

The title compound is twisted in such a way that the almost planar [Car—C(=O)—N(H)—C(H2] and [C(H2)—C(=O)N(H)—N=C(H)—Car] segments are inclined to on another by 77.36 (8)°, while the benzene rings are inclined to one another by 89.69 (9)°. In the crystal, molecules are linked via pairs of N—H⋯O hydrogen bonds, forming inversion dimers which are linked by O—H⋯O hydrogen bonds, involving the crystal water molecule, forming chains propagating along the a-axis direction.
Keywords: crystal structure, glycinyl hydrazone, monohydrate, hydrogen bonding
Abstract
The title compound, C17H16ClN3O2·H2O, an acylhydrazone derivative, contains a glycine moiety and two substituted benzene rings on either end of the chain. It crystallized as a monohydrate. The molecules adopt an E conformation with respect to the C=N double bond, as indicated by the N—N=C—C torsion angle of 179.38 (14)°. The molecule is twisted in such a way that the almost planar Car—C(=O)—N(H)—C(H2) and C(H2)—C(=O)N(H)—N=C—Car [r.m.s deviations = 0.009 and 0.025 Å, respectively] segments are inclined to on another by 77.36 (8)°, while the benzene rings are normal to one another, making a dihedral angle of 89.69 (9)°. In the crystal, the water molecule links three molecules through two O—H⋯O and one N—H⋯O hydrogen bonds. The molecules are linked via pairs of N—H⋯O hydrogen bonds, forming inversion dimers with an R 2 2(14) ring motif. The dimers are linked by O—H⋯O hydrogen bonds, involving two molecules of water, forming chains along [100], enclosing R 2 2(14) and R 2 2(18) ring motifs. The chains are linked through C—H⋯O interactions, forming sheets parallel to (010). Within the sheets, there are C—H⋯π and parallel slipped π–π stacking interactions present [inter-centroid distance = 3.6458 (12) Å].
Chemical context
N-Acylhydrazones have been reported to be promising in terms of their future potential as antibacterial drugs (Osorio et al., 2012 ▸). These predictions have provided a therapeutic pathway to develop new effective biologically active Schiff-base derivatives. N-Acylhydrazones may exist as Z/E geometrical isomers about the C=N double bond and as syn/anti amide conformers (Palla et al., 1986 ▸). The carbonyl group in the acylhydrazone provides the possibility for electron delocalization within the hydrazone moiety. The anti-TNF-α activity of glycinyl-hydrazone derivatives indicate that differences in the hydrophobicity of the imine-attached framework plays an important role. The study of conformational isomers of the amide unit of an N-methyl N-acylhydrazone derivative suggested that the amino spacer does not participate as a hydrogen-bond donor in the stabilization of the conformational isomers in solution (Lacerda et al., 2012 ▸). 
Prompted by the biological and structural importance of Schiff bases, as part of our structural studies (Gowda et al., 2000 ▸; Rodrigues et al., 2011 ▸; Jyothi & Gowda, 2004 ▸; Usha & Gowda, 2006 ▸; Purandara et al., 2015 ▸), we report herein on the synthesis, characterization and crystal structure of the title compound, (I), a new N-acylhydrazone derivative.
Structural commentary
The title compound crystallizes as a monohydrate (Fig. 1 ▸). The conformation of the N—H bond in the amide part is anti with respect to both the C=O bonds in the molecule, while the N—H bond in the hydrazone part is syn to both the C=O(hydrazone) and the C—H(imine) bonds. The C9—O2 bond length of 1.2251 (19) Å indicates that the molecule exists in the keto form in the solid state, and the C10—N3 bond length of 1.271 (2) Å confirms its significant double-bond character. The C9—N2 and N2—N3 bond distances of 1.351 (2) and 1.3771 (18) Å, respectively, indicate a significant delocalization of the π-electron density over the hydrazone portion of the molecule. Variations in the C—N bond lengths of 1.330 (2), 1.442 (2) and 1.351 (2) Å for C7—N1, C8—N1 and C9—N2, respectively, characterize mobility of the bridge and the integral flexibility of the –C(=O)–NH–CH2C(=O)–NH–N=CH– group connecting the two benzene rings. The molecule is twisted at atom C8, the C7—N1—C8—C9 torsion angle being 79.8 (2)°. The hydrazone part of the molecule is almost planar, with C9—N2—N3—C10 and N2—N3—C10—C11 torsion angles of −177.07 (15) and 179.38 (14)°, respectively. Further, the dihedral angle between the almost planar hydrazone segment (O2/N2/N3/C8–C11; maximum deviation of 0.029 (1) Å for atom N2) and the attached benzene ring (C11–C16) is 8.17 (6)°. The two benzene rings (C1–C6 and C11–C16) are orthogonal to each other, making a dihedral angle of 89.69 (9)°. The planar amide segment (O1/N1/C1/C7/C8; r.m.s. deviation = 0.009 Å) is inclined to the attached toluene ring (C1–C6) by 8.06 (9) Å.
Figure 1.
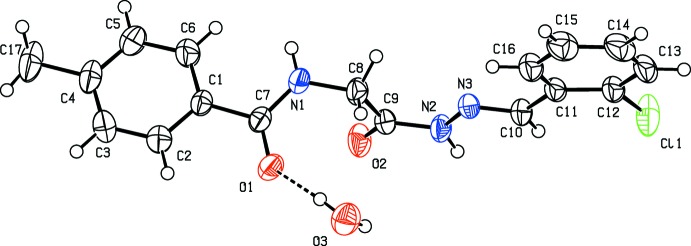
The molecular structure of the title compound, showing the atom labelling. Displacement ellipsoids are drawn at the 50% probability level.
Supramolecular features
In the crystal of (I), the amide carbonyl O-atom, O1, shows bifurcated hydrogen bonding (Table 1 ▸ and Fig. 2 ▸); one with the hydrazide hydrogen atom and the other with one of the hydrogen atoms of the water molecule (O3). The two hydrogen atoms of the water molecule are involved in hydrogen bonding with the O atoms of the amide carbonyl (O3—H31⋯O1) and glycine carbonyl (O3—H32⋯O2) groups of two different molecules of the title compound. The O atom is also involved in hydrogen bonding with the H atom of the carbonylamide group of a third symmetry-related molecule (N1—H1N⋯O3). A pair of N2—H2N⋯O1 intermolecular hydrogen bonds link the molecules, forming inversion dimers, with an 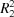 (14) ring motif. The dimers are further linked via hydrogen bonds involving the water molecule generating
(14) ring motif. The dimers are further linked via hydrogen bonds involving the water molecule generating 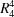 (14) and (18) ring motifs. Further, the N2—H2N⋯O1 and N1—H1N⋯O3 hydrogen bonds between the molecules of the main compound and water molecules translate into
(14) and (18) ring motifs. Further, the N2—H2N⋯O1 and N1—H1N⋯O3 hydrogen bonds between the molecules of the main compound and water molecules translate into 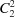 (6) chains along the a-axis direction (Table 1 ▸ and Fig. 2 ▸) The chains are linked by a C—H⋯O interaction, forming sheets parallel to (010). Within the sheets there are C—H⋯π, and parallel slipped π–π stacking interactions [Cg2⋯Cg2i = 3.6458 (12) Å; inter-planar distance = 3.4135 (8) Å, slippage = 1.281 Å; Cg2 is the centroid of ring C11–C16; symmetry code: (i) −x + 1, −y + 1, −z + 1] involving inversion-related chlorobenzene rings; see Fig. 3 ▸.
(6) chains along the a-axis direction (Table 1 ▸ and Fig. 2 ▸) The chains are linked by a C—H⋯O interaction, forming sheets parallel to (010). Within the sheets there are C—H⋯π, and parallel slipped π–π stacking interactions [Cg2⋯Cg2i = 3.6458 (12) Å; inter-planar distance = 3.4135 (8) Å, slippage = 1.281 Å; Cg2 is the centroid of ring C11–C16; symmetry code: (i) −x + 1, −y + 1, −z + 1] involving inversion-related chlorobenzene rings; see Fig. 3 ▸.
Table 1. Hydrogen-bond geometry (, ).
Cg1 is the centroid of the toluene ring C1C6.
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O3H31O1 | 0.84(2) | 2.13(2) | 2.897(2) | 152(3) |
| O3H32O2i | 0.86(2) | 1.92(2) | 2.772(2) | 174(3) |
| N1H1NO3ii | 0.84(2) | 2.15(2) | 2.941(2) | 158(2) |
| N2H2NO1i | 0.87(2) | 2.09(2) | 2.944(2) | 165(2) |
| C14H14O2iii | 0.93 | 2.57 | 3.404(2) | 150 |
| C15H15Cg1iii | 0.93 | 2.89 | 3.793(2) | 165 |
Symmetry codes: (i) 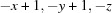 ; (ii)
; (ii) 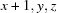 ; (iii)
; (iii) 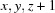 .
.
Figure 2.
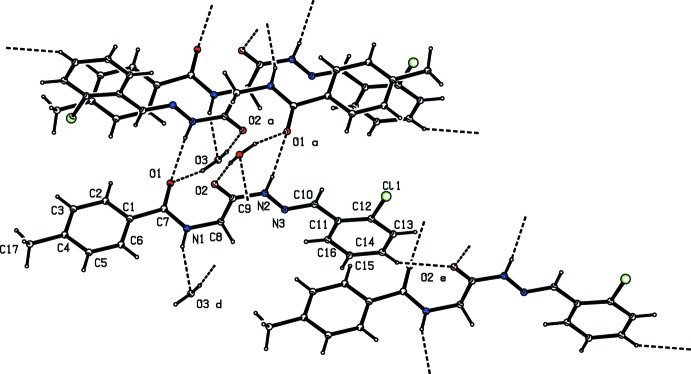
Hydrogen-bonding pattern in the title compound (see Table 1 ▸ for details). [Symmetry codes: (a) −x + 1, −y + 1, −z; (d) x + 1, y, z; (e) x, y, z + 1.]
Figure 3.

A view along the a axis of the crystal packing of the title compound. Hydrogen bonds are shown as dashed lines and C—H⋯π interactions are represented as red arrows (see Table 1 ▸ for further details).
Database survey
A search of the Cambridge Structural Database (Version 5.36, May 2015; Groom & Allen, 2014 ▸) for the fragment –NH–CH2–C(=O)–NH–N=CH–, yielded only one hit, namely N-(2-hydroxy-1-naphthylmethylene)-N′-(N-phenylglycyl)hydrazine (MEMTOO; Gudasi et al., 2006 ▸). A comparison of the structural details of the title compound, (I), with those of the recently published sulfonyl derivative, (E)-N-{2-[2-(3-chlorobenzylidene)hydrazinyl]-2-oxoethyl}-4-methylbenzenesulfonamide monohydrate (II) (Purandara et al., 2015 ▸), reveals the trans orientation of the amide group (C1–C7(=O1)N1) and hydrazone segment (N2–N3=C10–C11) with respect to the glycinyl C8—C9 bond in (I), as is evident from the N1—C8—C9—N2 torsion angle of 173.58 (15)°, in contrast to the cis orientation of the sulfonamide and hydrazone segments, with respect to the glycinyl C—C bond, observed in compound (II). In the structure of (I), the benzene ring (C1–C6) is almost coplanar with the amide group [dihedral angle = 8.21 (13)°]. This is in contrast to the L-shaped conformation (bent at the S atom) of the sulfonamide group with respect to the benzene ring in compound (II). The amide carbonyl O atom forms stronger O—H⋯O hydrogen bonds with the water H atoms than the sulfonyl O atom as observed in compound (II), indicating the stronger electron-withdrawing character of the amide group compared to the sulfonamide group.
Synthesis and crystallization
Triethylamine (0.03 mol) and 4-methylbenzoyl chloride (0.01 mol) were added to a stirred suspension of glycine ethylester hydrochloride (0.01 mol) in dichloromethane (50 ml) in an ice bath. The reaction mixture was stirred at room temperature for 20 h. After completion of the reaction, 2N hydrochloric acid (80 ml) was added slowly. The organic phase was separated and washed with water (30 ml), dried with anhydrous Na2SO4 and evaporated to yield the corresponding ester, N-(4-methylbenzoyl)glycine ethyl ester (L1). L1 (0.01 mol) was added in small portions to a stirred solution of 99% hydrazine hydrate (10 ml) in 30 ml ethanol. The mixture was refluxed for 6 h. After cooling to room temperature, the resulting precipitate was filtered, washed with cold water and dried to give N-(4-methylbenzoyl)-glycinyl hydrazide (L2). 2-Chlorobenzaldehyde (0.01 mol) and two drops of glacial acetic acid were added to L2 (0.01 mol) in anhydrous methanol (30 ml). The reaction mixture was refluxed for 8 h. After cooling, the precipitate was collected by vacuum filtration, washed with cold methanol and dried. It was recrystallized to constant melting point from methanol (479–480 K). Prism-like colourless single crystals of the title compound were grown from a solution in DMF by slow evaporation of the solvent.
The purity of the compound was checked by TLC and characterized by its IR spectrum. The characteristic absorptions observed are 3323.3, 3203.8, 1685.8, 1620.2 and 1566.2 cm−1 for the stretching bands of N—H (amide I), N—H (amide II), C=O(hydrazone), C=O(amide) and C=N, respectively. The characteristic 1H and 13C NMR spectra of the title compound are as follows: 1H NMR (400 MHz, DMSO-d6, δ p.p.m.): 2.36 (s, 3H), 4.01, 4.45 (2d, 2H, J = 5.8 Hz), 7.25 (d, 2H, Ar-H, J = 8.0 Hz), 7.33–7.40 (m, 2H, Ar-H), 7.42–7.45 (m, 1H, Ar-H), 7.81 (d, 2H, Ar-H), 7.97–7.99 (m, 1H, Ar-H), 8.39, 8.63 (2s, 1H), 8.54, 8.76 (2t, 1H, J = 5.7 Hz), 11.65, 11.73 (2s, 1H). 13C NMR (400 MHz, DMSO-d6, δ p.p.m.): 20.97, 40.74, 42.04, 126.60, 126.83, 127.28, 128.64, 129.66, 130.85, 131.35, 133.10, 139.45, 141.06, 142.70, 165.98, 166.54, 170.48.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The water H atoms and the NH H atoms were located in a difference Fourier map and refined with distances restraints: O—H = 0.85 (2), N—H = 0.86 (2) Å with U iso(H) = 1.5U eq(O) and 1.2U eq(N). The C-bound H atoms were positioned with idealized geometry and refined as riding atoms: C—H = 0.93–0.97 Å with U iso(H) = 1.5U eq(C) for methyl H atoms and 1.2U eq(C) for other H atoms.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C17H16ClN3O2H2O |
| M r | 347.79 |
| Crystal system, space group | Triclinic, P
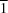
|
| Temperature (K) | 293 |
| a, b, c () | 6.9729(7), 10.642(1), 11.879(1) |
| , , () | 95.049(8), 100.324(9), 102.870(9) |
| V (3) | 837.88(14) |
| Z | 2 |
| Radiation type | Mo K |
| (mm1) | 0.25 |
| Crystal size (mm) | 0.50 0.40 0.32 |
| Data collection | |
| Diffractometer | Oxford Diffraction Xcalibur with Sapphire CCD detector |
| Absorption correction | Multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▸) |
| T min, T max | 0.886, 0.925 |
| No. of measured, independent and observed [I > 2(I)] reflections | 5538, 3393, 2829 |
| R int | 0.009 |
| (sin /)max (1) | 0.625 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.039, 0.103, 1.04 |
| No. of reflections | 3393 |
| No. of parameters | 230 |
| No. of restraints | 4 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| max, min (e 3) | 0.24, 0.33 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015011147/su5148sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015011147/su5148Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015011147/su5148Isup3.cml
CCDC reference: 1405614
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
HP thanks the Department of Science and Technology, Government of India, New Delhi, for a research fellowship under its INSPIRE Program and BTG thanks the University Grants Commission, Government of India, New Delhi, for a special grant under the UGC–BSR one-time grant to faculty. The authors also thank SAIF Panjab University for providing an NMR facility.
supplementary crystallographic information
Crystal data
| C17H16ClN3O2·H2O | Z = 2 |
| Mr = 347.79 | F(000) = 364 |
| Triclinic, P1 | Dx = 1.379 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.9729 (7) Å | Cell parameters from 3287 reflections |
| b = 10.642 (1) Å | θ = 3.1–27.7° |
| c = 11.879 (1) Å | µ = 0.25 mm−1 |
| α = 95.049 (8)° | T = 293 K |
| β = 100.324 (9)° | Prism, colourless |
| γ = 102.870 (9)° | 0.50 × 0.40 × 0.32 mm |
| V = 837.88 (14) Å3 |
Data collection
| Oxford Diffraction Xcalibur single crystal X-ray diffractometer with a Sapphire CCD detector | 3393 independent reflections |
| Radiation source: fine-focus sealed tube | 2829 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.009 |
| Rotation method data acquisition using ω scans | θmax = 26.4°, θmin = 3.1° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −7→8 |
| Tmin = 0.886, Tmax = 0.925 | k = −12→13 |
| 5538 measured reflections | l = −14→11 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.103 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0396P)2 + 0.4048P] where P = (Fo2 + 2Fc2)/3 |
| 3393 reflections | (Δ/σ)max < 0.001 |
| 230 parameters | Δρmax = 0.24 e Å−3 |
| 4 restraints | Δρmin = −0.33 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.60321 (11) | 0.19118 (5) | 0.47068 (5) | 0.0724 (2) | |
| O1 | 0.62994 (17) | 0.78208 (12) | −0.04768 (11) | 0.0451 (3) | |
| O2 | 0.7257 (2) | 0.49520 (12) | −0.03656 (10) | 0.0499 (3) | |
| N1 | 0.9290 (2) | 0.75016 (14) | 0.03466 (12) | 0.0417 (3) | |
| H1N | 1.054 (2) | 0.766 (2) | 0.0404 (17) | 0.050* | |
| N2 | 0.6958 (2) | 0.45136 (14) | 0.14178 (11) | 0.0395 (3) | |
| H2N | 0.617 (3) | 0.3748 (16) | 0.1142 (16) | 0.047* | |
| N3 | 0.7330 (2) | 0.49621 (14) | 0.25783 (11) | 0.0365 (3) | |
| C1 | 0.9132 (2) | 0.87432 (15) | −0.12558 (13) | 0.0340 (3) | |
| C2 | 0.8034 (3) | 0.93929 (18) | −0.19759 (16) | 0.0470 (4) | |
| H2 | 0.6703 | 0.9352 | −0.1934 | 0.056* | |
| C3 | 0.8883 (3) | 1.0105 (2) | −0.27602 (17) | 0.0543 (5) | |
| H3 | 0.8112 | 1.0537 | −0.3235 | 0.065* | |
| C4 | 1.0838 (3) | 1.01864 (17) | −0.28502 (15) | 0.0471 (4) | |
| C5 | 1.1929 (3) | 0.9537 (2) | −0.21334 (17) | 0.0551 (5) | |
| H5 | 1.3259 | 0.9581 | −0.2179 | 0.066* | |
| C6 | 1.1104 (3) | 0.8822 (2) | −0.13474 (17) | 0.0503 (5) | |
| H6 | 1.1879 | 0.8390 | −0.0876 | 0.060* | |
| C7 | 0.8130 (2) | 0.79820 (15) | −0.04311 (13) | 0.0349 (3) | |
| C8 | 0.8517 (3) | 0.67330 (17) | 0.11810 (14) | 0.0418 (4) | |
| H8A | 0.7536 | 0.7113 | 0.1479 | 0.050* | |
| H8B | 0.9608 | 0.6755 | 0.1822 | 0.050* | |
| C9 | 0.7542 (2) | 0.53317 (16) | 0.06684 (13) | 0.0365 (4) | |
| C10 | 0.6807 (2) | 0.41135 (17) | 0.32262 (14) | 0.0383 (4) | |
| H10 | 0.6205 | 0.3256 | 0.2906 | 0.046* | |
| C11 | 0.7154 (2) | 0.44844 (17) | 0.44785 (13) | 0.0365 (4) | |
| C12 | 0.6834 (3) | 0.35608 (18) | 0.52341 (15) | 0.0426 (4) | |
| C13 | 0.7134 (3) | 0.3916 (2) | 0.64139 (15) | 0.0517 (5) | |
| H13 | 0.6894 | 0.3284 | 0.6899 | 0.062* | |
| C14 | 0.7787 (3) | 0.5207 (2) | 0.68642 (16) | 0.0558 (5) | |
| H14 | 0.7984 | 0.5451 | 0.7656 | 0.067* | |
| C15 | 0.8151 (3) | 0.6143 (2) | 0.61429 (16) | 0.0539 (5) | |
| H15 | 0.8613 | 0.7016 | 0.6448 | 0.065* | |
| C16 | 0.7828 (3) | 0.57766 (19) | 0.49659 (15) | 0.0453 (4) | |
| H16 | 0.8069 | 0.6415 | 0.4487 | 0.054* | |
| C17 | 1.1791 (4) | 1.0976 (2) | −0.36898 (19) | 0.0688 (6) | |
| H17A | 1.0858 | 1.1420 | −0.4073 | 0.103* | |
| H17B | 1.2140 | 1.0409 | −0.4252 | 0.103* | |
| H17C | 1.2981 | 1.1602 | −0.3279 | 0.103* | |
| O3 | 0.3605 (2) | 0.76894 (15) | 0.11275 (13) | 0.0555 (4) | |
| H31 | 0.452 (3) | 0.799 (3) | 0.078 (2) | 0.083* | |
| H32 | 0.340 (4) | 0.6869 (17) | 0.094 (2) | 0.083* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.1083 (5) | 0.0530 (3) | 0.0572 (3) | 0.0101 (3) | 0.0233 (3) | 0.0258 (2) |
| O1 | 0.0388 (6) | 0.0496 (7) | 0.0445 (7) | 0.0007 (5) | 0.0129 (5) | 0.0100 (5) |
| O2 | 0.0698 (9) | 0.0503 (7) | 0.0277 (6) | 0.0064 (6) | 0.0134 (6) | 0.0093 (5) |
| N1 | 0.0383 (7) | 0.0470 (8) | 0.0364 (7) | −0.0009 (6) | 0.0082 (6) | 0.0159 (6) |
| N2 | 0.0474 (8) | 0.0396 (8) | 0.0278 (7) | 0.0010 (6) | 0.0081 (6) | 0.0094 (6) |
| N3 | 0.0372 (7) | 0.0458 (8) | 0.0270 (6) | 0.0077 (6) | 0.0077 (5) | 0.0115 (6) |
| C1 | 0.0395 (8) | 0.0314 (8) | 0.0285 (7) | 0.0016 (6) | 0.0088 (6) | 0.0038 (6) |
| C2 | 0.0442 (10) | 0.0510 (10) | 0.0485 (10) | 0.0116 (8) | 0.0115 (8) | 0.0165 (8) |
| C3 | 0.0641 (12) | 0.0526 (11) | 0.0495 (11) | 0.0152 (9) | 0.0112 (9) | 0.0241 (9) |
| C4 | 0.0656 (12) | 0.0367 (9) | 0.0349 (9) | −0.0020 (8) | 0.0169 (8) | 0.0061 (7) |
| C5 | 0.0463 (10) | 0.0709 (13) | 0.0527 (11) | 0.0093 (9) | 0.0227 (9) | 0.0204 (10) |
| C6 | 0.0454 (10) | 0.0651 (12) | 0.0476 (10) | 0.0167 (9) | 0.0155 (8) | 0.0250 (9) |
| C7 | 0.0384 (8) | 0.0322 (8) | 0.0303 (8) | −0.0005 (6) | 0.0090 (6) | 0.0028 (6) |
| C8 | 0.0467 (9) | 0.0454 (9) | 0.0290 (8) | 0.0003 (7) | 0.0072 (7) | 0.0117 (7) |
| C9 | 0.0379 (8) | 0.0433 (9) | 0.0288 (8) | 0.0075 (7) | 0.0077 (6) | 0.0115 (7) |
| C10 | 0.0424 (9) | 0.0427 (9) | 0.0322 (8) | 0.0093 (7) | 0.0112 (7) | 0.0126 (7) |
| C11 | 0.0328 (8) | 0.0508 (10) | 0.0308 (8) | 0.0137 (7) | 0.0102 (6) | 0.0153 (7) |
| C12 | 0.0396 (9) | 0.0558 (10) | 0.0377 (9) | 0.0141 (8) | 0.0123 (7) | 0.0196 (8) |
| C13 | 0.0470 (10) | 0.0817 (15) | 0.0340 (9) | 0.0208 (10) | 0.0120 (8) | 0.0276 (9) |
| C14 | 0.0490 (11) | 0.0920 (16) | 0.0288 (9) | 0.0226 (10) | 0.0071 (8) | 0.0091 (9) |
| C15 | 0.0540 (11) | 0.0652 (13) | 0.0408 (10) | 0.0157 (9) | 0.0067 (8) | 0.0015 (9) |
| C16 | 0.0478 (10) | 0.0527 (11) | 0.0382 (9) | 0.0135 (8) | 0.0112 (7) | 0.0140 (8) |
| C17 | 0.0966 (17) | 0.0565 (12) | 0.0513 (12) | −0.0029 (12) | 0.0318 (12) | 0.0176 (10) |
| O3 | 0.0576 (8) | 0.0586 (8) | 0.0554 (8) | 0.0145 (7) | 0.0223 (7) | 0.0109 (7) |
Geometric parameters (Å, º)
| Cl1—C12 | 1.740 (2) | C6—H6 | 0.9300 |
| O1—C7 | 1.240 (2) | C8—C9 | 1.516 (2) |
| O2—C9 | 1.2251 (19) | C8—H8A | 0.9700 |
| N1—C7 | 1.330 (2) | C8—H8B | 0.9700 |
| N1—C8 | 1.442 (2) | C10—C11 | 1.467 (2) |
| N1—H1N | 0.842 (15) | C10—H10 | 0.9300 |
| N2—C9 | 1.351 (2) | C11—C16 | 1.386 (3) |
| N2—N3 | 1.3771 (18) | C11—C12 | 1.397 (2) |
| N2—H2N | 0.873 (15) | C12—C13 | 1.385 (3) |
| N3—C10 | 1.271 (2) | C13—C14 | 1.373 (3) |
| C1—C2 | 1.379 (2) | C13—H13 | 0.9300 |
| C1—C6 | 1.383 (2) | C14—C15 | 1.381 (3) |
| C1—C7 | 1.496 (2) | C14—H14 | 0.9300 |
| C2—C3 | 1.384 (3) | C15—C16 | 1.382 (3) |
| C2—H2 | 0.9300 | C15—H15 | 0.9300 |
| C3—C4 | 1.371 (3) | C16—H16 | 0.9300 |
| C3—H3 | 0.9300 | C17—H17A | 0.9600 |
| C4—C5 | 1.373 (3) | C17—H17B | 0.9600 |
| C4—C17 | 1.510 (2) | C17—H17C | 0.9600 |
| C5—C6 | 1.380 (2) | O3—H31 | 0.840 (17) |
| C5—H5 | 0.9300 | O3—H32 | 0.856 (17) |
| C7—N1—C8 | 122.85 (15) | H8A—C8—H8B | 107.9 |
| C7—N1—H1N | 121.7 (14) | O2—C9—N2 | 121.16 (16) |
| C8—N1—H1N | 115.4 (14) | O2—C9—C8 | 122.74 (14) |
| C9—N2—N3 | 119.90 (14) | N2—C9—C8 | 116.08 (14) |
| C9—N2—H2N | 118.6 (13) | N3—C10—C11 | 120.13 (16) |
| N3—N2—H2N | 120.7 (13) | N3—C10—H10 | 119.9 |
| C10—N3—N2 | 115.65 (14) | C11—C10—H10 | 119.9 |
| C2—C1—C6 | 117.83 (15) | C16—C11—C12 | 116.92 (16) |
| C2—C1—C7 | 118.58 (15) | C16—C11—C10 | 121.14 (15) |
| C6—C1—C7 | 123.59 (15) | C12—C11—C10 | 121.94 (16) |
| C1—C2—C3 | 121.00 (17) | C13—C12—C11 | 121.76 (18) |
| C1—C2—H2 | 119.5 | C13—C12—Cl1 | 117.92 (14) |
| C3—C2—H2 | 119.5 | C11—C12—Cl1 | 120.32 (14) |
| C4—C3—C2 | 121.22 (18) | C14—C13—C12 | 119.63 (17) |
| C4—C3—H3 | 119.4 | C14—C13—H13 | 120.2 |
| C2—C3—H3 | 119.4 | C12—C13—H13 | 120.2 |
| C3—C4—C5 | 117.71 (16) | C13—C14—C15 | 120.05 (17) |
| C3—C4—C17 | 121.75 (19) | C13—C14—H14 | 120.0 |
| C5—C4—C17 | 120.53 (19) | C15—C14—H14 | 120.0 |
| C4—C5—C6 | 121.76 (18) | C14—C15—C16 | 119.8 (2) |
| C4—C5—H5 | 119.1 | C14—C15—H15 | 120.1 |
| C6—C5—H5 | 119.1 | C16—C15—H15 | 120.1 |
| C5—C6—C1 | 120.49 (17) | C15—C16—C11 | 121.87 (17) |
| C5—C6—H6 | 119.8 | C15—C16—H16 | 119.1 |
| C1—C6—H6 | 119.8 | C11—C16—H16 | 119.1 |
| O1—C7—N1 | 122.19 (14) | C4—C17—H17A | 109.5 |
| O1—C7—C1 | 120.78 (15) | C4—C17—H17B | 109.5 |
| N1—C7—C1 | 117.03 (14) | H17A—C17—H17B | 109.5 |
| N1—C8—C9 | 112.26 (14) | C4—C17—H17C | 109.5 |
| N1—C8—H8A | 109.2 | H17A—C17—H17C | 109.5 |
| C9—C8—H8A | 109.2 | H17B—C17—H17C | 109.5 |
| N1—C8—H8B | 109.2 | H31—O3—H32 | 102 (3) |
| C9—C8—H8B | 109.2 | ||
| C9—N2—N3—C10 | −177.07 (15) | N3—N2—C9—O2 | 178.83 (15) |
| C6—C1—C2—C3 | −0.3 (3) | N3—N2—C9—C8 | −2.4 (2) |
| C7—C1—C2—C3 | −179.62 (17) | N1—C8—C9—O2 | −7.6 (2) |
| C1—C2—C3—C4 | 0.2 (3) | N1—C8—C9—N2 | 173.58 (15) |
| C2—C3—C4—C5 | −0.1 (3) | N2—N3—C10—C11 | 179.38 (14) |
| C2—C3—C4—C17 | −179.09 (19) | N3—C10—C11—C16 | 7.7 (2) |
| C3—C4—C5—C6 | 0.1 (3) | N3—C10—C11—C12 | −171.98 (16) |
| C17—C4—C5—C6 | 179.14 (19) | C16—C11—C12—C13 | 1.3 (2) |
| C4—C5—C6—C1 | −0.3 (3) | C10—C11—C12—C13 | −179.00 (16) |
| C2—C1—C6—C5 | 0.3 (3) | C16—C11—C12—Cl1 | −178.83 (13) |
| C7—C1—C6—C5 | 179.62 (17) | C10—C11—C12—Cl1 | 0.9 (2) |
| C8—N1—C7—O1 | 1.4 (3) | C11—C12—C13—C14 | −0.8 (3) |
| C8—N1—C7—C1 | −179.01 (15) | Cl1—C12—C13—C14 | 179.30 (15) |
| C2—C1—C7—O1 | 7.7 (2) | C12—C13—C14—C15 | −0.3 (3) |
| C6—C1—C7—O1 | −171.59 (17) | C13—C14—C15—C16 | 0.9 (3) |
| C2—C1—C7—N1 | −171.90 (16) | C14—C15—C16—C11 | −0.4 (3) |
| C6—C1—C7—N1 | 8.8 (2) | C12—C11—C16—C15 | −0.7 (3) |
| C7—N1—C8—C9 | 79.8 (2) | C10—C11—C16—C15 | 179.63 (17) |
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the toluene ring C1–C6.
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H31···O1 | 0.84 (2) | 2.13 (2) | 2.897 (2) | 152 (3) |
| O3—H32···O2i | 0.86 (2) | 1.92 (2) | 2.772 (2) | 174 (3) |
| N1—H1N···O3ii | 0.84 (2) | 2.15 (2) | 2.941 (2) | 158 (2) |
| N2—H2N···O1i | 0.87 (2) | 2.09 (2) | 2.944 (2) | 165 (2) |
| C14—H14···O2iii | 0.93 | 2.57 | 3.404 (2) | 150 |
| C15—H15···Cg1iii | 0.93 | 2.89 | 3.793 (2) | 165 |
Symmetry codes: (i) −x+1, −y+1, −z; (ii) x+1, y, z; (iii) x, y, z+1.
References
- Gowda, B. T., Kumar, B. H. A. & Fuess, H. (2000). Z. Naturforsch. Teil A Phys. Sci. 55, 721–728.
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Gudasi, K. B., Patil, M. S., Vadavi, R. S., Shenoy, R. V., Patil, S. A. & Nethaji, M. (2006). Transition Met. Chem. 31, 580–585.
- Jyothi, K. & Gowda, B. T. (2004). Z. Naturforsch. Teil A Phys. Sci. 59, 64–68.
- Lacerda, R. B., da Silva, L. L., de Lima, C. K. F., Miguez, E., Miranda, A. L. P., Laufer, S. A., Barreiro, E. J. & Fraga, C. A. M. (2012). PLoS One, 7, e46925. [DOI] [PMC free article] [PubMed]
- Osorio, T. M., Monache, F. D., Chiaradia, L. D., Mascarello, A., Stumpf, T. R., Zanetti, C. R., Silveira, D. B., Barardi, C. R. M., Smania, E. de F. A., Viancelli, A., Garcia, L. A. T., Yunes, R. A., Nunes, R. J. & Smânia, A. Jr (2012). Bioorg. Med. Chem. Lett. 22, 225–230.
- Oxford Diffraction (2009). CrysAlis CCD and CrysAlis RED. Oxford Diffraction Ltd, Yarnton, England.
- Palla, G., Predieri, G., Domiano, P., Vignali, C. & Turner, W. (1986). Tetrahedron, 42, 3649–3654.
- Purandara, H., Foro, S. & Gowda, B. T. (2015). Acta Cryst. E71, 602–605. [DOI] [PMC free article] [PubMed]
- Rodrigues, V. Z., Foro, S. & Gowda, B. T. (2011). Acta Cryst. E67, o2179. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Usha, K. M. & Gowda, B. T. (2006). J. Chem. Sci. 118, 351–359.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015011147/su5148sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015011147/su5148Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015011147/su5148Isup3.cml
CCDC reference: 1405614
Additional supporting information: crystallographic information; 3D view; checkCIF report