

Abstract
Background
Vibrio parahaemolyticus is a common pathogen infecting humans and marine animals; this pathogen has become a major concern of marine food products and trade. In this study, V. parahaemolyticus isolated from sewage was exposed to different culture conditions and analyzed by isobaric tag for relative and absolute quantitation (iTRAQ) based reversed-phase liquid chromatography-tandem mass spectrometry (LC-MS/MS) technique. Our goal is to gain further insights into the proteomics of V. parahaemolyticus, particularly differentially expressed proteins closely correlated with growth conditions and pathogenicity associated proteins.
Results
In this study, a total of 2,717 proteins including numerous membrane proteins were significantly identified, and 616 proteins displayed significant differential expression under different conditions. Of them, 12 proteins mainly participating in metabolism showed the most elastic expression differentiation between different culture conditions. Some membrane proteins such as type I secretion outer membrane protein, TolC, lipoprotein, efflux system proteins iron-regulated protein A and putaive Fe-regulated protein B, ferric siderophore receptor homolog and several V. parahaemolyticus virulence-associated proteins were differentially regulated under different conditions. Some differentially regulated proteins were analyzed and confirmed at gene expression level by quantitative real time polymerase chain reaction (qRT-PCR).
Conclusions
Proteomics analysis results revealed the characteristics of V. parahaemolyticus proteome expression, provided some promising biomarkers related with growth conditions, the results likely advance insights into the mechanism involved in the response of V. parahaemolyticus to different conditions. Some virulence-associated proteins were discovered to be differentially expressed under different conditions.
Electronic supplementary material
The online version of this article (doi:10.1186/s12953-015-0075-4) contains supplementary material, which is available to authorized users.
Keywords: Vibrio parahaemolyticus, Quantitative proteomics, iTRAQ, Biomarker, Pathogenicity
Introduction
Vibrio parahaemolyticus is a common opportunistic pathogen infecting humans and marine animals; this pathogen causes food-borne gastroenteritis, occasional wound infection, and sepsis in immune-compromised patients, as well as great losses in crustacean and fish aquaculture. V. parahaemolyticus has been considered as a significant public health concern and sanitary risk in the production and trade of seafood worldwide because this species is abundant in marine products [1]. Numerous cases of V. parahaemolyticus infection have been reported in East Asia, South East Asia, North America, and others [2–7]; as such, V. parahaemolyticus has been recognized as pandemic. In 2004, a highly virulent strain caused a major outbreak with more than 1,000 cases in Chile [8].
V. parahaemolyticus is widely distributed in estuarine, marine, and coastal environments [9]. Majid Alipour et al. [10] detected 62 (20.3 %) V. parahaemolyticus strains from 300 seawater and sediment samples in the southern coast of the Caspian Sea. Cabrera-Garcia et al. [11] reported that 15 % of the seawater samples in the Gulf of Mexico contained V. parahaemolyticus. We also isolated V. parahaemolyticus strains multiple times in sewage and adjacent seawaters of Dalian, China, in different seasons.
Almost all of the clinical V. parahaemolyticus isolates exhibit β-hemolysis on Wagatsuma agar, and this phenomenon is known as Kanagawa phenomenon, which is induced by thermostable direct hemolysin (TDH) produced by V. parahaemolyticus and has been considered a crucial marker that distinguishes pathogenic strains from non-pathogenic strains [12]. Since 1996, “pandemic clones” mainly belonging to sero type O3:K6, have caused gastroenteritis outbreaks in India and other parts of the world. More than 50 % of V. parahaemolyticus strains isolated from patients in India are of sero type O3:K6 [2, 8]. V. parahaemolyticus outbreak rapidly spread to other countries in Asia, South America, North America, Africa, and Europe, resulting in a pandemic that affected numerous individuals [13–16]. In 1998, a new highly virulent strain was responsible for a large gastroenteritis outbreak in Galveston Bay, Texas [1].
Although V. parahaemolyticus has been recognized for many years, the response of this species to different environments at a proteome level remains unclear. Proteomics aims to monitor global proteins in a cell or an organism, reveal plasticity in terms of development and environment, and evaluate gene expression, protein–protein interactions, and correlation between proteome expression and environment. Advances in proteomics based on tandem MS and applications of isobaric peptide and protein labeling for relative quantification provide promising tools to discover biomarkers and elucidate the molecular regulatory mechanisms underlying responses to different environments. In this study, V. parahaemolyticus was subjected to proteomic analysis by iTRAQ labeling to identify differentially expressed proteins upon exposure to different growth environments. iTRAQ is a powerful tool to relatively and absolutely quantify proteins, and has been extensively applied to proteome analysis since this technology was invented in 2004 [17–22].
To the best of our knowledge, this study is the first to perform a detailed proteomic analysis of V. parahaemolyticus in different growth environments.
Results and discussion
Protein identification
A total of 2,717 proteins or approximately 60 % of the 4,832 predicted Open Reading Frames in the genome (V. parahaemolyticus, RIMD2210633) were significantly identified from 70,197 MS/MS spectra and 28,006 peptides using 1 % false discovery rate (FDR) as cutoff in the triplicate independent experiments. The result showed a very wide coverage of the protein identification method utilized in this study. The MS/MS spectra of representative peptides belonging to two differentially expressed proteins are shown in Fig. 1.
Fig. 1.
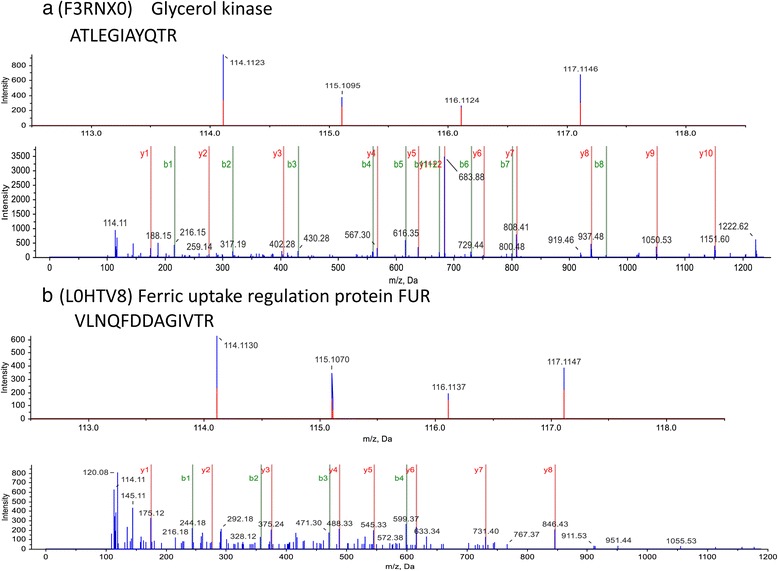
Representative MS/MS spectra of peptides from differentially expressed proteins. a. ATLEGIAYQTR peptide belongs to glycerol kinase. b. VLNQFDDAGIVTR peptide belongs to ferric uptake regulation protein FUR. The spectra indicate the relative intensity of reporter ions (VPP/114, VPE/115, VPX/116, and VPW/117) from MS/MS fragmentation
Among 2,717 proteins, 2,110 were analyzed in term of their gene ontology (GO) annotations in UniProtKB database (http://www.uniprot.org/) [23]. Based on GO classification, their molecular functions were mainly displayed in catalytic activity with approximately 44 % of all molecular functions, binding function takes up 35.50 %, transporter activity takes up 7.10 %, and nucleic acid binding transcription factor activity takes up 3.85 % (Fig. 2).
Fig. 2.
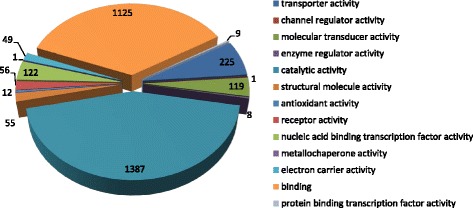
Gene ontology enrichment analysis of molecular function of the identified proteins. The classification of the molecular function of the identified proteins is based on a UniProt KB search and KEGG pathway analysis
Analysis of biological processes revealed that most proteins were involved in metabolic processes accounting for 30.90 % of all biological processes (1330/4303), 29.37 % of cellular processes, 5.25 % of regulation of biological processes, and 4.72 % of responses to stimuli (Fig. 3). In addition, more than 500 proteins (approximately 20 % of the proteins identified in this study) were not assigned annotated function.
Fig. 3.
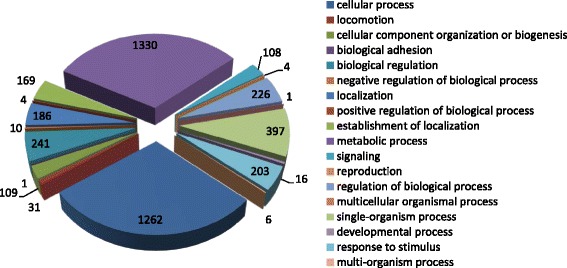
Gene ontology enrichment analysis of biological process of the identified proteins. The classification of the biological process of the identified proteins is based on a UniProt KB search and KEGG pathway analysis
In this study, numerous membrane proteins were significantly identified, they included 1 putative porins OmpU; 2 TolC family (outer membrane protein TolC, " type I secretion outer membrane protein TolC); 4 OmpA family proteins (outer membrane protein OmpA, OmpA family protein OmpA, outer membrane protein OmpA, outer membrane protein A); 4 Iron-regulated proteins (Iron-regulated outer membrane virulence protein homolog, iron-regulated protein A, putaive Fe-regulated protein B, Iron-regulated virulence regulatory protein homolog); 2 receptor proteins (cyclic AMP receptor protein, outer membrane protein OmpK); 13 transport proteins (heme transport protein HutA, Long-chain fatty acid transport protein, ferrous iron transport protein B, biopolymer transport protein ExbB-related protein, magnesium and cobalt transport protein CorA, Putative transport protein, etc.); 9 polysaccharide-related proteins (putative polysaccharide export-related protein, polysaccharide biosynthesis/export protein, putative polysaccharide export-related protein, etc.); 27 lipoprotein (lipoprotein, thiamin biosynthesis lipoprotein ApbE, apolipoprotein N-acyltransferase, etc.); 11efflux pump proteins (ABC- type multidrug efflux pump, Putative multidrug efflux membrane fusion protein, RND multidrug efflux transporter, putative Co Zn Cd efflux system membrane fusion protein, putative cation efflux system, glutathione-regulated potassium-efflux system protein KefB, etc.); 3 pilins(pilin protein MshA, type 4 prepilin-like proteins leader peptide-processing enzyme, pilin protein MshA).
We also discovered several types of secretion system proteins, they were 2 type I secretion (type I secretion outer membrane protein TolC, putative transport protein); 2 type II secretion (type II secretion system protein L, type II secretion system protein K); 7 type III secretion system (T3SS) protein (YscC, T3SS ATPase, T3SS cytoplasmic protein YscL, Putative translocation protein in T3SS, etc.) and 2 type VI secretion protein (type VI secretion ATPase, ClpV1 family and type VI secretion protein, VC_A0110 family) as well as 11 general secretion pathway proteins.
Other virulence related proteins such as three hemolysins(GN VP0730, VP2536, VPA0257) and regulator proteins Hfq and rsmA were also significantly identified.
Protein quantification and differentiation analysis
Proteins with iTRAQ ratios <0.50 or >2.0, p < 0.05 were considered significantly different in protein quantification. Following these criteria, a total of 2,643 proteins, or more than 97 % of the identified proteins were accurately quantified by iTRAQ labeling coupled with RP–LC interfaced with Triple 5600 mass spectrometer (AB sciex) (iTRAQ ratios of the MS/MS spectra of representative peptides belonging to two differentially expressed proteins are illustrated in Fig. 1).
A total of 616 proteins were significantly regulated in VPP, VPE, and VPX compared with VPW. The numbers of the differentially expressed proteins under different conditions were listed in Table 1. An additional file showed the GO enrichment analysis of differentially regulated proteins in VPP, VPE, VPX, and VPW (see Additional file 1: Table S1 and S2).
Table 1.
Number of the differentially expressed proteins of V. parahaemolyticus among different samples
| Comparison | Number of differentially expressed proteins | ||
|---|---|---|---|
| Total number | Up-regulated | Down-regulated | |
| VPP(114)/VPW(117) | 97 | 47 | 50 |
| VPE(115)/VPW(117) | 281 | 147 | 134 |
| VPX(116)/VPW(117) | 400 | 184 | 216 |
| VPE(115)/ VPP(114) | 331 | 178 | 153 |
| VPX(116)/ VPP(114) | 448 | 197 | 251 |
| VPE(115)/ VPX(116) | 567 | 331 | 236 |
Only 97 proteins were differentially expressed in VPP compared with VPW, by contrast, 281 proteins were significantly altered in VPE compared with VPW and 400 proteins displayed significant differential abundance between VPX and VPW. The result indicated that V. parahaemolyticu presented highly variable protein expression and growing environment had a direct effect on protein expression of V. parahaemolyticus.
Some protein was regulated only in some condition. Of the 616 significantly regulated proteins, 33 proteins were regulated only in VPP compared with VPW (15 were up-regulated, 18 were down-regulated); 113 were only found to altered VPE versus VPW (65 up-regulated, 48 down-regulated); and the abundance of 232 proteins was differently regulated in VPX (112 up-regulated, 120 down-regulated) (Fig. 4). The detailed information of the proteins was listed in Additional files 2, 3 and 4. These results further indicated that the detected V. parahaemolyticus required different factors to transport various ions, nutrients, and other metabolites across the outer membranes under different growth conditions; V. parahaemolyticus also required specific signal pathways that respond to various environmental stimuli. In another study, V. splendidus was reported to forms distinct populations in different ecological niches in marine environment [24]. Keymer et al. [25] also demonstrated that a non-homogeneous marine environment stimulates the formation of diverse populations of V. cholerae strains.
Fig. 4.
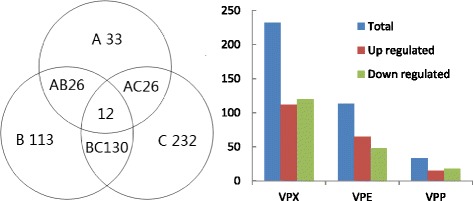
Numbers of unique differentially expressed proteins under different conditions. In the Venn diagram, A, B, and C indicate the number of significantly unique regulated proteins in VPP, VPE, and VPX compared with VPW, respectively; AB, AC, and BC indicate the overlap of the proteins simultaneously differentially expressed under the two conditions compared with VPW; 12 indicates the number of the proteins simultaneously differentially expressed in VPP, VPE, and VPX compared with VPW;. The column diagram indicates the number of up-regulated and down-regulated unique proteins in VPP, VPE, and VPX
Among the differentially expressed proteins, 12 were differentially regulated in VPP, VPE, and VPX compared with VPW without exception (Table 2). The abundance of most of these proteins was increased in VPP, VPE, and VPX compared with VPW. Function analysis results showed that the proteins with catalytic activity, including glycerol kinase (GN glpK; key enzyme in the regulation of glycerol uptake and metabolism), isocitrate dehydrogenase (GN VIPARAQ4037_2971), NADP-dependent, putative tricarboxylic transport TctC (GN VP1651), putative acyltransferase (GN VP10329_09192), and diaminobutyrate-pyruvate transaminase, mainly participate in the metabolism of carbohydrates, lipids, and proteins. The 12 differently regulated proteins exhibited the most elastic expression adapting to different growth conditions, they are promising biomarkers to monitor environment.
Table 2.
Differentially expressed proteins in VPP, VPE, and VPX compared with VPW
| Accession | Protein name | Changes in the relative abundance of proteins | ||||||
|---|---|---|---|---|---|---|---|---|
| VPP /VPWa | P Valueb | VPE/VPW | P Value | VPX/VPW | P Value | GO | ||
| F3RNX0 | Glycerol kinase | 2.109↑c | 0.048 | 0.425 | 0.007 | 0.233 | 2.0E-04 | glycerol catabolic process |
| E1DA96 | Isocitrate dehydroge nase, ADP-dependent | 2.399↑ | 0.026 | 2.377↑ | 1.38E-06 | 0.071 | 1.1E-06 | tricarboxylic acid cycle |
| L0HV77 | 30S ribosomal protein S4 | 0.308 | 0.006 | 2.679↑ | 0.007 | 0.302 | 4.0 E-04 | translation |
| F3RYI1 | putative acyltransferase | 2.858↑ | 0.001 | 0.154 | 0.007 | 2.884↑ | 7.61E-05 | acetyl-CoA C-acetyltransferase activity |
| E1EMA1 | Carbamoyl-phosphate synthase small chain | 2.535↑ | 0.049 | 2.655↑ | 0.022 | 7.656↑ | 1.70E-06 | glutamine catabolic process |
| L0I0Y7 | Ribosomal protein S6 modification protein | 0.092 | 5.71E-06 | 0.291 | 9.22E-05 | 0.461 | 0.002 | cellular protein modification process |
| Q87SB9 | ATP-dependent RNA helicase SrmB ATP | 2.965↑ | 0.048 | 3.981↑ | 0.004 | 6.081↑ | 0.001 | ATP-dependent helicaseactivity |
| Q87P98 | Amino acid ABC transporter, periplasmic amino acid-binding protein | 4.093↑ | 0.001 | 0.191 | 0.001 | 9.462↑ | 3.95E-06 | transporter activity |
| F3RQ38 | Diaminobutyrate-pyruvate transaminase and L-2,4-diaminobutyrate decarboxylase | 0.217 | 0.021 | 2.535↑ | 0.001 | 0.146 | 0.014 | carboxylic acid metabolic process |
| Q87P67 | Putative tricarboxylic transport TctC | 4.325↑ | 0.003 | 4.325↑ | 0.002 | 2.992↑ | 0.013 | outer membrane-bound periplasmic space |
| L0HXG6 | ABC- type antimicrobial peptide transport system, permease component | 6.252↑ | 0.016 | 2.489↑ | 0.041 | 15.136↑ | 0.008 | integral to membrane |
| Q87PV1 | Uncharacterized protein | 3.664↑ | 0.048 | 10.568↑ | 0.012 | 3.565↑ | 0.014 | protein serine/threonine kinase activity |
aiTRAQ ratio indicates the relative quantification of the differentially expressed proteins
bStatistical analysis of iTRAQ ratio was performed using unpaired t-test
cUp arrows indicate an increase in protein expression
Functional analysis of the proteins with different abundance showed the regulated proteins could be clustered into 4 major groups: 1) metabolic proteins; 2) proteins involved in transcription and translation; 3) membrane-associated proteins including proteins involved in transport, antibiotic efflux system, secretion system, outer membrane proteins, etc.; 4) virulence factors including proteins involved in iron acquisition, secreted protease, etc. In this paper, we would pay emphasis on analysis of the latter three groups.
A number of proteins related to DNA replication, cell division and transcriptional regulation were observed to be differentially regulated in some conditions. Most of proteins participated in chromosome structure, DNA replication, and transcription were down-regulated in VPP (Additional file 2: Table S3). However, 20 translation-related proteins were present at increased abundance in VPE, including peptide chain release factor 1, translation initiation factor IF-2, elongation factor Ts, elongation factor Tu, 30S ribosomal proteins (S9, S10, S8, S11, S13, S6, and S18), and 50S ribosomal proteins (L6,L9, L13, L31, and L19) (Additional file 3: Table S4). 30S small ribosomal subunit proteins and 50S large ribosomal subunit proteins constitute ribosome and exhibit different functions in protein synthesis.
Surface proteins can directly participate in microbial virulence by facilitating pathogen dissemination via interactions with host factors. For example, outer membrane proteins are located at host–bacterial interface and are important for host immune responses and as targets for drug therapy [26]. Drug efflux pumps can participate in drug resistance to multiple antimicrobials through export drugs, and also serve other functions in bacteria. Recently, antibiotic-resistant strains of the bacterium from clinical and environmental sources have been frequently reported [27, 28]. Investigation of secretion systems is often critical to understanding the virulence mechanisms of bacterial pathogens. It was estimated that as high as 30–40 % of proteins were secreted or localized to the cell envelope. So far, seven different secretion systems, type I-VII, have been described in bacteria [29, 30]. These secretion systems release factors that modulate the host environment to favor bacterial fitness and or virulence.
In this study, numerous membrane proteins were identified and some membrane proteins were differentially regulated in some condition. VPX displayed highly elevated abundance of type I secretion outer membrane protein TolC, lipoprotein, apolipoprotein N-acyltransferase, long-chain fatty acid transport protein, magnesium and cobalt transport protein CorA, but a putative polysaccharide export-related protein displayed reduced. Moreover, a number of efflux system proteins, including putative multidrug efflux membrane fusion protein, ABC transporter, periplasmic substrate-binding protein, phosphate ABC transporter, and permease protein PstA were present at increased abundance in VPX. And chemotaxis proteins, such as putative chemotaxis transducer (GN VPA0562), methyl-accepting chemotaxis proteins (GN VP1904, VIPARK5030_0929, VPBB_A0559, VPA0511), and magnesium and cobalt transport protein CorA were up-regulated similarly.
Many proteins involved in V. parahaemolyticus virulence were significantly up-regulated in VPX; this result could be attributed to the blood component of blood agar, which suggested that the pathogenicity of V. parahaemolyticus is likely enhanced in blood after infecting animals. Nevertheless, further studies should be conducted to elucidate the underlying mechanisms.
Iron availability was reported to play a major role in the virulence of bacterium [31]. We discovered that four iron-regulated proteins, iron-regulated outer membrane virulence protein homolog (GN VP2602), iron-regulated protein A (GN VPBB_1761), putative Fe-regulated protein B (GN VPA0664) and putative iron compound receptor (GN VPA1435) and four transport proteins including ferric siderophore receptor homolog (GN VPA1657), heme transport protein HutA (GN VPBB_A0827), ferric vibrioferrin receptor (GN VPA1656) and ferric aerobactin receptor(GN iutA) were significantly decreased in VPE compared with VPP, VPW and VPX, namely they were significantly increased relatively in VPP, VPW and VPX (Fig. 5). Pyridoxamine 5'-phosphate oxidase-related putative heme iron utilization protein (GN VPBB_A0398) also was down-regulated. Only iron-containing alcohol dehydrogenase (GN VPA0829) was significantly up-regulated. Other proteins involved in iron maintenance, including ferrous iron transport protein B (GN VP0858); ferric uptake regulatory protein, bacterioferritin (GN VP2768), co-migratory protein(GN A79_2683) and ferritin (GN VP0077) was increased to some extent but not to a significantly different degree.
Fig. 5.
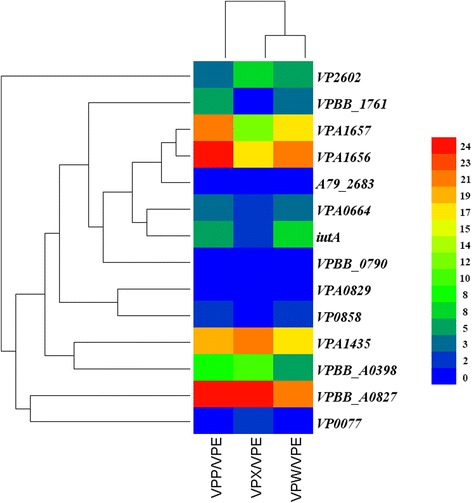
Differential expression of iron-responsive proteins in VPP, VPE, and VPW compared with VPE. Red indicates higher expression, blue indicates lower expression, and graphic symbols indicate the ratio change of expression between the strains. The proteins were grouped depending on their expression level
Ferric uptake regulatory protein Fur (GN VPBB_0790) containing sequence-specific DNA binding transcription factor is the master regulator as a repressor of iron acquisition-related genes [32], Miyamoto et al. [33] showed that the Fur protein regulates the expression of the virulence-associated orthologous groups in the family Vibrionaceae. In this study, increased Fur level was consistent with decreased abundances observed for several iron acquisition proteins in VPE (Fig. 5).
Iron acquisition via siderophore production is critical for successful colonization and for providing bacterium with a distinct competitive advantage over other pathogens [20]. In this study, protein expression of V. parahaemolyticus displayed a unique feature on the FeCl3 agar because of the ferric component. We discovered that the down-regulated proteins in VPE were involved in iron uptake, while up-regulated proteins were involved in iron storage. The results suggesting V. parahaemolyticus was adapted to an iron-rich environment, the species can maintain intracellular iron concentration at a relatively stable level by self-adjusting mechanisms. In other study, Pseudomonas aeruginosa was observed to be adapted to an iron-limited environment [20].
Several proteases, including the proteases of insulinase family protein, protease IV, protease HtpX, secreted microbial collagenase and putative membrane-associated Zn-dependent protease were evidently increased in VPX; however, ATP-dependent Clp protease were decreased. Protease is a virulence determinant [20], which suggested VPX may possess stronger virulence.
Information about the subcellular localization and organization of secretion system as well as identification and functional characterization of their substrates are key steps toward understanding these intricate systems. The role of type VI secretion system in virulence, symbiosis, biofilm formation, and stress response has been documented in several bacteria [34, 35]. In this study, type VI secretion protein-VC_A0110 family protein (GN VIPARK5030_1375) was up-regulated in VPE.
T3SS is possessed by gram-negative bacteria, especially those animal and plant pathogens, e.g., Yersinia, Shigella, Salmonella, Pseudomonas and Escherichia species [36, 37]. The T3SS secretes and translocates effector proteins into the cytosol of eukaryotic cells, thus contributing to bacterial virulence against the host [36], and V. parahaemolyticus was first reported to contain T3SS [38]. Previous study has showed that T3SS expression was effected by environmental factors [39], but the 7 T3SS proteins including Spa33, ATPase YscC and YscL, etc., were not significantly differentially expressed in this study.
Three hemolysin proteins including putative hemolysins (GN VP0730, GN VP2536) and hemolysin (GN VPA0257) were identified, although TDH production was reported to be affected by environmental factors, such as pH, temperature, and chemicals [40, 41], but significant differences in abundance have not been observed between the four culture conditions in this study. TDH and TDH-related hemolysin (TRH) are considered as the main virulence factors of V. parahaemolyticus, and pathogenetic V. parahaemolyticus strains can contain TDH, TRH, or both [12, 42, 43]. However, the presence of pathogenic populations of V. parahaemolyticus in environmental samples is generally low [3]. Presence of the V. parahaemolyticusstrains containing TDH could be accounted for human fecal contamination in seawater from the sewage at the coast of Dalian. This information may be considered to prevent sanitary problems that may affect human health.
Two important regulator proteins Hfq and RsmA were successfully identified, they are two RNA binding proteins and major post-transcriptional regulators of gene expression. Increasing evidence shows that Hfq and its dependent sRNAs play a fundamental role in the regulation of stress response and pathogenesis [44]. Nakano et al. [45] investigated the effect of Hfq on the expression of virulence-associated genes including TDH using an hfq deletion mutant and observed that Hfq could depress the expression of TDH and may be involved in the pathogenicity of V. parahaemolyticus. RsmA (Ribosomal RNA small subunit methyltransferase A, GN rsmA) is the homologue of E. coli CsrA, was reported to play an important role in regulation of virulence and biocontrol factor production in Pseudomonas aeruginosa and P. fluorescens [46], and was examined to affect expression of some functional proteins including efflux transporters, outer membrane proteins, sigma factors, and stress response proteins in in Serratia sp. ATCC 39006 [17]. In this study, the two proteins were not significantly differentially expressed, however, Hfq was elevated to 1.318–1.542 folds in VPP, VPE and VPX compared with VPW, while the putative hemolysin was reduced to 0.597–0.614 fold, which was consistent with Nakano’s discovery.
Correlation of expression between proteins and genes
In order to analyze the correlation between protein expression and gene expression and to confirm the accuracy of iTRAQ ratio obtained from the present proteomics study, we analyzed genes expression of 50 differentially expressed proteins in VPX compared with VPW by qRT-PCR. Data from triplicate experiments were analyzed for statistical significance by the unpaired t-test (two side test), with p < 0.05 being considered statistically significant. When the fold change of gene expression > 1.2 and P <0.05 indicated that the genes were regarded as regulated. Part results were showed as fold means ± standard deviation (SD) in Fig. 6.
Fig. 6.
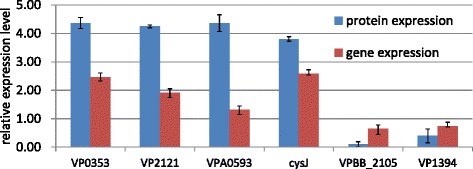
Comparison of iTRAQ result and qRT-PCR analysis. iTRAQ ratios of regulated proteins in VPX compared with VPW were confirmed by qRT-PCR analyses. The comparison of the fold changes showed the directions of protein regulation and gene regulation were the same for the 6 representative proteins. The former 4 proteins were up-regulated at both protein level and RNA level, and the latter 2 proteins were down-regulated. Blue bars values represent average protein expression ± SD and red bars values represent average gene expression ± SD from three independent experiments
Comparison of the fold changes showed the direction of change was the same for 39 proteins, about 80 % of detected proteins, at the level of both RNA and protein, which gave a positive Pearson correlation (Additional file 5: Table S6), and 9 proteins showed no significant correlation between the protein production and the mRNA transcript. However, 2 proteins, magnesium and cobalt transport protein CorA and alcohol dehydrogenase, iron-dependent presented negative correlation, which suggested a post-transcriptional effect on regulation. These results indicated that the iTRAQ ratios in present study are almost consistent with quantitative results obtained by qRT-PCR.
Conclusions
This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in V. parahaemolyticus under different conditions. The results confirmed that V. parahaemolyticus presented a unique protein profile that indicated the adaptive mechanisms of this species to different environments. This profile could also provide promissing candidate proteins to detect environmental changes by using V. parahaemolyticus. Numerous membrane proteins such as type I secretion outer membrane protein TolC, lipoprotein, efflux system proteins and virulence associated proteins were characterized, some of them were significantly regulated. This study would advance our understanding of the evolution and pathogenicity of this food-borne pathogen.
Materials and methods
Source, isolation and culture of V. parahaemolyticus
V. parahaemolyticus strain was isolated from a sewage outlet in Dalian, China and selectively identified using thiosulfate-citrate-bile-sucrose agar medium (Haibo, China). The identity of V. parahaemolyticus was confirmed by 16S rRNA gene sequence analysis and analytical profile index tests. The identified and characterized pure culture was streaked onto sterile sewage agar (0.5 % beef extract, 1 % tryptone, 2 % agar, sterilized sewage) and seawater agar (0.5 % beef extract, 1 % tryptone, 2 % agar, sterilized seawater), the former mimics bacterial growth in sewage water and the latter mimics growth in seawater environment. In view of the hemolysis function of V. parahaemolyticus and requirement of ferric ion in metabolism, blood agar (0.5 % beef extract, 1 % tryptone, 2 % agar, 2 % sodium chloride, 5 % anticoagulant goat blood) and FeCl3 agar (0.5 % beef extract, 1 % tryptone, 2 % agar, 1 mmol/L FeCl3, sterilized seawater) were chosen to culture bacterium. Bacterium was cultured on every medium in triplicate. After the cultures were incubated at 37 °C for 12 h, the colonies on the four media were collected and named as VPW, VPP, VPE, and VPX matching sewage agar, seawater agar, FeCl3 agar and blood agar, respectively. The bacterial cultures were maintained at −80 °C for further analysis.
Quantitative proteomics by iTRAQ and LC-MS/MS
Total protein extraction and quantification
Total protein was extracted from the colonies as described previously [20]; protein concentration was determined with Bradford colorimetric method (Bio-Rad) according to standard protocol. At least 100 μg of protein from each sample was lyophilized using a speed vacuum system (Martin Christ, Germany).
iTRAQ labeling and strong cation exchange fractionation
iTRAQ labeling involves denaturation, reduction, trypsin digestion, and labeling. Protein samples were reduced using a reducing reagent (AB Sciex, PN: 4381664) for 1 h at 60 °C according to iTRAQ protocol. Cysteine was blocked using cysteine blocking reagent (AB Sciex, PN: 4381664) for 10 min at room temperature and then digested with trypsin (AB Sciex, PN: 4370285) overnight at 37 °C. iTRAQ (AB Sciex,PN:4381664) labeling of peptides was conducted according to the manufacturer’s protocol. In brief, peptides from V. parahaemolyticus colonies VPP, VPE, VPX, and VPW were labeled with iTRAQ reagents containing the reporters 114, 115, 116, and 117, respectively. Every sample was labeled in triplicate. Labeling was carried out for 2 h at room temperature. After labeling was performed, the peptides from the four colonies were pooled and fractionated by strong cation exchange chromatography on Durashell-C18 (4.6 mm × 250 mm, 5 μm 100 Å; Agela, Catalog Number: DC952505-0) to remove the remaining iTRAQ reagent and other reagents. The peptides were separated and eluted using a linear gradient of 0 to 70 % ACN for 70 min at a flow rate of 0.8 mL/min. Eluted peptides were monitored using a diode array detector at a wave length range of 200 nm to 400 nm. The eluted peptides were then collected at an interval of 1 min beginning at the first 5 min; a total of 48 fractionated components were collected per sample. The components were reduced to dryness by using a speed vacuum centrifuge.
Mass spectrometry data analysis and protein quantification
Mass spectrometry analysis was performed using a reversed-phase liquid chromatography system (Eksigent) interfaced with Triple TOF™ 5600mass spectrometer (AB Sciex). MS/MS data were acquired by online analysis of peptides eluted using 5 to 80 % acetonitrile in 0.1 % formic acid for 100 min with a flow rate of 300 nL/min. MS/MS spectra were obtained in a data-dependent manner from m/z 350 to 1250 for TOF MS scan and from m/z 100 to1500 for product ion scan; these scans targeted the 10 most abundant ions in each survey scan with an accumulation period of 0.1 s and a dynamic exclusion period of 25 s.
MS data were analyzed by ProteinPilot 4.5(AB SCIEX, Foster City), which includes Paragon algorithm for identifying peptides and Pro Group algorithm for summarizing proteins. Search parameters were set as iTRAQ labeling at N-terminus and lysine residues, cysteine modification by Methylmethanethiosulfonate as fixed modifications, and trypsin as a protease. Proteins identified with 1 % false discovery rate (FDR) as determined by Pro Group algorithm were used for further analysis. The MS/MS data of 48 SCX fractions generated by LC–MS/MS analysis were searched against a database of predicted proteins reported for V. parahaemolyticusby using ProteinPilot 4.5 software. FDR was analyzed using PSPEP software in ProteinPilot 4.5. A threshold of 1 % FDR was used to identify and quantify proteins.
Proteins with a ratio <0.50 or >2.0were considered to be differentially expressed, and data from triplicate experiments were analyzed for statistical significance by unpaired t- test. In all cases, p < 0.05 was considered significant, namely, iTRAQ ratios <0.50 or >2.0 with p < 0.05 were considered significantly different in protein quantification.
Gene ontology analysis of proteins and pathogenicity analysis
The identified proteins were subjected to GO analysis by using the analytical system of Generic Gene Ontology Term Finder (http://go.princeton.edu/cgi-bin/GO Term Finder) [47]. The fundamental functions of proteins were counted and analyzed; differentially expressed proteins were enriched into different functions with p ≤0.05 as threshold in the chi square test. The biological pathways of the proteins were acquired from the KEGG Pathway database (http://www.genome.ad.jp/kegg/) [48] coupled with UniProtKB annotation (UniProtKB database, http://www.uniprot.org/) [23]. The abundance of virulence-related proteins was analyzed to reveal the effect of growth conditions on the pathogenicity of V. parahaemolyticus strain.
qRT-PCR analysis
Total RNA extraction
Total RNA was extracted from VPE and VPW when V. parahaemolyticus was cultured for 12 h at 37 °C. RNA was extracted using the hot phenol method as previously described [49]. Precipitated RNA was re-suspended in 100 μL H2O and treated with DNase I (Roche Diagnostics, Basel, Switzerland), according to the manufacturer’s protocol. The RNA was incubated at 37 °C for 30 min. The concentration of the RNA was verified by absorbance measurements at 260 nm and the purity was verified by its A260/280 ratio with a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies Inc, USA), the integrity was verified by electrophoresis separation using the RNA Nano kit (Agilent Technologies).
qRT-PCR
Regulation of the gene expression matching 50 regulated proteins in VPX compared with VPW was analyzed by qRT-PCR. 4 μg of total RNA was transcribed to cDNA using oligodT primers and SuperScript II Reverse Transcriptase Kit as recommended (Invitrogen). qRT-PCR reaction was performed in triplicate using the ABI Prism 7900HT sequence detection system (Applied Biosystems) with SYBR® Premix Ex TaqTM II according to the manufacturer’s protocols. Oligonucleotide primers for quantitative PCR are listed in Additional file 5: Table S6. Reaction mixture contained 10 μL SYBR® Premix Ex TaqTM II 2×, 1 μL (10 μM) of reverse and forward primer, and 2.5 μL of cDNA in a final volume of 25 μL. The thermal amplification was performed as follow: initial denaturation at 95 °C for 1 min, followed by 40 cycles with 30 s at 95 °C, 15 s annealing at 55 °C and 30s at 72 °C, followed by a single fluorescence measurement. Relative gene expression was obtained using 16S rRNA as the control with mRNA/16S rRNA = 1 in the VPW. Gene expression data obtained from PCR reaction were evaluated using Q-Gene.
Acknowledgments
This study was supported by the Chinese Special Fund for the Non-profit Marine Research(Grant No.201105007-4), the Major Research Program of Science and Technology, Zhejiang Province (Grant No.2006C13089), the Funding Project of Science and Technology Bureau, Ningbo (Grant No.2008C50027), and the Opening Foundation of the State Key Laboratory of Offshore Area Environment, State Oceanic Administration of China (Grant No.201103).
Abbreviations
- iTRAQ
isobaric tags for relative and absolute quantitation
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- qRT-PCR
quantitative real time polymerase chain reaction
- TDH
thermostable direct hemolysin
- MS
Mass spectrometry
- FDR
false discovery rate
- GO
Gene Ontology
- T3SS
type III secretion system
- TRH
TDH-related hemolysin
- SD
standard deviation
Additional files
GO enrichment analysis of proteins with differential expressions. Table S2. GO enrichment analysis of proteins with differential expressions (biological process). (XLSX 14 kb)
Unique differentially expressed proteins in VPP compared with VPW. (XLSX 21 kb)
Unique differentially expressed proteins in VPE compared with VPW. (DOCX 39 kb)
Unique differentially expressed proteins in VPX compared with VPW. (DOCX 65 kb)
The correlation of expression between proteins and genes. (XLSX 16 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WY performed the experiments and data analysis, and composed the draft of the manuscript. DD conceived and supervised experimental work. CZ performed data acquisition and interpretation. JZ contributed to the proteomic analysis. XS participated in the supervision of the study and were involved in manuscript editing. All authors have read and approved the final manuscript.
Contributor Information
Wenxin Yang, Email: ywxin168@163.coml.
Dewen Ding, Email: 13941164513@139.com.
Chundan Zhang, Email: zhangchundan@nbu.edu.cn.
Jun Zhou, Email: 237602544@qq.com.
Xiurong Su, Email: suxiurong@nbu.edu.cn.
References
- 1.DePaola A, Nordstrom JL, Dalsgaard A, Forslund A, Oliver J, Bates T, Bourdage KL, Gulig PA. Analysis of Vibrio vulnificus from market oysters and septicemia cases for virulence markers. Appl Environ Microbiol. 2003;69:4006–4011. doi: 10.1128/AEM.69.7.4006-4011.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okuda J, Ishibashi M, Hayakawa E, Nishino T, Takeda Y, Mukhopadhyay AK, Garg S, Bhattacharya SK, Nair GB, Nishibuchi M. Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J Clin Microbiol. 1997;35:3150–3155. doi: 10.1128/jcm.35.12.3150-3155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez-Urtaza J, Lozano-Leon A, DePaola A, Ishibashi M, Shimada K, Nishibuchi M, Liebana E. Characterization of pathogenic Vibrio parahaemolyticus isolates from clinical sources in Spain and comparison with Asian and North American pandemic isolates. J Clin Microbiol. 2004;42:4672–4678. doi: 10.1128/JCM.42.10.4672-4678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ansaruzzaman M, Lucas M, Deen JL, Bhuiyan NA, Wang XY, Safa A, Sultana M, Chowdhury A, Nair GB, Sack DA, von Seidlein L, Puri MK, Ali M, Chaignat CL, Clemens JD. Pandemic serovars(O3:K6 and O4:K68) of Vibrio parahaemolyticus associated with diarrhea in Mozambique: spread of the pandemic into the African continent. J Clin Microbiol. 2005;43:2559–2562. doi: 10.1128/JCM.43.6.2559-2562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Escalona N, Cachicas V, Acevedo C, Rioseco ML, Vergara JA, Cabello F, Romero J, Espejo RT. Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg Infect Dis. 2005;11:129–131. doi: 10.3201/eid1101.040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sujeewa AKW, Norrakiah AS, Laina M. Prevalence of toxic genes of Vibrio parahaemolyticus in shrimps (Penaeus monodon) and culture environment. Int Food Res J. 2009;16:89–95. [Google Scholar]
- 7.Paydar M, Teh CS, Thong KL. Prevalence and characterisation of potentially virulent Vibrio parahaemolyticus in seafood in Malaysia using conventional methods, PCR and REP-PCR. Food Control. 2013;32:13–18. doi: 10.1016/j.foodcont.2012.11.034. [DOI] [Google Scholar]
- 8.Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus sero type O3:K6 and its serovariants. Clin Microbiol Rev. 2007;20:39–48. doi: 10.1128/CMR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joseph SW, Colwell RR, Kaper JB. Vibrio parahaemolyticus and related halophilic Vibrios. Crit Rev Microbiol. 1982;10:77–124. doi: 10.3109/10408418209113506. [DOI] [PubMed] [Google Scholar]
- 10.Alipour M, Issazadeh K, Soleimani J. Isolation and identification of Vibrio parahaemolyticus from seawater and sediment samples in the southern coast of the Caspian Sea. Comp Clin Pathol. 2014;23:129–133. doi: 10.1007/s00580-012-1583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabrera-Garcia ME, Vazquez-Salinas C, Q-R EI. Serologic and molecular characterization of Vibrio parahaemolyticus strains isolated from seawater and fish products of the Gulf of Mexico. Appl Environ Microbiol. 2004;70:6401–6407. doi: 10.1128/AEM.70.11.6401-6406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishibuchi M, Kaper JB. Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect Immun. 1992;63:2093–2099. doi: 10.1128/iai.63.6.2093-2099.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishibuchi M, Fasano A, Russell RG, Kaper JB. Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect Immun. 1992;60:3539–3545. doi: 10.1128/iai.60.9.3539-3545.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bag PK, Nandi S, Bhadra RK, Ramamurthy T, Bhattacharya SK, Nishibuchi M, et al. Clonal diversity among recently emerged strains of Vibrio parahaemolyticus O3:K6 associated with pandemic spread. J Clin Microbiol. 1999;37:2354–2357. doi: 10.1128/jcm.37.7.2354-2357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto C, Okuda J, Ishibashi M, Iwanaga M, Garg P, Rammamurthy T, Wong HC, Depaola A, Kim YB, Albert MJ, Nishibuchi M. Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J Clin Microbiol. 2000;38:578–585. doi: 10.1128/jcm.38.2.578-585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nair GB, Hormazábal JC. The Vibrio parahaemolyticus pandemic. Rev Chilena Infectol. 2005;22:125–130. doi: 10.4067/s0716-10182005000200002. [DOI] [PubMed] [Google Scholar]
- 17.Wilf NM, Reid AJ, Ramsay JP, Williamson NR, Croucher NJ, Gatto L, et al. RNA-seq reveals the RNA binding proteins,Hfq and RsmA, play various roles in virulence,antibiotic production and genomic flux in Serratia sp. ATCC 39006. BMC Genomics. 2013;14:822–838. doi: 10.1186/1471-2164-14-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prasad TSK, Keerthikumar S, Chaerkady R, Kandasamy K, Renuse S, Marimuthu A, et al. Comparative Proteomic Analysis of Candida albicans and Candida glabrata. Clin Proteom. 2010;6:163–174. doi: 10.1007/s12014-010-9057-9. [DOI] [Google Scholar]
- 19.Hanl B, Chen S, Dai S, Yan N, Wan T. Isobaric Tags for Relative and Absolute Quantification based Comparative Proteomics Reveals the Features of Plasma Membrane-Associated Proteomes of Pol len Grains and Pollen Tubes from Lilium davidii. J Integr Plant Biol. 2010;52:1043–1059. doi: 10.1111/j.1744-7909.2010.00996.x. [DOI] [PubMed] [Google Scholar]
- 20.Hare NJ, Solis N, Harmer C, Marzook NB, Rose B, Harbour C, et al. Proteomic profiling of Pseudomonas aeruginosa AES-1R, PAO1 and A14 reveals potential virulence determinants associated with a transmissible cystic fibrosis-associated strain. BMC Microbiol. 2012;12:16–29. doi: 10.1186/1471-2180-12-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho NR, Lettieri T. Proteomic analysis of the marine diatom Thalassiosira pseudonana upon exposure to benzo(a)pyrene. BMC Genomics. 2011;12:159–169. doi: 10.1186/1471-2164-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.UniProtKB database. [http://www.uniprot.org/].
- 24.Hunt DE, David LA, Gevers D, Preheim SP, Alm E, Polz MF. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science. 2008;320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 25.Keymer DP, Miller MC, Schoolnik GK, Boehm AB. Genomic and phenotypic diversity of coastal Vibrio cholerae strains is linked to environmental factors. Appl Environ Microbiol. 2007;73:3705–3714. doi: 10.1128/AEM.02736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khushiramani R, Girisha SK, Bhowmick PP, Karunasagar I, Karunasagar I. Prevalence of different outer membrane proteins in isolates of Aeromonas species. World J Microbiol Biotechnol. 2008;24(10):2263–2268. doi: 10.1007/s11274-008-9740-4. [DOI] [Google Scholar]
- 27.Xiong XP, Wang C, Ye MZ, Yang TC, Peng XX, Li H. Differentially Expressed Outer Membrane Proteins of Vibrio alginolyticus in Response to Six types of Antibiotics. Mar Biotechnol. 2010;12:686–695. doi: 10.1007/s10126-009-9256-4. [DOI] [PubMed] [Google Scholar]
- 28.Tai DT, Thuy AV, Nhi NTN, Ngoc NTK, Lan NTP. Virulence and antimicrobial resistance characteristics of Vibrio parahaemolyticus isolated from environment, food and clinical samples in the south of Vietnam, 2010. BMC Proc. 2011;5(Suppl 1):94–95. doi: 10.1186/1753-6561-5-s1-p94. [DOI] [Google Scholar]
- 29.Economou ACP, Fernandez RC, Palmer T, Plano GV, Pugsley AP. Secretion by numbers: protein traffic in prokaryotes. Mol Microbiol. 2006;62:308–319. doi: 10.1111/j.1365-2958.2006.05377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng TTTB, Setubal JC. Protein secretion systems in bacterial-host associations, and their description in the gene ontology. BMC Microbiol. 2009;9(Suppl1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gulig PABK, Starks AM. Molecular pathogenesis of Vibrio vulnificus. J Microbiol. 2005;43:118–131. [PubMed] [Google Scholar]
- 32.Cornelis P, Matthijs S, Van Oeffelen L. Iron uptake regulation in Pseudomonas aeruginosa. Biometals. 2009;22:15–22. doi: 10.1007/s10534-008-9193-0. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto KKK, Ikebayashi S, Tsuchiya T, Yamamoto S, Tsujibo H. Proteomic analysis of Vibrio vulnificus M2799 grown under ironrepleted and iron-depleted conditions. Microb Pathog. 2009;46(3):171–177. doi: 10.1016/j.micpath.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Rice EW, Johnson CJ, Clark RM, Fox KR, Reasoner DJ, Dunnigan ME, Panigrahi P, Johnson JA, Morris JG., Jr Chlorine and survival of “rugose” Vibrio cholerae. Lancet. 1992;340:740. doi: 10.1016/0140-6736(92)92289-R. [DOI] [PubMed] [Google Scholar]
- 35.Weber B, Hasic M, Chen C, Wai SN, Milton DL. Type VI secretion modulates quorum sensing and stress response in Vibrio anguillarum. Environ Microbiol. 2009;11:3018–3028. doi: 10.1111/j.1462-2920.2009.02005.x. [DOI] [PubMed] [Google Scholar]
- 36.Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry RDFJ. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita AKY, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 39.Henke JMBB. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J Bacteriol. 2004;186:3794–3805. doi: 10.1128/JB.186.12.3794-3805.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong HC, Lee YS. Regulation of iron on bacterial growth and production of thermostable direct hemolysin by Vibrio parahaemolyticus in intraperitoneal infected mice. Microbiol Immunol. 1994;38:367–371. doi: 10.1111/j.1348-0421.1994.tb01792.x. [DOI] [PubMed] [Google Scholar]
- 41.Osawa R, Yamai S. Production of thermostable direct hemolysin by Vibrio parahaemolyticus enhanced by conjugated bile acids. Appl Environ Microbiol. 1996;62:3023–3025. doi: 10.1128/aem.62.8.3023-3025.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Honda T, Ni YX, Miwatani T. Purification and characterization of a hemolysin produced by a clinical isolate of Kanagawa phenomenon-negative Vibrio parahaemolyticus and related to the thermostable direct hemolysin. Infect Immun. 1988;56:961–965. doi: 10.1128/iai.56.4.961-965.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Honda T, Iida T. The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct heamolysin and related heamolysins. Rev Med Microbiol. 1993;4:106–113. doi: 10.1097/00013542-199304000-00006. [DOI] [Google Scholar]
- 44.Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr Opin Microbiol. 2010;13(1):24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Nakano M, Takahashi A, Su Z, Harada N, Mawatari K, Nakaya Y. Hfq regulates the expression of the thermostable direct hemolysin gene in Vibrio parahaemolyticus. BMC Microbiol. 2008;8:155–163. doi: 10.1186/1471-2180-8-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lapouge K, Schubert M, Allain FH-T, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67(2):241–253. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 47.Generic Gene Ontology(Go)Term Finder. [http://go.princeton.edu/cgi-bin/GO Term Finder].
- 48.KEGG Pathway Database. [http://www.genome.ad.jp/kegg/].
- 49.Mattatall NRSK. Salmonella typhimurium LT2 possesses three distinct 23S rRNA intervening sequences. J Bacteriol. 1996;178(8):2272–2278. doi: 10.1128/jb.178.8.2272-2278.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]