

Abstract
The title compound, C17H27NO2S, exhibits a distorted geometry of the aromatic ring with elongated bonds at the ipso-C atom. The S atom deviates from the aromatic ring plane by 0.393 (4) Å. Similar to this, the adjacent isopropyl groups are bent out of the aromatic ring plane by −0.125 (4) and −0.154 (4) Å. Even the distant isopropyl group in para-position to the sulfonyl moiety shows a slight deviation from the ring plane of 0.111 (5) Å. These distortions, which are caused by the bulky substituents, can also be observed in related sulfonylaziridine structures.
Keywords: crystal structure, aziridine, triisopropylbenzenesulfonyl, consecutive ring-opening reactions
Related literature
For the crystal structure of a related phenyl-substituted compound, see: Golz et al. (2014 ▸). For a discussion of the geometry of the triisopropylbenzenesulfonyl moiety, see: Sandrock et al. (2004 ▸). For a discussion of the pyramidalized geometry of N-sulfonylamides, see: Ohwada et al. (1998 ▸). By regioselective ring opening reactions, countless nitrogen-containing compounds are accessible, see: Stamm (1999 ▸); Schneider (2009 ▸). For consecutive ring-opening reactions of aziridines by triethylamine, see: Golz & Strohmann (2015 ▸). In some cases, the three-membered aziridine ring is further activated by electron-withdrawing groups (Hu, 2004 ▸) to increase its reactivity.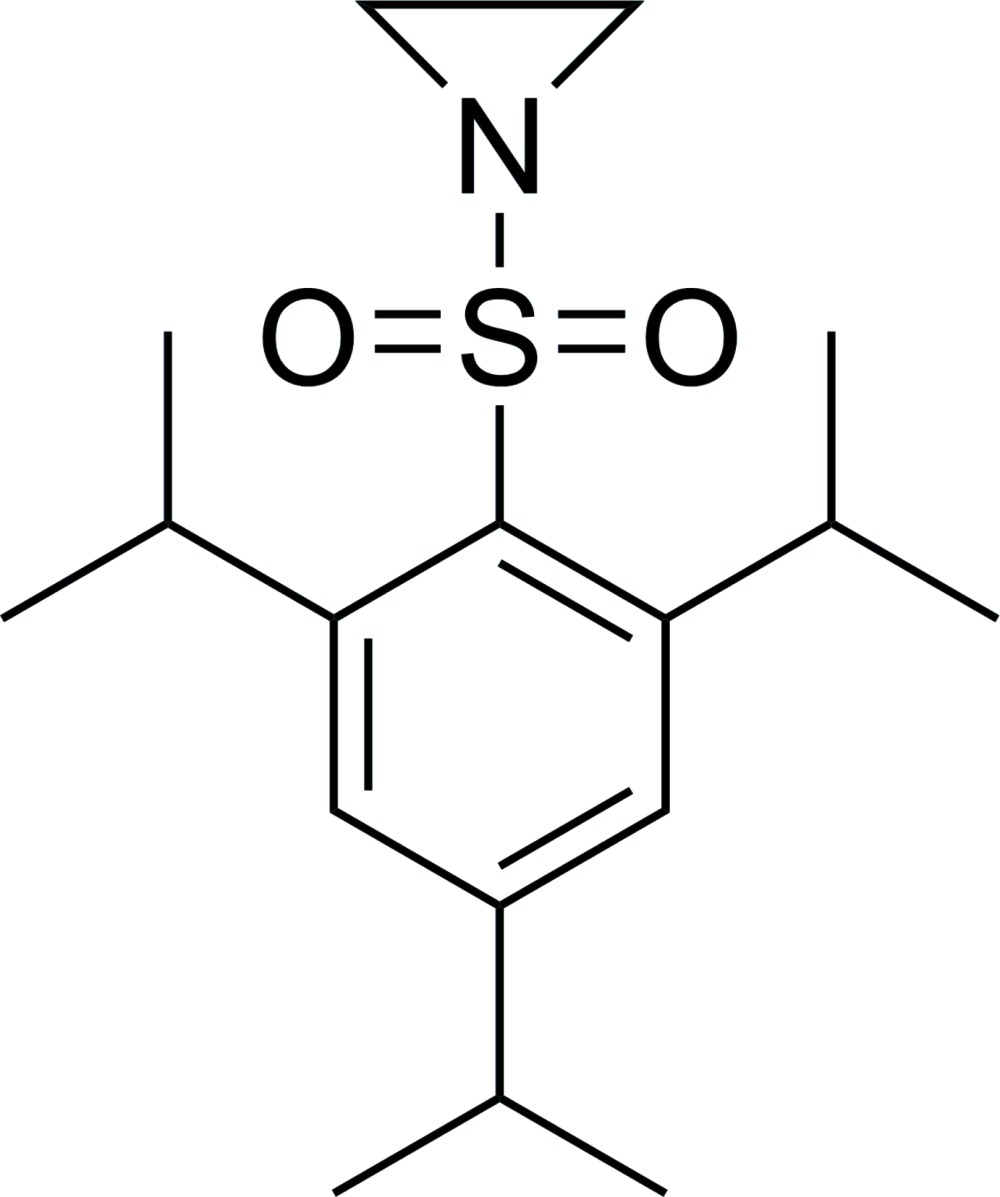
Experimental
Crystal data
C17H27NO2S
M r = 309.45
Monoclinic,
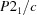
a = 6.2679 (8) Å
b = 17.5289 (18) Å
c = 16.3890 (13) Å
β = 100.331 (10)°
V = 1771.5 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.19 mm−1
T = 173 K
0.33 × 0.25 × 0.01 mm
Data collection
Agilent Xcalibur Sapphire3 diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2013 ▸) T min = 0.782, T max = 1.000
9521 measured reflections
3449 independent reflections
2479 reflections with I > 2σ(I)
R int = 0.049
Refinement
R[F 2 > 2σ(F 2)] = 0.053
wR(F 2) = 0.141
S = 1.07
3449 reflections
196 parameters
H-atom parameters constrained
Δρmax = 0.33 e Å−3
Δρmin = −0.58 e Å−3
Data collection: CrysAlis PRO (Agilent, 2013 ▸); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▸); program(s) used to refine structure: SHELXL2014 (Sheldrick, 2015 ▸); molecular graphics: OLEX2 (Dolomanov et al., 2009 ▸); software used to prepare material for publication: OLEX2.
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015010221/fk2088sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010221/fk2088Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010221/fk2088Isup3.cml
. DOI: 10.1107/S2056989015010221/fk2088fig1.tif
Molecular structure of the title compound with anisotropic displacement ellipsoids drawn at 50% probability level.
CCDC reference: 1403377
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
We are grateful to the Deutsche Forschungsgemeinschaft (DFG) for financial support.
supplementary crystallographic information
S1. Structural commentary
Aziridines are interesting and versatile building blocks in synthetic chemistry due to their high ring strain. By regioselective ring opening reactions, countless nitrogen-containing compounds are accessible (Stamm, 1999; Schneider, 2009). For example, the aziridine ring can be opened by triethylamine (Golz & Strohmann, 2015). In some cases, the three-membered aziridine ring is further activated by electron-withdrawing groups (Hu, 2004) to increase its reactivity.
The title compound, C17H27NO2S, is a representative of the class of activated aziridines, as it contains a triisopropylbenzene substituted sulfonyl ester attached to the nitrogen atom. In the aromatic ring, the bulky substituents lead to a distortion of its geometry. This is expressed by the increased bond lengths and out-of-plane bent substituents around the benzene ring. At the ipso-carbon, the bonds C10–C11 and C10–C6 are slightly elongated to 1.410 (3) Å. In contrast, the other bonds of the aromatic ring exhibit usual lengths [C4–C15 1.374 (3) Å, C5–C61.380 (3) Å, C11–C15 1.388 (3) Å, C4–C5 1.389 (3) Å]. The sulfonyl group as well as the adjacent isopropyl groups bend out of the aromatic plane. This is caused by steric repulsion between the isopropyl groups and the sulfonyl oxygen atoms. The sulfur atom deviates from the mean aromatic ring plane by 0.393 (4) Å. The carbon atoms C7 and C12 show distances of -0.125 (4) Å and -0.154 (4) Å, respectively (see Table 5). A similar distortion can also be observed at the isopropyl group in para position to the sulfonyl moiety. Here, C1 has a distance to the aromatic ring plane of 0.111 (5) Å, thus being distorted in the same direction as the sulfur atom. This is caused by steric repulsion between the C1 isopropyl group and the adjacent isopropyl groups in ortho position in respect of the sulfonyl moiety.
S2. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1.
Hydrogen atoms were located from difference Fourier maps, refined at idealized positions riding on the carbon atoms with isotropic displacement parameters Uiso(H) = 1.2Ueq(C) or 1.5Ueq(–CH3) and aromatic C–H = 0.95 Å, primary C–H = 0.98 Å, secondary C–H = 0.99 Å, tertiary C–H = 1.00 Å. All CH3 hydrogen atoms were allowed to rotate but not to tip. Aziridine protons could be located from difference Fourier maps, but were refined as idealized CH2 groups.
Figures
Fig. 1.
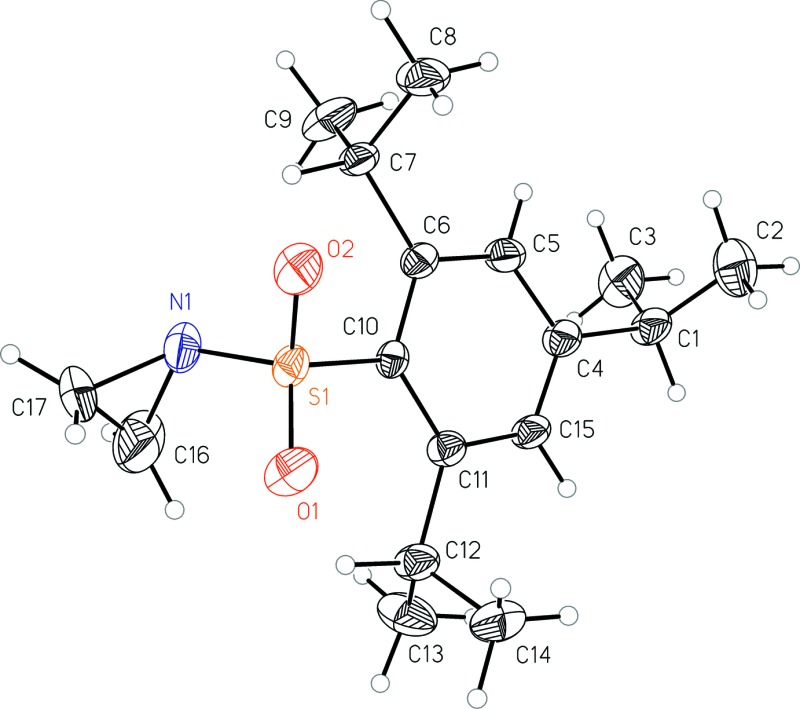
Molecular structure of the title compound with anisotropic displacement ellipsoids drawn at 50% probability level.
Crystal data
| C17H27NO2S | F(000) = 672 |
| Mr = 309.45 | Dx = 1.160 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.2679 (8) Å | Cell parameters from 2277 reflections |
| b = 17.5289 (18) Å | θ = 3.3–29.2° |
| c = 16.3890 (13) Å | µ = 0.19 mm−1 |
| β = 100.331 (10)° | T = 173 K |
| V = 1771.5 (3) Å3 | Plate, colourless |
| Z = 4 | 0.33 × 0.25 × 0.01 mm |
Data collection
| Agilent Xcalibur Sapphire3 diffractometer | 3449 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2479 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.049 |
| Detector resolution: 16.0560 pixels mm-1 | θmax = 26.0°, θmin = 2.5° |
| ω scans | h = −7→7 |
| Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2013) | k = −15→21 |
| Tmin = 0.782, Tmax = 1.000 | l = −20→20 |
| 9521 measured reflections |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.053 | H-atom parameters constrained |
| wR(F2) = 0.141 | w = 1/[σ2(Fo2) + (0.0473P)2 + 0.8936P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.07 | (Δ/σ)max < 0.001 |
| 3449 reflections | Δρmax = 0.33 e Å−3 |
| 196 parameters | Δρmin = −0.58 e Å−3 |
Special details
| Experimental. Absorption correction: CrysAlisPro, Agilent Technologies, Version 1.171.36.28 (release 01-02-2013 CrysAlis171 .NET) (compiled Feb 1 2013,16:14:44) Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.71473 (10) | 0.20619 (4) | 0.37952 (4) | 0.0245 (2) | |
| C10 | 0.7527 (4) | 0.26810 (13) | 0.29748 (14) | 0.0205 (5) | |
| C15 | 0.8227 (4) | 0.28863 (14) | 0.16069 (15) | 0.0272 (6) | |
| H15 | 0.8229 | 0.2705 | 0.1061 | 0.033* | |
| C6 | 0.8105 (4) | 0.34434 (14) | 0.31802 (14) | 0.0246 (6) | |
| C5 | 0.8842 (4) | 0.38836 (15) | 0.25903 (15) | 0.0297 (6) | |
| H5 | 0.9273 | 0.4394 | 0.2726 | 0.036* | |
| C11 | 0.7462 (4) | 0.24050 (14) | 0.21624 (15) | 0.0243 (6) | |
| C4 | 0.8981 (4) | 0.36110 (15) | 0.18058 (15) | 0.0279 (6) | |
| O1 | 0.7219 (3) | 0.12858 (10) | 0.35547 (11) | 0.0370 (5) | |
| O2 | 0.8625 (3) | 0.22687 (11) | 0.45315 (10) | 0.0360 (5) | |
| N1 | 0.4729 (4) | 0.23217 (13) | 0.39562 (14) | 0.0345 (6) | |
| C17 | 0.3491 (5) | 0.17365 (19) | 0.4317 (2) | 0.0500 (9) | |
| H17A | 0.4185 | 0.1234 | 0.4451 | 0.060* | |
| H17B | 0.2536 | 0.1908 | 0.4702 | 0.060* | |
| C16 | 0.2886 (6) | 0.1945 (3) | 0.3447 (2) | 0.0701 (12) | |
| H16A | 0.1543 | 0.2246 | 0.3280 | 0.084* | |
| H16B | 0.3190 | 0.1572 | 0.3029 | 0.084* | |
| C12 | 0.6577 (5) | 0.16388 (14) | 0.18281 (15) | 0.0312 (6) | |
| H12 | 0.5872 | 0.1391 | 0.2261 | 0.037* | |
| C7 | 0.7975 (4) | 0.38301 (14) | 0.40041 (15) | 0.0290 (6) | |
| H7 | 0.7291 | 0.3463 | 0.4347 | 0.035* | |
| C1 | 0.9900 (5) | 0.41027 (15) | 0.11913 (15) | 0.0331 (7) | |
| H1 | 0.9766 | 0.3811 | 0.0660 | 0.040* | |
| C13 | 0.4860 (6) | 0.17400 (18) | 0.10515 (18) | 0.0489 (9) | |
| H13A | 0.3751 | 0.2099 | 0.1166 | 0.073* | |
| H13B | 0.4184 | 0.1246 | 0.0887 | 0.073* | |
| H13C | 0.5539 | 0.1940 | 0.0602 | 0.073* | |
| C3 | 0.8614 (5) | 0.48402 (17) | 0.10107 (18) | 0.0457 (8) | |
| H3A | 0.7095 | 0.4719 | 0.0787 | 0.068* | |
| H3B | 0.9223 | 0.5142 | 0.0605 | 0.068* | |
| H3C | 0.8695 | 0.5133 | 0.1525 | 0.068* | |
| C14 | 0.8399 (5) | 0.11176 (17) | 0.1661 (2) | 0.0471 (8) | |
| H14A | 0.9167 | 0.1361 | 0.1259 | 0.071* | |
| H14B | 0.7787 | 0.0631 | 0.1436 | 0.071* | |
| H14C | 0.9415 | 0.1026 | 0.2180 | 0.071* | |
| C2 | 1.2285 (5) | 0.42692 (19) | 0.1493 (2) | 0.0496 (9) | |
| H2A | 1.2459 | 0.4567 | 0.2006 | 0.074* | |
| H2B | 1.2856 | 0.4560 | 0.1068 | 0.074* | |
| H2C | 1.3083 | 0.3788 | 0.1597 | 0.074* | |
| C8 | 1.0244 (5) | 0.40142 (17) | 0.44750 (16) | 0.0407 (8) | |
| H8A | 1.1099 | 0.3544 | 0.4564 | 0.061* | |
| H8B | 1.0137 | 0.4243 | 0.5012 | 0.061* | |
| H8C | 1.0951 | 0.4374 | 0.4151 | 0.061* | |
| C9 | 0.6542 (6) | 0.45350 (17) | 0.38647 (18) | 0.0476 (8) | |
| H9A | 0.7228 | 0.4919 | 0.3562 | 0.071* | |
| H9B | 0.6351 | 0.4744 | 0.4401 | 0.071* | |
| H9C | 0.5125 | 0.4396 | 0.3541 | 0.071* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0289 (4) | 0.0256 (4) | 0.0205 (3) | 0.0015 (3) | 0.0085 (3) | 0.0032 (3) |
| C10 | 0.0213 (13) | 0.0238 (13) | 0.0168 (11) | 0.0008 (10) | 0.0048 (10) | 0.0025 (10) |
| C15 | 0.0372 (16) | 0.0275 (14) | 0.0178 (12) | −0.0032 (12) | 0.0077 (11) | −0.0036 (10) |
| C6 | 0.0284 (14) | 0.0279 (14) | 0.0174 (12) | −0.0012 (11) | 0.0039 (11) | −0.0011 (10) |
| C5 | 0.0401 (17) | 0.0269 (14) | 0.0228 (13) | −0.0082 (12) | 0.0077 (12) | −0.0035 (11) |
| C11 | 0.0283 (14) | 0.0233 (13) | 0.0212 (12) | 0.0003 (11) | 0.0043 (11) | −0.0005 (10) |
| C4 | 0.0358 (16) | 0.0297 (15) | 0.0190 (12) | −0.0034 (12) | 0.0068 (11) | 0.0003 (11) |
| O1 | 0.0590 (14) | 0.0250 (10) | 0.0292 (10) | 0.0063 (9) | 0.0137 (10) | 0.0051 (8) |
| O2 | 0.0431 (13) | 0.0460 (12) | 0.0184 (9) | −0.0037 (9) | 0.0036 (9) | 0.0068 (8) |
| N1 | 0.0286 (13) | 0.0448 (14) | 0.0326 (12) | −0.0001 (11) | 0.0128 (11) | 0.0007 (11) |
| C17 | 0.0423 (19) | 0.053 (2) | 0.063 (2) | −0.0197 (15) | 0.0294 (17) | −0.0063 (17) |
| C16 | 0.0294 (19) | 0.123 (4) | 0.059 (2) | −0.014 (2) | 0.0122 (17) | −0.030 (2) |
| C12 | 0.0473 (18) | 0.0251 (14) | 0.0221 (13) | −0.0042 (12) | 0.0088 (12) | −0.0015 (11) |
| C7 | 0.0442 (17) | 0.0251 (14) | 0.0197 (12) | −0.0084 (12) | 0.0115 (12) | −0.0048 (11) |
| C1 | 0.0504 (18) | 0.0296 (15) | 0.0224 (13) | −0.0063 (13) | 0.0152 (13) | −0.0008 (11) |
| C13 | 0.060 (2) | 0.0442 (19) | 0.0360 (16) | −0.0173 (16) | −0.0084 (16) | −0.0013 (14) |
| C3 | 0.060 (2) | 0.0423 (19) | 0.0371 (16) | −0.0001 (16) | 0.0160 (16) | 0.0155 (14) |
| C14 | 0.059 (2) | 0.0336 (17) | 0.0517 (19) | −0.0011 (15) | 0.0181 (17) | −0.0145 (15) |
| C2 | 0.047 (2) | 0.055 (2) | 0.0497 (19) | −0.0071 (16) | 0.0182 (16) | 0.0124 (16) |
| C8 | 0.056 (2) | 0.0432 (18) | 0.0215 (13) | −0.0145 (15) | 0.0028 (14) | −0.0060 (13) |
| C9 | 0.070 (2) | 0.0402 (18) | 0.0353 (16) | 0.0090 (16) | 0.0172 (16) | −0.0109 (14) |
Geometric parameters (Å, º)
| S1—C10 | 1.777 (2) | C7—H7 | 1.0000 |
| S1—O1 | 1.4193 (19) | C7—C8 | 1.526 (4) |
| S1—O2 | 1.4300 (19) | C7—C9 | 1.520 (4) |
| S1—N1 | 1.649 (2) | C1—H1 | 1.0000 |
| C10—C6 | 1.410 (3) | C1—C3 | 1.524 (4) |
| C10—C11 | 1.410 (3) | C1—C2 | 1.517 (4) |
| C15—H15 | 0.9500 | C13—H13A | 0.9800 |
| C15—C11 | 1.388 (3) | C13—H13B | 0.9800 |
| C15—C4 | 1.374 (4) | C13—H13C | 0.9800 |
| C6—C5 | 1.380 (3) | C3—H3A | 0.9800 |
| C6—C7 | 1.526 (3) | C3—H3B | 0.9800 |
| C5—H5 | 0.9500 | C3—H3C | 0.9800 |
| C5—C4 | 1.389 (3) | C14—H14A | 0.9800 |
| C11—C12 | 1.518 (4) | C14—H14B | 0.9800 |
| C4—C1 | 1.516 (3) | C14—H14C | 0.9800 |
| N1—C17 | 1.473 (3) | C2—H2A | 0.9800 |
| N1—C16 | 1.456 (4) | C2—H2B | 0.9800 |
| C17—H17A | 0.9900 | C2—H2C | 0.9800 |
| C17—H17B | 0.9900 | C8—H8A | 0.9800 |
| C17—C16 | 1.455 (5) | C8—H8B | 0.9800 |
| C16—H16A | 0.9900 | C8—H8C | 0.9800 |
| C16—H16B | 0.9900 | C9—H9A | 0.9800 |
| C12—H12 | 1.0000 | C9—H9B | 0.9800 |
| C12—C13 | 1.523 (4) | C9—H9C | 0.9800 |
| C12—C14 | 1.526 (4) | ||
| O1—S1—C10 | 111.10 (11) | C8—C7—H7 | 107.9 |
| O1—S1—O2 | 115.43 (12) | C9—C7—C6 | 110.6 (2) |
| O1—S1—N1 | 112.65 (12) | C9—C7—H7 | 107.9 |
| O2—S1—C10 | 109.23 (11) | C9—C7—C8 | 112.1 (2) |
| O2—S1—N1 | 105.67 (12) | C4—C1—H1 | 107.8 |
| N1—S1—C10 | 101.77 (11) | C4—C1—C3 | 111.1 (2) |
| C6—C10—S1 | 117.51 (17) | C4—C1—C2 | 111.3 (2) |
| C6—C10—C11 | 120.9 (2) | C3—C1—H1 | 107.8 |
| C11—C10—S1 | 121.32 (19) | C2—C1—H1 | 107.8 |
| C11—C15—H15 | 118.3 | C2—C1—C3 | 110.9 (2) |
| C4—C15—H15 | 118.3 | C12—C13—H13A | 109.5 |
| C4—C15—C11 | 123.4 (2) | C12—C13—H13B | 109.5 |
| C10—C6—C7 | 125.5 (2) | C12—C13—H13C | 109.5 |
| C5—C6—C10 | 117.8 (2) | H13A—C13—H13B | 109.5 |
| C5—C6—C7 | 116.7 (2) | H13A—C13—H13C | 109.5 |
| C6—C5—H5 | 118.6 | H13B—C13—H13C | 109.5 |
| C6—C5—C4 | 122.7 (2) | C1—C3—H3A | 109.5 |
| C4—C5—H5 | 118.6 | C1—C3—H3B | 109.5 |
| C10—C11—C12 | 126.2 (2) | C1—C3—H3C | 109.5 |
| C15—C11—C10 | 117.1 (2) | H3A—C3—H3B | 109.5 |
| C15—C11—C12 | 116.6 (2) | H3A—C3—H3C | 109.5 |
| C15—C4—C5 | 117.5 (2) | H3B—C3—H3C | 109.5 |
| C15—C4—C1 | 121.6 (2) | C12—C14—H14A | 109.5 |
| C5—C4—C1 | 120.9 (2) | C12—C14—H14B | 109.5 |
| C17—N1—S1 | 116.0 (2) | C12—C14—H14C | 109.5 |
| C16—N1—S1 | 116.1 (2) | H14A—C14—H14B | 109.5 |
| C16—N1—C17 | 59.5 (2) | H14A—C14—H14C | 109.5 |
| N1—C17—H17A | 117.8 | H14B—C14—H14C | 109.5 |
| N1—C17—H17B | 117.8 | C1—C2—H2A | 109.5 |
| H17A—C17—H17B | 114.9 | C1—C2—H2B | 109.5 |
| C16—C17—N1 | 59.7 (2) | C1—C2—H2C | 109.5 |
| C16—C17—H17A | 117.8 | H2A—C2—H2B | 109.5 |
| C16—C17—H17B | 117.8 | H2A—C2—H2C | 109.5 |
| N1—C16—H16A | 117.7 | H2B—C2—H2C | 109.5 |
| N1—C16—H16B | 117.7 | C7—C8—H8A | 109.5 |
| C17—C16—N1 | 60.8 (2) | C7—C8—H8B | 109.5 |
| C17—C16—H16A | 117.7 | C7—C8—H8C | 109.5 |
| C17—C16—H16B | 117.7 | H8A—C8—H8B | 109.5 |
| H16A—C16—H16B | 114.8 | H8A—C8—H8C | 109.5 |
| C11—C12—H12 | 107.9 | H8B—C8—H8C | 109.5 |
| C11—C12—C13 | 110.9 (2) | C7—C9—H9A | 109.5 |
| C11—C12—C14 | 111.0 (2) | C7—C9—H9B | 109.5 |
| C13—C12—H12 | 107.9 | C7—C9—H9C | 109.5 |
| C13—C12—C14 | 111.1 (2) | H9A—C9—H9B | 109.5 |
| C14—C12—H12 | 107.9 | H9A—C9—H9C | 109.5 |
| C6—C7—H7 | 107.9 | H9B—C9—H9C | 109.5 |
| C8—C7—C6 | 110.4 (2) | ||
| S1—C10—C6—C5 | 166.7 (2) | C5—C6—C7—C8 | −67.6 (3) |
| S1—C10—C6—C7 | −13.6 (3) | C5—C6—C7—C9 | 57.1 (3) |
| S1—C10—C11—C15 | −166.5 (2) | C5—C4—C1—C3 | −58.7 (4) |
| S1—C10—C11—C12 | 15.3 (4) | C5—C4—C1—C2 | 65.4 (3) |
| S1—N1—C17—C16 | −106.4 (3) | C11—C10—C6—C5 | −7.3 (4) |
| S1—N1—C16—C17 | 106.1 (2) | C11—C10—C6—C7 | 172.4 (3) |
| C10—S1—N1—C17 | 154.1 (2) | C11—C15—C4—C5 | −3.9 (4) |
| C10—S1—N1—C16 | 87.0 (2) | C11—C15—C4—C1 | 177.0 (3) |
| C10—C6—C5—C4 | 1.6 (4) | C4—C15—C11—C10 | −1.5 (4) |
| C10—C6—C7—C8 | 112.8 (3) | C4—C15—C11—C12 | 176.8 (2) |
| C10—C6—C7—C9 | −122.6 (3) | O1—S1—C10—C6 | −162.51 (19) |
| C10—C11—C12—C13 | 125.2 (3) | O1—S1—C10—C11 | 11.4 (3) |
| C10—C11—C12—C14 | −110.8 (3) | O1—S1—N1—C17 | 35.1 (2) |
| C15—C11—C12—C13 | −52.9 (3) | O1—S1—N1—C16 | −32.0 (3) |
| C15—C11—C12—C14 | 71.0 (3) | O2—S1—C10—C6 | −34.0 (2) |
| C15—C4—C1—C3 | 120.4 (3) | O2—S1—C10—C11 | 139.9 (2) |
| C15—C4—C1—C2 | −115.5 (3) | O2—S1—N1—C17 | −91.8 (2) |
| C6—C10—C11—C15 | 7.2 (4) | O2—S1—N1—C16 | −158.9 (2) |
| C6—C10—C11—C12 | −170.9 (2) | N1—S1—C10—C6 | 77.4 (2) |
| C6—C5—C4—C15 | 3.8 (4) | N1—S1—C10—C11 | −108.7 (2) |
| C6—C5—C4—C1 | −177.1 (3) | C7—C6—C5—C4 | −178.1 (3) |
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: FK2088).
References
- Agilent (2013). CrysAlis PRO. Oxford Diffraction Ltd, Yarnton, Oxfordshire, England.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Golz, C., Preut, H. & Strohmann, C. (2014). Acta Cryst. E70, o153. [DOI] [PMC free article] [PubMed]
- Golz, C. & Strohmann, C. (2015). Acta Cryst E71, 564–566. [DOI] [PMC free article] [PubMed]
- Hu, X. E. (2004). Tetrahedron, 60, 2701–2743.
- Ohwada, T., Okamoto, I., Shudo, K. & Yamaguchi, K. (1998). Tetrahedron Lett. 39, 7877–7880.
- Sandrock, P. B., Meyers, C. Y., Rath, N. P. & Robinson, P. D. (2004). Acta Cryst. E60, o544–o546.
- Schneider, C. (2009). Angew. Chem. Int. Ed. 48, 2082–2084.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Stamm, H. (1999). J. Prakt. Chem. 341, 319–331.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015010221/fk2088sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010221/fk2088Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010221/fk2088Isup3.cml
. DOI: 10.1107/S2056989015010221/fk2088fig1.tif
Molecular structure of the title compound with anisotropic displacement ellipsoids drawn at 50% probability level.
CCDC reference: 1403377
Additional supporting information: crystallographic information; 3D view; checkCIF report