild type) in order to create PMKH1 (PM7002 minC::Kanr). The complementing strain was created by introducing pMinCDE (pKNG101 plus minCDE) into PMKH1. (B) The minC::Kanr mutation was also introduced into PMKM1 (PM7002 rcsB::Strr) in order to create PMKH4 (PM7002 minC::Kanr rcsB::Strr). All 5 strains were grown overnight in LB broth containing the appropriate antibiotics and then equilibrated to the same optical density. To investigate their abilities to swarm, 4-μl droplets of the equilibrated inocula were spotted onto the same plate, and the migration distance was measured every 30 min. The averages of four measurements are shown for PM7002, PMKH1, PMKM1, PMKH4, and PMKH5. The error bars indicate standard deviations.
Since the minC::Kanr mutation in both wild-type and rcsB::Strr mutant backgrounds resulted in deficient swarming, the basis for this defect was examined by (i) analyzing the cell morphology at the swarm fronts, (ii) determining the levels of flagellin expressed in wild-type and minC::Kanr mutant strains, and (iii) examining the nucleoid distribution in wild-type and minC::Kanr mutant cells using DAPI staining. The PM7002 swarm front consisted of highly elongated, multinucleate swarm cells on the exterior and rod-shaped cells in the interior (Fig. 6A). In contrast, the minC::Kanr mutant swarm front was a heterologous population of swollen and slightly elongated rod-shaped cells and minicells (Fig. 6A). The altered swarmer cell morphology at the swarm fronts of the minC::Kanr mutant could be complemented by reintroducing the minCDE operon in single copy (Fig. 6A). The levels of flagellin (FlaA) detected by Western blotting were approximately 2-fold lower in the minC::Kanr mutant at T5 (Fig. 6B). However, complementation with pMinCDE did not restore flagellin expression in the minC::Kanr mutant. In both the minC::Kanr mutant and the complemented strain, the reduced flagellin likely contributes to the reduction in swarming. The basis for the ability of pMinCDE to complement the defect in cell morphology but not the reduced flagellin expression is unknown. There are no genes immediately downstream of minCDE in P. mirabilis that might have decreased expression due to a polar effect of the minC::Kanr mutation or that could have been affected by introducing pMinCDE into the chromosome at the min locus. The lack of complementation with respect to swarming may have resulted from expression differences that failed to mimic the physiological level of MinCDE required for normal swarming but were able to rescue the morphological defect. For example, in E. coli, the min operon must be present at 1 to 10 times the physiological level in order to complement a min deletion mutant (32). Lastly, in both wild-type and minC::Kanr mutant cells, multiple chromosomes typical of swarmer cells were present and were similar in appearance in each strain (Fig. 6C).
FIG 6.
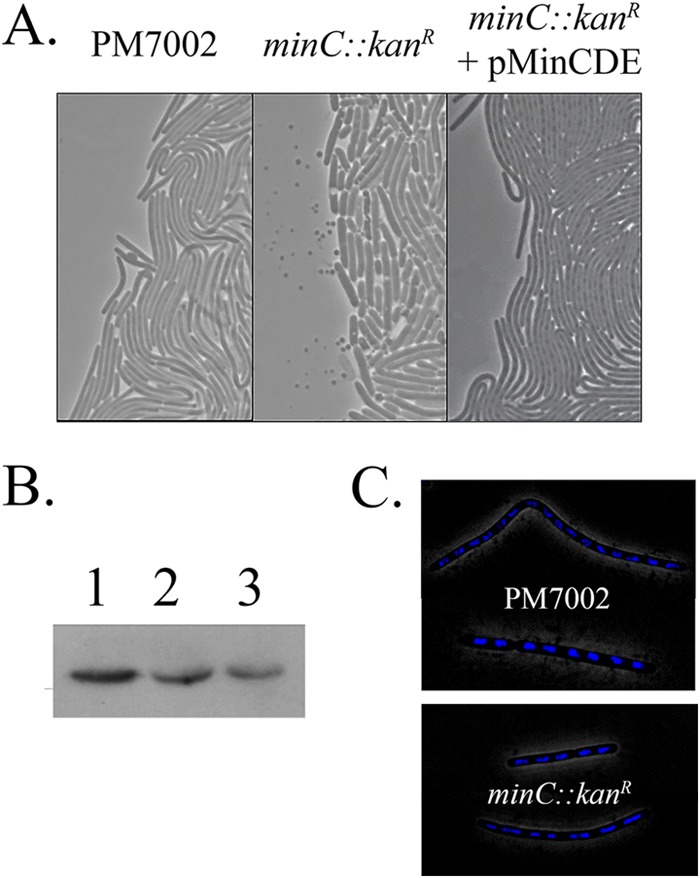
The minC::Kanr mutation causes an atypical swarm front. (A) Stationary-phase cultures of PM7002 (wild type), PMKH1 (PM7002 minC::Kanr), and PMKH5 (PM7002 minC::Kanr plus pMinCDE) grown overnight in LB broth were equilibrated to the same optical density and then spotted onto 1.5% LB agar contained in a Coverwell imaging chamber. Once the cells began to swarm, phase-contrast images of PM7002 (left), PMKH1 (center), and PMKH5 (right) were taken of the swarm front at ×1,000 magnification. (B) Western blot analysis of flagellin (FlaA) expression in PM7002 wild-type (lane 1), PM7002 minC::Kanr (lane 2), and PM7002 minC::Kanr plus pMinCDE (lane 3) cells harvested after 5 h growth on LB agar plates. (C) DAPI staining of chromosomes in PM7002 (wild-type) and PMKH1 (PM7002 minC::Kanr) swarmer cells.
DISCUSSION
Previous studies have demonstrated that mutations that inactivate the Rcs phosphorelay in P. mirabilis result in hyperswarming on agar surfaces and constitutive swarmer cell formation in liquid media, a normally nonpermissive condition (10, 13, 14). The basis for the hyperswarming phenotype is due, in part, to the overexpression of the flhDC operon, which then activates genes involved in the production of flagella. However, overexpression of flhDC alone does not result in cell elongation, and therefore, the Rcs phosphorelay must also regulate genes involved in swarmer cell elongation (10). Since rcs mutants constitutively elongate to swarmer cells in liquid media, this condition was chosen to identify RcsB-regulated genes that contributed to cell elongation by using RNA-Seq analysis. Based on this analysis, we chose to focus on two genes, minC and minD, that were activated by the Rcs phosphorelay and encoded components of the MinCDE cell division inhibition complex. Interestingly, a third gene, minE, which is typically cotranscribed with minC and minD, did not show up in the RNA-Seq analysis as RcsB regulated. However, our work demonstrated that minE was cotranscribed with minCD and thus that the three genes formed an operon. The inability to identify minE in the RNA-Seq analysis may have been due to reduced sensitivity of the technique compared to qRT-PCR, which showed a 2.4-fold reduction in the rcsB mutant (Fig. 1C). To confirm the activation of the minCDE operon by RcsB, a combination of transcriptional lacZ fusions and real-time PCR was used. In addition, purified RcsB was capable of binding the minCDE promoter region, indicating the regulation was direct. A possible RcsB binding site (TAAGAATAATCCTA) was present immediately upstream of the −35 region of the promoter (Fig. 1B). Additional studies, including mutagenesis of this putative RcsB binding site or demonstrating its ability to confer RcsB-dependent regulation on a heterologous promoter, will be needed to confirm the role of the sequence in RcsB binding.
Identification of the MinCDE cell division inhibition complex as regulated by the Rcs phosphorelay prompted our investigation of its function in swarmer cell elongation and swarming motility. We hypothesized that the expression of minCDE had to be tightly regulated during swarmer cell differentiation. During swarmer cell differentiation, there is a sharp increase in minCDE expression approximately 2 h after contact with an agar surface (T2), and the increased levels of MinCDE likely contributed to the inhibition of cell division (Fig. 3). Consistent with this, we have observed that even slight increases in minCDE expression from an IPTG-inducible promoter resulted in a block in cell division and the formation of highly elongated cells (data not shown). Then, as the activity of the Rcs phosphorelay begins to decrease at T3, the resulting decrease in levels of MinCDE would partially alleviate the block in cell division and allow cells to prepare for the consolidation phase, when they divide again. Consistent with this, a minC mutant was unable to elongate normally during swarmer cell differentiation and formed shorter cells than the wild type (Fig. 6). In addition, the minC mutant did not swarm as efficiently as the wild type (Fig. 5). This decrease in swarming was likely due to the decreased flagellin expression and the abnormal morphology in the minC mutant, which may affect the ability of the cells to form multicellular rafts. For example, it has been shown that a ccmA mutant in P. mirabilis produces curved cells that cannot align properly to form rafts during swarming. The mutant is able to differentiate into elongated, C-shaped swarmer cells but is unable to swarm (34).
To our knowledge, this is the first study to investigate the contribution of a cell division system to swarmer cell differentiation in P. mirabilis. In other bacteria, a minC mutation was found to reduce motility in Helicobacter pylori, but the mutant was highly elongated in liquid culture, which the authors hypothesized was the cause of reduced motility (35). In contrast, a mutation in a minE homolog in the Gram-positive bacterium Clostridium perfringens resulted in hypermotility and hyperelongation (36). In P. mirabilis, a null allele in minC, the proprietor of cell division inhibition in the Min system, did not induce hyperelongation in liquid culture or on solid surfaces in our study. The difference seen between these studies and ours highlights the underlying complexities and varying roles of the Min system in different organisms.
ACKNOWLEDGMENTS
We are grateful to Robert Belas for providing the FlaA antibody.
This work was supported by a Merit Review award from the Department of Veterans Affairs and by a Research Career Scientist award (P.N.R.). K.E.H. is supported by the Burroughs Wellcome Fund's Molecules to Mankind program.
REFERENCES
- 1.Hoeniger JFM. 1965. Development of flagella by Proteus mirabilis. J Gen Microbiol 40:29–42. doi: 10.1099/00221287-40-1-29. [DOI] [Google Scholar]
- 2.Jones BV, Young R, Mahenthiralingam E, Stickler DJ. 2004. Ultrastructure of Proteus mirabilis swarmer cell rafts and role of swarming in catheter associated urinary tract infection. Infect Immun 72:3941–3950. doi: 10.1128/IAI.72.7.3941-3950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauprich O, Matsushita M, Weijer CJ, Siegert F, Esipov SE, Shapiro JA. 1996. Periodic phenomena in Proteus mirabilis swarm colony development. J Bacteriol 178:6525–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams FD, Schwarzhoff RH. 1978. Nature of the swarming phenomenon in Proteus. Annu Rev Microbiol 32:101–122. doi: 10.1146/annurev.mi.32.100178.000533. [DOI] [PubMed] [Google Scholar]
- 5.Armbruster CE, Mobley HLT. 2012. Merging mythology and morphology: the multifaceted lifestyle of Proteus mirabilis. Nat Rev Microbiol 10:743–754. doi: 10.1038/nrmicro2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rather PN. 2005. Swarmer cell differentiation in Proteus mirabilis. Environ Microbiol 7:1065–1073. doi: 10.1111/j.1462-2920.2005.00806.x. [DOI] [PubMed] [Google Scholar]
- 7.Stout V, Gottesman S. 1990. RcsB and RcsC: a two-component regulator of capsule synthesis in Escherichia coli. J Bacteriol 172:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda S, Fujisawa Y, Matsubara M, Aiba H, Mizuno T. 2001. A novel feature of the multistep phosphorelay in Escherichia coli: a revised model of the RcsC→YojN→RcsB signalling pathway implicated in capsular synthesis and swarming behavior. Mol Microbiol 40:440–450. doi: 10.1046/j.1365-2958.2001.02393.x. [DOI] [PubMed] [Google Scholar]
- 9.Gervais FG, Drapeau GR. 1992. Identification, cloning, and characterization of rcsF, a new regulator gene for exopolysaccharide synthesis that suppresses the division mutation ftsZ84 in Escherichia coli K-12. J Bacteriol 174:8016–8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemmer KM, Rather PN. 2007. Regulation of flhDC expression in Proteus mirabilis. Res Microbiol 158:295–302. doi: 10.1016/j.resmic.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Claret L, Hughes C. 2000. Functions of the subunits of the FlhD2C2 transcriptional master regulator of bacterial flagellum biosynthesis and swarming. J Mol Biol 303:467–478. doi: 10.1006/jmbi.2000.4149. [DOI] [PubMed] [Google Scholar]
- 12.Pearson MM, Rasko DA, Smith SN, Mobley HLT. 2010. Transcriptome of swarming Proteus mirabilis. Infect Immun 78:2834–2845. doi: 10.1128/IAI.01222-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belas R, Schneider R, Melch M. 1998. Characterization of Proteus mirabilis precocious swarming mutants: identification of rsbA, encoding a regulator of swarming behavior. J Bacteriol 180:6126–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liaw SJ, Lia HC, Ho SW, Luh KT, Wang WB. 2001. Characterisation of p-nitrophenylglycerol-resistant Proteus mirabilis super-swarming mutants. J Med Microbiol 50:1039–1048. [DOI] [PubMed] [Google Scholar]
- 15.Belas R. 1997. Proteus mirabilis and other swarming bacteria, p 183–219. In Shapiro J, Dworkin M (ed), Bacteria as multicellular organisms. Oxford University Press, New York, NY. [Google Scholar]
- 16.Sturgill G, Rather PN. 2004. Evidence that putrescine acts as an extracellular signal required for swarming in Proteus mirabilis. Mol Microbiol 51:437–446. doi: 10.1046/j.1365-2958.2003.03835.x. [DOI] [PubMed] [Google Scholar]
- 17.Carballès F, Bertrand C, Bouche JP, Cam K. 1999. Regulation of Escherichia coli cell division genes ftsA and ftsZ by the two-component system rcsC-rcsB. Mol Microbiol 34:442–450. doi: 10.1046/j.1365-2958.1999.01605.x. [DOI] [PubMed] [Google Scholar]
- 18.Gervais FG, Phoenix P, Drapeau GR. 1992. The rcsB gene, a positive regulator of colonic acid biosynthesis in Escherichia coli, is also an activator of ftsZ expression. J Bacteriol 174:3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Detweiler CS, Monack DM, Brodsky IE, Mathew H, Falkow S. 2003. virK, somA and rcsC are important for systemic Salmonella enterica serovar Typhimurium infection and cationic peptide resistance. Mol Microbiol 48:385–400. doi: 10.1046/j.1365-2958.2003.03455.x. [DOI] [PubMed] [Google Scholar]
- 20.Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanié-Cornet MP, Gutierrez C, Cam K. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol Microbiol 49:823–832. [DOI] [PubMed] [Google Scholar]
- 21.Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with cell division in Escherichia coli. Nature 354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 22.Ward JE, Lutkenhaus J. 1985. Overproduction of FtsZ induces minicell formation in E. coli. Cell 42:941–949. doi: 10.1016/0092-8674(85)90290-9. [DOI] [PubMed] [Google Scholar]
- 23.de Boer PA, Crossley RE, Rothfield LI. 1990. Central role for the Escherichia coli minC gene product in two different cell division-inhibition systems. Proc Natl Acad Sci U S A 87:1129–1133. doi: 10.1073/pnas.87.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Boer PA, Crossley RE, Rothfield LI. 1992. Roles of MinC and MinD in the site-specific septation block mediated by the MinCDE system of Escherichia coli. J Bacteriol 174:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Z, Gogol EP, Lutkenhaus J. 2002. Dynamic assembly of MinD on phospholipid vesicles regulated by ATP and MinE. Proc Natl Acad Sci U S A 99:6761–6766. doi: 10.1073/pnas.102059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z, Lutkenhaus J. 2001. Topological regulation of cell division in E. coli spatiotemporal oscillation of MinD requires stimulation of its ATPase by MinE and phospholipid. Mol Cell 7:1337–1343. doi: 10.1016/S1097-2765(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 27.Zhao CR, de Boer PA, Rothfield LI. 1995. Proper placement of the Escherichia coli division site requires two functions that are associated with different domains of the MinE protein. Proc Natl Acad Sci U S A 92:4313–4317. doi: 10.1073/pnas.92.10.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adler HI, Fisher WD, Cohen A, Hardigree AA. 1967. Miniature Escherichia coli cells deficient in DNA. Proc Natl Acad Sci U S A 57:321–326. doi: 10.1073/pnas.57.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farinha MA, Kropinski AM. 1990. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol 172:3496–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JH. 1972. Experiments in molecular genetics. Cold Springs Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.de Boer PA, Crossley RE, Rothfield LI. 1988. Isolation and properties of minB, a complex genetic locus involved in correct placement of the division site in Escherichia coli. J Bacteriol 170:2106–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Boer PA, Crossley RE, Rothfield LI. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- 34.Hay NA, Tipper DJ, Gygi D, Hughes C. 1999. A novel membrane protein influencing cell shape and multicellular swarming of Proteus mirabilis. J Bacteriol 181:2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiou P, Luo C, Chang K, Lin N. 2013. Maintenance of the cell morphology by MinC in Helicobacter pylori. PLoS One 8:e71208. doi: 10.1371/journal.pone.0071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, McCord KD, Howarth J, Popham DL, Jensen RV, Melville SB. 2014. Hypermotility in Clostridium perfringens strain SM101 is due to spontaneous mutations in genes linked to cell division. J Bacteriol 196:2405–2412. doi: 10.1128/JB.01614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manoil C, Beckwith J. 1985. TnphoA: a transposon probe for protein export signals. Proc Natl Acad Sci U S A 82:8129–8133. doi: 10.1073/pnas.82.23.8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mobley HLT, Warren JW. 1987. Urease-positive bacteriuria and obstruction of long-term urinary catheters. J Clin Microbiol 25:2216–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaniga K, Delor I, Cornelis GR. 1991. A wide-host-range suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene in Yersinia enterocolitica. Gene 109:137–141. doi: 10.1016/0378-1119(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 41.Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]