

Abstract
In the title compound, C15H18N2O3S, the hydropyrimidine ring adopts a sofa conformation with the methine C atom as the flap. The benzene ring is almost perpendicular to the mean plane of the hydropyrimidine ring, making a dihedral angle of 85.51 (8)°, and the methoxy O atom lies over the centre of the pyrimidine ring. In the crystal, weak N—H⋯S interactions form a zigzag chain running along the b-axis direction.
Keywords: crystal structure, Biginelli reactions, dihyropyrimidinones, three-component reactions, N—H⋯S interactions
Related literature
For syntheses of dihydropyrimidinones and their analogous, see: Biginelli (1893 ▸); Varala et al. (2003 ▸); Gohain et al. (2004 ▸); Ahmed et al. (2009 ▸). For biological activities of hydropyrimidinones, see: Salehi et al. (2006 ▸); Singh et al. (2010 ▸); Hed et al. (2009 ▸); Russowsky et al. (2007 ▸); Shah et al. (2009 ▸). For the synthesis of the title compound, see: Ahmed et al. (2012 ▸).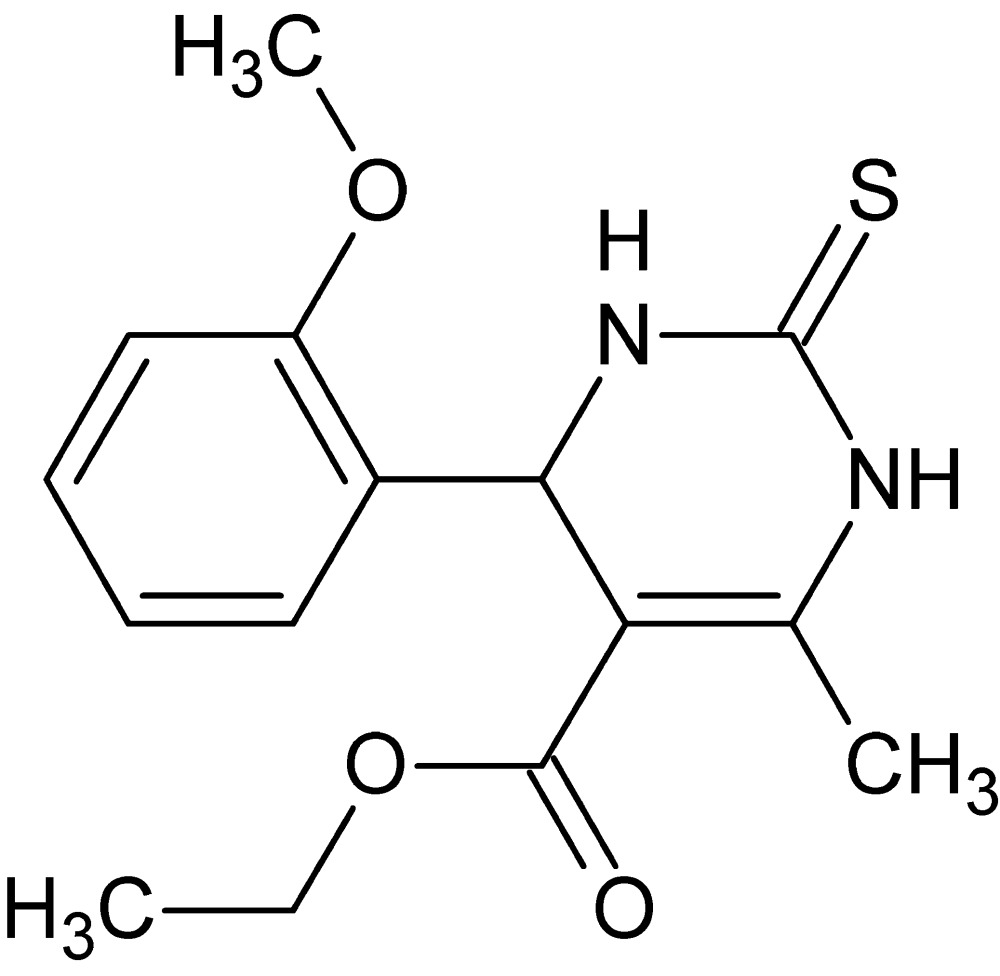
Experimental
Crystal data
C15H18N2O3S
M r = 306.37
Triclinic,
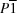
a = 7.9791 (2) Å
b = 8.2031 (2) Å
c = 11.8405 (3) Å
α = 81.987 (1)°
β = 87.975 (1)°
γ = 80.850 (1)°
V = 757.60 (3) Å3
Z = 2
Cu Kα radiation
μ = 2.00 mm−1
T = 150 K
0.25 × 0.21 × 0.12 mm
Data collection
Bruker D8 VENTURE PHOTON 100 CMOS diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2014 ▸) T min = 0.73, T max = 0.79
9145 measured reflections
2929 independent reflections
2773 reflections with I > 2σ(I)
R int = 0.021
Refinement
R[F 2 > 2σ(F 2)] = 0.039
wR(F 2) = 0.101
S = 1.08
2929 reflections
193 parameters
H-atom parameters constrained
Δρmax = 0.43 e Å−3
Δρmin = −0.26 e Å−3
Data collection: APEX2 (Bruker, 2014 ▸); cell refinement: SAINT (Bruker, 2014 ▸); data reduction: SAINT; program(s) used to solve structure: SHELXT (Sheldrick, 2015a ▸); program(s) used to refine structure: SHELXL2014 (Sheldrick, 2015b ▸); molecular graphics: DIAMOND (Brandenburg & Putz, 2012 ▸); software used to prepare material for publication: SHELXL2014.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015010026/is5401sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010026/is5401Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010026/is5401Isup3.cml
. DOI: 10.1107/S2056989015010026/is5401fig1.tif
The molecular structure of the title compound showing labeling scheme and 50% probability ellipsoids.
. DOI: 10.1107/S2056989015010026/is5401fig2.tif
A section of the chain formed by N—H⋯S hydrogen bonds (dashed lines).
b . DOI: 10.1107/S2056989015010026/is5401fig3.tif
A packing diagram viewed along the b axis. N—H⋯S interactions are shown as dotted lines.
CCDC reference: 1402530
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H1AS1i | 0.91 | 2.46 | 3.3539(13) | 167 |
| N2H2AS1ii | 0.91 | 2.58 | 3.4327(14) | 157 |
Symmetry codes: (i) 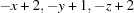 ; (ii)
; (ii) 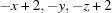 .
.
Acknowledgments
The support of NSF–MRI grant No. 1228232 for the purchase of the diffractometer and Tulane University for support of the Tulane Crystallography Laboratory are gratefully acknowledged.
supplementary crystallographic information
S1. Comment
Dihydropyrimidin-2(1H)-one scaffold compounds are an important class of substances in organic and medicinal chemistry. Aryl-substituted 3, 4-dihydropyrimidin-2(1H)-ones and their sulfur analogue have been reported to possess diverse range of pharmacological activity (Salehi et al., 2006) such as anticancer, anti HIV, antibacterial, antimalarial, antihypertensive, sedative, hypnotics, anticonvulsant, antithyroid,antihistaminic agents and antibiotics (Singh et al., 2010; Hed et al., 2009; Russowsky et al., 2007; Shah et al., 2009). This stimulated the invention of a wide range of synthetic methods for their preparation and chemical transformations. In recent years, several modified procedures have been reported to improve the efficiency of the Biginelli dihydropyrimidine synthesis (Biginelli, 1893) by using different catalysts e.g. Lewis acids (Varala et al., 2003; Gohain et al., 2004) or by using basic condition via phase transfer catalysis (Ahmed et al., 2009). In this context, we report in this study the crystal structure of the title compound.
In the title compound (Fig. 1), the plane of the benzene ring is almost parallel to the C1···N2 vector with the methoxy oxygen atom (O1) lying over the centre of the pyrimidine ring. The pyrimidine ring has Cremer-Pople puckering parameters Q = 0.201 (2) Å, θ = 62.2 (5)° and φ = 42.9 (2)°. In the crystal, weak N—H···S interactions (Table 1) form a chain running parallel to the b axis (Figs. 2 & 3).
S2. Experimental
The title compound was prepared according to our reported method (Ahmed et al., 2012). Colourless crystals suitable for X-ray analysis were grown from ethanol (m.p. 473–475 K, yield 98%).
S3. Refinement
H-atoms attached to C were placed in calculated positions (C—H = 0.95–1.00 Å), while those attached to N were placed in a difference map and their parameters adjusted to give N—H = 0.91 Å. All were included as riding contributions with isotropic displacement parameters 1.2 or 1.5 times those of the attached atoms.
Figures
Fig. 1.
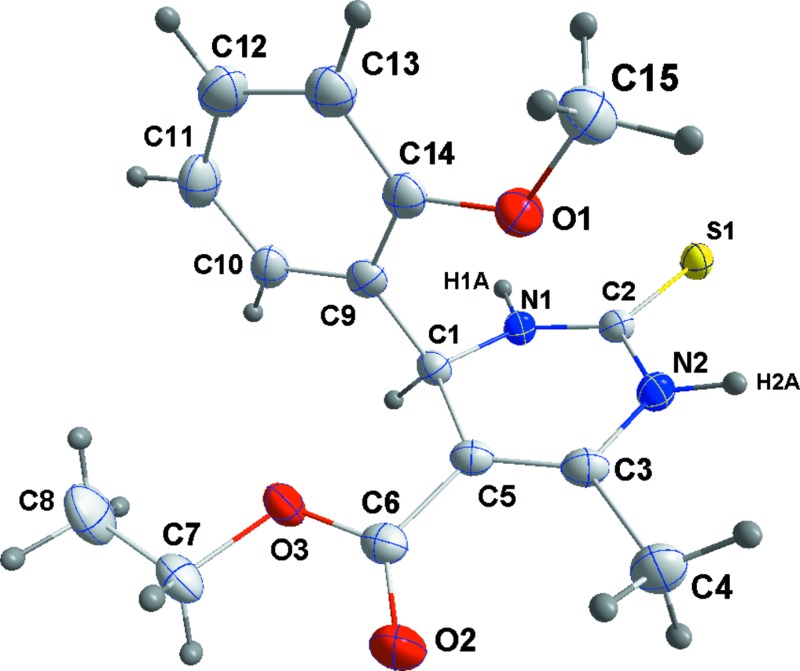
The molecular structure of the title compound showing labeling scheme and 50% probability ellipsoids.
Fig. 2.
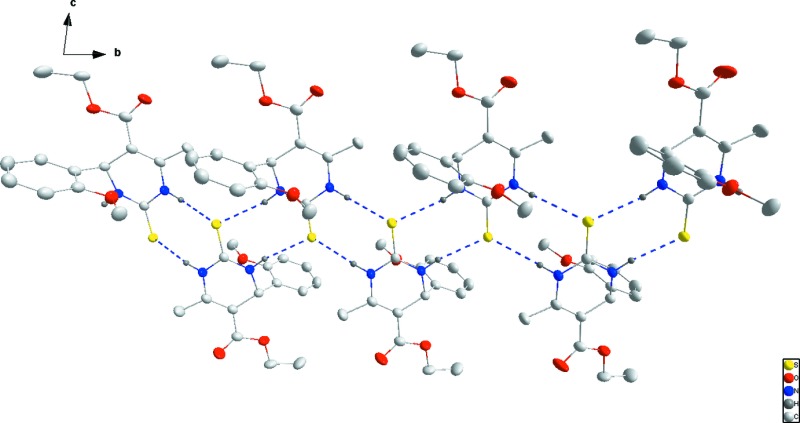
A section of the chain formed by N—H···S hydrogen bonds (dashed lines).
Fig. 3.
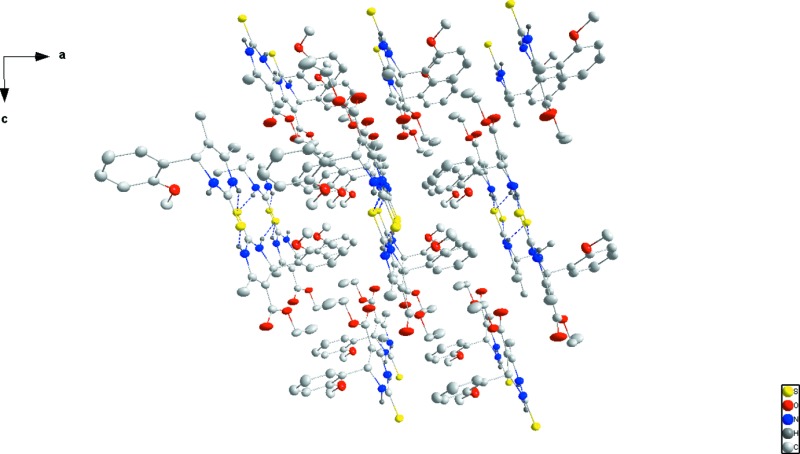
A packing diagram viewed along the b axis. N—H···S interactions are shown as dotted lines.
Crystal data
| C15H18N2O3S | Z = 2 |
| Mr = 306.37 | F(000) = 324 |
| Triclinic, P1 | Dx = 1.343 Mg m−3 |
| a = 7.9791 (2) Å | Cu Kα radiation, λ = 1.54178 Å |
| b = 8.2031 (2) Å | Cell parameters from 7936 reflections |
| c = 11.8405 (3) Å | θ = 3.8–72.2° |
| α = 81.987 (1)° | µ = 2.00 mm−1 |
| β = 87.975 (1)° | T = 150 K |
| γ = 80.850 (1)° | Thick plate, colourless |
| V = 757.60 (3) Å3 | 0.25 × 0.21 × 0.12 mm |
Data collection
| Bruker D8 VENTURE PHOTON 100 CMOS diffractometer | 2929 independent reflections |
| Radiation source: INCOATEC IµS micro–focus source | 2773 reflections with I > 2σ(I) |
| Mirror monochromator | Rint = 0.021 |
| Detector resolution: 10.4167 pixels mm-1 | θmax = 72.2°, θmin = 3.8° |
| ω scans | h = −9→9 |
| Absorption correction: multi-scan (SADABS; Bruker, 2014) | k = −10→10 |
| Tmin = 0.73, Tmax = 0.79 | l = −14→14 |
| 9145 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: mixed |
| wR(F2) = 0.101 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0487P)2 + 0.4254P] where P = (Fo2 + 2Fc2)/3 |
| 2929 reflections | (Δ/σ)max = 0.001 |
| 193 parameters | Δρmax = 0.43 e Å−3 |
| 0 restraints | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. H-atoms attached to carbon were placed in calculated positions (C—H = 0.95 - 0.98 Å) while those attached to nitrogen were placed in locations derived from a difference map and their parameters adjusted to give N—H = 0.91 Å. All were included as riding contributions with isotropic displacement parameters 1.2 - 1.5 times those of the attached atoms. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 1.08339 (5) | 0.22865 (4) | 1.02799 (3) | 0.02400 (13) | |
| O1 | 0.64475 (15) | 0.28798 (15) | 0.86937 (10) | 0.0312 (3) | |
| O2 | 0.8568 (2) | 0.26940 (18) | 0.47910 (11) | 0.0494 (4) | |
| O3 | 0.75790 (16) | 0.52294 (15) | 0.52331 (9) | 0.0304 (3) | |
| N1 | 0.96037 (16) | 0.42742 (15) | 0.84338 (11) | 0.0213 (3) | |
| H1A | 0.9617 | 0.5109 | 0.8864 | 0.026* | |
| N2 | 0.99441 (17) | 0.14788 (16) | 0.83198 (11) | 0.0240 (3) | |
| H2A | 1.0080 | 0.0431 | 0.8710 | 0.029* | |
| C1 | 0.85934 (19) | 0.47576 (19) | 0.73835 (13) | 0.0216 (3) | |
| H1 | 0.9084 | 0.5685 | 0.6916 | 0.026* | |
| C2 | 1.00503 (19) | 0.27318 (19) | 0.89394 (13) | 0.0209 (3) | |
| C3 | 0.9414 (2) | 0.1739 (2) | 0.71936 (13) | 0.0247 (3) | |
| C4 | 0.9585 (3) | 0.0158 (2) | 0.66684 (15) | 0.0351 (4) | |
| H4A | 0.8722 | 0.0271 | 0.6082 | 0.053* | |
| H4B | 0.9429 | −0.0770 | 0.7259 | 0.053* | |
| H4C | 1.0717 | −0.0061 | 0.6320 | 0.053* | |
| C5 | 0.88282 (19) | 0.3296 (2) | 0.66990 (13) | 0.0230 (3) | |
| C6 | 0.8339 (2) | 0.3648 (2) | 0.54883 (14) | 0.0277 (3) | |
| C7 | 0.6888 (2) | 0.5701 (2) | 0.40933 (14) | 0.0331 (4) | |
| H7A | 0.6051 | 0.4980 | 0.3959 | 0.040* | |
| H7B | 0.7806 | 0.5588 | 0.3512 | 0.040* | |
| C8 | 0.6050 (3) | 0.7480 (3) | 0.40272 (18) | 0.0486 (5) | |
| H8A | 0.5172 | 0.7580 | 0.4625 | 0.073* | |
| H8B | 0.5529 | 0.7841 | 0.3278 | 0.073* | |
| H8C | 0.6900 | 0.8183 | 0.4136 | 0.073* | |
| C9 | 0.6761 (2) | 0.5437 (2) | 0.76670 (13) | 0.0255 (3) | |
| C10 | 0.6100 (2) | 0.7098 (2) | 0.72635 (15) | 0.0304 (4) | |
| H10 | 0.6797 | 0.7785 | 0.6817 | 0.036* | |
| C11 | 0.4425 (3) | 0.7744 (2) | 0.75156 (17) | 0.0382 (4) | |
| H11 | 0.3972 | 0.8861 | 0.7228 | 0.046* | |
| C12 | 0.3434 (2) | 0.6753 (2) | 0.81827 (16) | 0.0370 (4) | |
| H12 | 0.2298 | 0.7205 | 0.8357 | 0.044* | |
| C13 | 0.4043 (2) | 0.5117 (2) | 0.86057 (15) | 0.0320 (4) | |
| H13 | 0.3342 | 0.4452 | 0.9068 | 0.038* | |
| C14 | 0.5720 (2) | 0.4459 (2) | 0.83384 (13) | 0.0269 (3) | |
| C15 | 0.5435 (2) | 0.1762 (2) | 0.93170 (16) | 0.0361 (4) | |
| H15A | 0.4988 | 0.2205 | 1.0013 | 0.054* | |
| H15B | 0.6133 | 0.0671 | 0.9521 | 0.054* | |
| H15C | 0.4489 | 0.1642 | 0.8845 | 0.054* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0304 (2) | 0.0195 (2) | 0.0222 (2) | −0.00308 (15) | −0.00427 (14) | −0.00340 (14) |
| O1 | 0.0296 (6) | 0.0277 (6) | 0.0355 (6) | −0.0073 (5) | 0.0044 (5) | 0.0011 (5) |
| O2 | 0.0744 (10) | 0.0443 (8) | 0.0274 (7) | 0.0095 (7) | −0.0101 (6) | −0.0165 (6) |
| O3 | 0.0391 (7) | 0.0314 (6) | 0.0209 (6) | −0.0055 (5) | −0.0058 (5) | −0.0026 (5) |
| N1 | 0.0255 (6) | 0.0180 (6) | 0.0217 (6) | −0.0048 (5) | −0.0032 (5) | −0.0046 (5) |
| N2 | 0.0309 (7) | 0.0181 (6) | 0.0238 (6) | −0.0034 (5) | −0.0014 (5) | −0.0056 (5) |
| C1 | 0.0237 (7) | 0.0218 (7) | 0.0202 (7) | −0.0052 (6) | −0.0020 (6) | −0.0033 (6) |
| C2 | 0.0202 (7) | 0.0206 (7) | 0.0227 (7) | −0.0045 (6) | 0.0013 (6) | −0.0044 (6) |
| C3 | 0.0261 (8) | 0.0261 (8) | 0.0241 (8) | −0.0069 (6) | 0.0019 (6) | −0.0084 (6) |
| C4 | 0.0497 (11) | 0.0273 (9) | 0.0309 (9) | −0.0059 (8) | −0.0026 (8) | −0.0121 (7) |
| C5 | 0.0235 (7) | 0.0256 (8) | 0.0217 (7) | −0.0063 (6) | 0.0014 (6) | −0.0067 (6) |
| C6 | 0.0278 (8) | 0.0332 (9) | 0.0233 (8) | −0.0059 (7) | 0.0010 (6) | −0.0066 (6) |
| C7 | 0.0370 (9) | 0.0430 (10) | 0.0199 (8) | −0.0104 (8) | −0.0046 (7) | 0.0000 (7) |
| C8 | 0.0683 (14) | 0.0410 (11) | 0.0346 (10) | −0.0088 (10) | −0.0160 (10) | 0.0062 (8) |
| C9 | 0.0268 (8) | 0.0292 (8) | 0.0222 (7) | −0.0048 (6) | −0.0031 (6) | −0.0081 (6) |
| C10 | 0.0335 (9) | 0.0276 (8) | 0.0289 (8) | 0.0007 (7) | −0.0053 (7) | −0.0055 (7) |
| C11 | 0.0398 (10) | 0.0284 (9) | 0.0436 (10) | 0.0035 (8) | −0.0066 (8) | −0.0041 (8) |
| C12 | 0.0352 (9) | 0.0377 (10) | 0.0367 (9) | 0.0019 (8) | 0.0000 (7) | −0.0094 (8) |
| C13 | 0.0320 (9) | 0.0368 (9) | 0.0278 (8) | −0.0069 (7) | −0.0017 (7) | −0.0049 (7) |
| C14 | 0.0294 (8) | 0.0286 (8) | 0.0233 (8) | −0.0044 (7) | −0.0036 (6) | −0.0049 (6) |
| C15 | 0.0366 (10) | 0.0323 (9) | 0.0386 (10) | −0.0106 (8) | 0.0047 (8) | 0.0033 (8) |
Geometric parameters (Å, º)
| S1—C2 | 1.6969 (15) | C5—C6 | 1.476 (2) |
| O1—C14 | 1.349 (2) | C7—C8 | 1.498 (3) |
| O1—C15 | 1.430 (2) | C7—H7A | 0.9900 |
| O2—C6 | 1.205 (2) | C7—H7B | 0.9900 |
| O3—C6 | 1.340 (2) | C8—H8A | 0.9800 |
| O3—C7 | 1.4541 (19) | C8—H8B | 0.9800 |
| N1—C2 | 1.322 (2) | C8—H8C | 0.9800 |
| N1—C1 | 1.4783 (18) | C9—C14 | 1.397 (2) |
| N1—H1A | 0.9098 | C9—C10 | 1.403 (2) |
| N2—C2 | 1.3580 (19) | C10—C11 | 1.395 (3) |
| N2—C3 | 1.391 (2) | C10—H10 | 0.9500 |
| N2—H2A | 0.9098 | C11—C12 | 1.376 (3) |
| C1—C5 | 1.522 (2) | C11—H11 | 0.9500 |
| C1—C9 | 1.523 (2) | C12—C13 | 1.382 (3) |
| C1—H1 | 1.0000 | C12—H12 | 0.9500 |
| C3—C5 | 1.347 (2) | C13—C14 | 1.403 (2) |
| C3—C4 | 1.501 (2) | C13—H13 | 0.9500 |
| C4—H4A | 0.9800 | C15—H15A | 0.9800 |
| C4—H4B | 0.9800 | C15—H15B | 0.9800 |
| C4—H4C | 0.9800 | C15—H15C | 0.9800 |
| C14—O1—C15 | 118.77 (14) | C8—C7—H7A | 110.4 |
| C6—O3—C7 | 116.45 (13) | O3—C7—H7B | 110.4 |
| C2—N1—C1 | 125.30 (12) | C8—C7—H7B | 110.4 |
| C2—N1—H1A | 117.1 | H7A—C7—H7B | 108.6 |
| C1—N1—H1A | 114.7 | C7—C8—H8A | 109.5 |
| C2—N2—C3 | 123.59 (14) | C7—C8—H8B | 109.5 |
| C2—N2—H2A | 116.2 | H8A—C8—H8B | 109.5 |
| C3—N2—H2A | 119.5 | C7—C8—H8C | 109.5 |
| N1—C1—C5 | 108.79 (12) | H8A—C8—H8C | 109.5 |
| N1—C1—C9 | 110.68 (12) | H8B—C8—H8C | 109.5 |
| C5—C1—C9 | 115.38 (12) | C14—C9—C10 | 118.77 (16) |
| N1—C1—H1 | 107.2 | C14—C9—C1 | 121.76 (15) |
| C5—C1—H1 | 107.2 | C10—C9—C1 | 119.46 (15) |
| C9—C1—H1 | 107.2 | C11—C10—C9 | 120.32 (17) |
| N1—C2—N2 | 117.28 (14) | C11—C10—H10 | 119.8 |
| N1—C2—S1 | 122.70 (11) | C9—C10—H10 | 119.8 |
| N2—C2—S1 | 119.94 (12) | C12—C11—C10 | 119.55 (17) |
| C5—C3—N2 | 119.62 (14) | C12—C11—H11 | 120.2 |
| C5—C3—C4 | 127.50 (15) | C10—C11—H11 | 120.2 |
| N2—C3—C4 | 112.89 (14) | C11—C12—C13 | 121.84 (17) |
| C3—C4—H4A | 109.5 | C11—C12—H12 | 119.1 |
| C3—C4—H4B | 109.5 | C13—C12—H12 | 119.1 |
| H4A—C4—H4B | 109.5 | C12—C13—C14 | 118.62 (17) |
| C3—C4—H4C | 109.5 | C12—C13—H13 | 120.7 |
| H4A—C4—H4C | 109.5 | C14—C13—H13 | 120.7 |
| H4B—C4—H4C | 109.5 | O1—C14—C9 | 115.19 (15) |
| C3—C5—C6 | 121.63 (14) | O1—C14—C13 | 123.92 (15) |
| C3—C5—C1 | 120.93 (14) | C9—C14—C13 | 120.89 (16) |
| C6—C5—C1 | 117.42 (14) | O1—C15—H15A | 109.5 |
| O2—C6—O3 | 122.34 (16) | O1—C15—H15B | 109.5 |
| O2—C6—C5 | 126.96 (16) | H15A—C15—H15B | 109.5 |
| O3—C6—C5 | 110.70 (13) | O1—C15—H15C | 109.5 |
| O3—C7—C8 | 106.67 (14) | H15A—C15—H15C | 109.5 |
| O3—C7—H7A | 110.4 | H15B—C15—H15C | 109.5 |
| C2—N1—C1—C5 | −25.4 (2) | C3—C5—C6—O3 | 171.37 (14) |
| C2—N1—C1—C9 | 102.32 (16) | C1—C5—C6—O3 | −6.8 (2) |
| C1—N1—C2—N2 | 16.9 (2) | C6—O3—C7—C8 | 177.69 (16) |
| C1—N1—C2—S1 | −166.47 (11) | N1—C1—C9—C14 | −61.89 (18) |
| C3—N2—C2—N1 | 0.7 (2) | C5—C1—C9—C14 | 62.18 (19) |
| C3—N2—C2—S1 | −176.02 (12) | N1—C1—C9—C10 | 116.92 (15) |
| C2—N2—C3—C5 | −6.0 (2) | C5—C1—C9—C10 | −119.01 (16) |
| C2—N2—C3—C4 | 173.96 (15) | C14—C9—C10—C11 | −1.1 (2) |
| N2—C3—C5—C6 | 176.60 (14) | C1—C9—C10—C11 | −179.90 (15) |
| C4—C3—C5—C6 | −3.3 (3) | C9—C10—C11—C12 | 1.3 (3) |
| N2—C3—C5—C1 | −5.3 (2) | C10—C11—C12—C13 | −0.7 (3) |
| C4—C3—C5—C1 | 174.72 (16) | C11—C12—C13—C14 | −0.3 (3) |
| N1—C1—C5—C3 | 18.7 (2) | C15—O1—C14—C9 | −175.65 (14) |
| C9—C1—C5—C3 | −106.35 (17) | C15—O1—C14—C13 | 4.3 (2) |
| N1—C1—C5—C6 | −163.15 (13) | C10—C9—C14—O1 | −179.93 (14) |
| C9—C1—C5—C6 | 71.79 (18) | C1—C9—C14—O1 | −1.1 (2) |
| C7—O3—C6—O2 | 5.8 (2) | C10—C9—C14—C13 | 0.1 (2) |
| C7—O3—C6—C5 | −174.41 (13) | C1—C9—C14—C13 | 178.93 (14) |
| C3—C5—C6—O2 | −8.9 (3) | C12—C13—C14—O1 | −179.41 (16) |
| C1—C5—C6—O2 | 172.98 (18) | C12—C13—C14—C9 | 0.5 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···S1i | 0.91 | 2.46 | 3.3539 (13) | 167 |
| N2—H2A···S1ii | 0.91 | 2.58 | 3.4327 (14) | 157 |
Symmetry codes: (i) −x+2, −y+1, −z+2; (ii) −x+2, −y, −z+2.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: IS5401).
References
- Ahmed, B., Khan, R. A., Habibullah & Keshari, M. (2009). Tetrahedron Lett. 50, 2889–2892.
- Ahmed, E. A., Mohamed, M. A. A. & El-Saghier, A. M. M. (2012). J. Am. Sci. 8, 815–818.
- Biginelli, P. (1893). Gazz. Chim. Ital. 23, 360–416.
- Brandenburg, K. & Putz, H. (2012). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (2014). APEX2, SAINT and SADABS. Bruker AXS, Inc., Madison, Wisconsin, USA.
- Gohain, M., Prajapati, D. & Sandhu, J. S. (2004). Synlett, pp. 0235–0238.
- Hed, L. C., Sharma, R., Pareek, C. & Chaudhari, P. B. (2009). Eur. J. Chem. 6, 770–774.
- Russowsky, D., Benvenutti, E. V., Roxo, G. S. & Grasel, F. (2007). Lett. Org. Chem. 4, 39–42.
- Salehi, P., Dabiri, M., Khosropour, A. R. & Roozbehniya, P. (2006). J. Iran. Chem. Soc. 3, 98–104.
- Shah, T. B., Gupte, A., Patel, M. R., Chaudhari, V. S., Patel, H. & Patel, V. C. (2009). Indian J. Chem. Sect. B, 48, 88–96.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Singh, O. M., Devi, N. S., Devi, L. R. & Khumanthem, N. (2010). Int. J. Drug Des. Discov. 1, 258–264.
- Varala, R., Alam, M. M. & Adapa, S. R. (2003). Synlett, pp. 67–70.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015010026/is5401sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010026/is5401Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010026/is5401Isup3.cml
. DOI: 10.1107/S2056989015010026/is5401fig1.tif
The molecular structure of the title compound showing labeling scheme and 50% probability ellipsoids.
. DOI: 10.1107/S2056989015010026/is5401fig2.tif
A section of the chain formed by N—H⋯S hydrogen bonds (dashed lines).
b . DOI: 10.1107/S2056989015010026/is5401fig3.tif
A packing diagram viewed along the b axis. N—H⋯S interactions are shown as dotted lines.
CCDC reference: 1402530
Additional supporting information: crystallographic information; 3D view; checkCIF report