

Abstract
In the title compound, C5N6, all the atoms are approximately coplanar. In the crystal, molecules are packed with short contact distances of 2.885 (2) (between the diazo N atom connected to the ring and a cyano N atom on a neighboring molecule) and 3.012 (2) Å (between the terminal diazo N atom and an N atom of a neighboring imidazole ring).
Keywords: crystal structure, diazo, imidazole, carbonitrile
Related literature
For synthesis of the title compound, see: Lu & Just (2001 ▸); Sheppard & Webster (1973 ▸). Few diazo-containing molecules have been isolated, and of these, only a small number have been examined by X-ray diffraction, see: Daidone et al. (2005 ▸); Dippold et al. (2012 ▸). The majority of these compounds are found as diazonium ions, rather than the neutral diazo species, see: Bugg et al. (1964 ▸).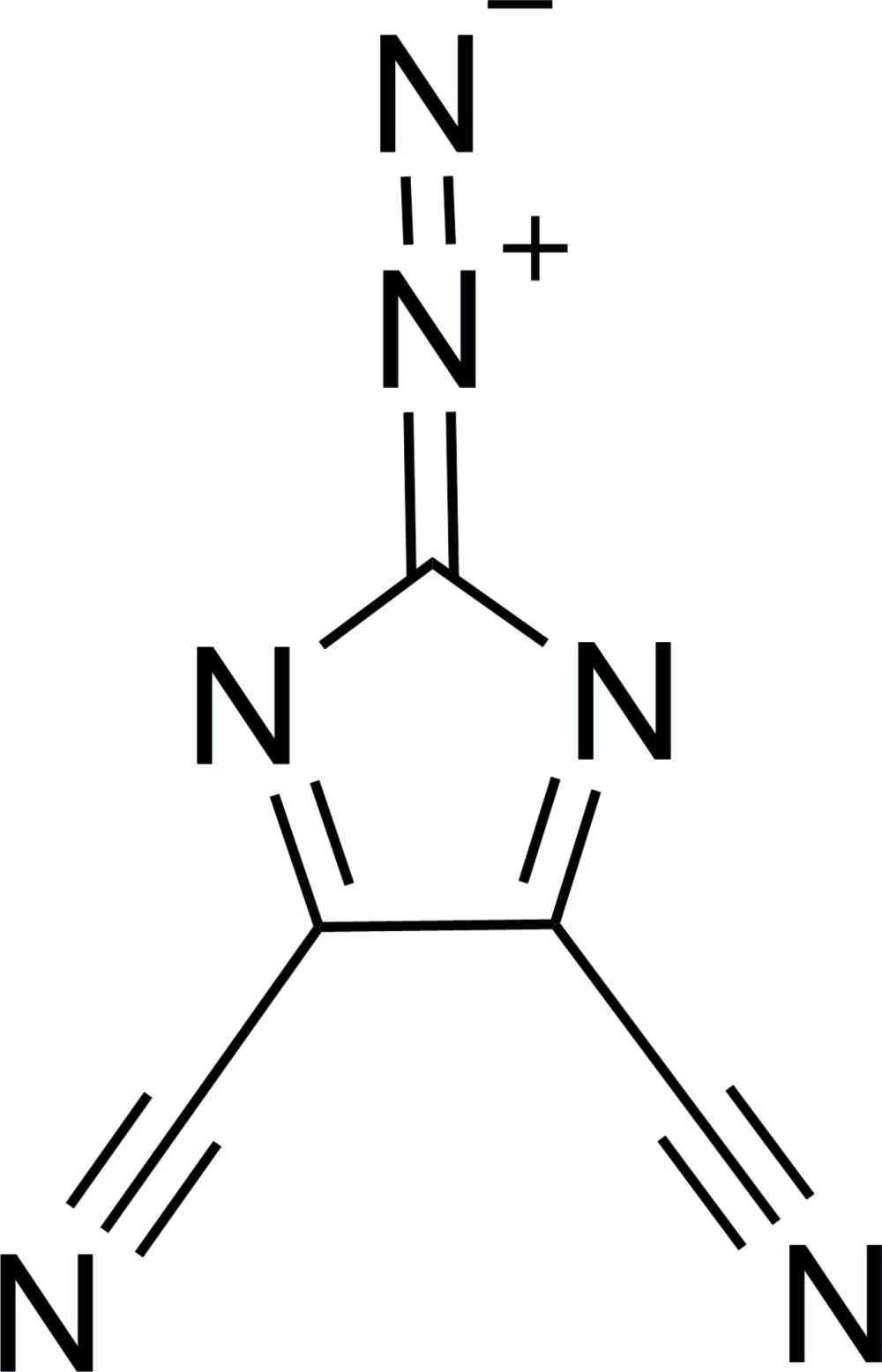
Experimental
Crystal data
C5N6
M r = 144.11
Trigonal,
a = 8.0746 (3) Å
c = 16.7315 (6) Å
V = 944.73 (8) Å3
Z = 6
Mo Kα radiation
μ = 0.11 mm−1
T = 150 K
0.37 × 0.30 × 0.08 mm
Data collection
Bruker SMART APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2008 ▸) T min = 0.930, T max = 0.991
9178 measured reflections
1289 independent reflections
1269 reflections with I > 2σ(I)
R int = 0.019
Refinement
R[F 2 > 2σ(F 2)] = 0.021
wR(F 2) = 0.058
S = 1.09
1289 reflections
100 parameters
Δρmax = 0.14 e Å−3
Δρmin = −0.11 e Å−3
Data collection: APEX2 (Bruker, 2009 ▸); cell refinement: APEX2 and SAINT (Bruker, 2009 ▸); data reduction: SAINT (Bruker, 2009 ▸) and XPREP (Bruker, 2008 ▸); program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▸); program(s) used to refine structure: SHELXTL; molecular graphics: ORTEP-3 for Windows (Farrugia, 2012 ▸) and Mercury (Macrae et al., 2008 ▸); software used to prepare material for publication: CHEMDRAW Ultra (Cambridge Soft, 2014 ▸).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015010944/cq2016sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010944/cq2016Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010944/cq2016Isup3.cdx
Supporting information file. DOI: 10.1107/S2056989015010944/cq2016Isup4.cml
. DOI: 10.1107/S2056989015010944/cq2016fig1.tif
The molecular structure of the title compound (displacement ellipsoids are drawn at the 50% probability level).
b . DOI: 10.1107/S2056989015010944/cq2016fig2.tif
A packing diagram of the title compound viewed along the b axis. Close contacts are represented by dashed lines.
CCDC reference: 1056377
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
Crystallographic studies were supported in part by the Office of Naval Research (ONR) (Award No. N00014-15-WX-0-0149). The authors would like to thank the Joint Munitions Technology Development Program for funding this work. Los Alamos National Laboratory is operated by Los Alamos National Security (LANS, LLC) under contract No. DE-AC52-06 N A25396 for the US Department of Energy (LA-UR-15-23325).
supplementary crystallographic information
S1. Comment
Diazo moieties are ubiquitious in organic chemistry due to their ability to act as as outstanding leaving groups in substitution reactions. This same reactivity also leads to their instability, and most are prone to decomposition, often violently. For this reason, few diazo-containing molecules have been isolated, and of these, only a small number have been examined by X-ray diffraction [Daidone et al. (2005), Dippold et al. (2012)]. The majority of these compounds are found as diazonium ions, rather than the neutral diazo species [Bugg et al. (1964)]. In contrast, the title compound has a neutral diazo moiety on C2 and is unusual in that it contains only carbon and nitrogen.
S2. Experimental
Caution! This compound can explode from slight friction, impact, or thermal shock. Although the explosion is not powerful, it is recommended that this material is only prepared in less than 5 gram quantities, handled wet, and not confined in any way.
The title compound was prepared by the literature method (Lu and Just, 2001). Crystals were obtained by slow evaporation of a dilute aqueous solution of the title compound.
S3. Refinement
The Flack parameter initially refined to 0.3 (5). Before the final refinement cycle, this parameter was reset to zero and the Friedel pairs merged.
Figures
Fig. 1.
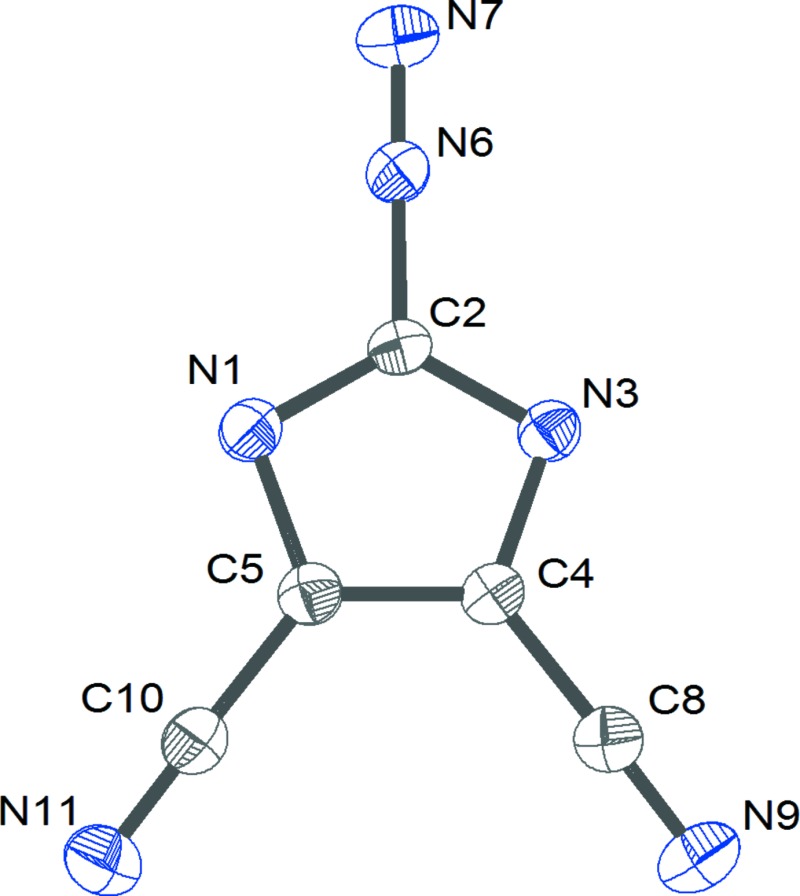
The molecular structure of the title compound (displacement ellipsoids are drawn at the 50% probability level).
Fig. 2.
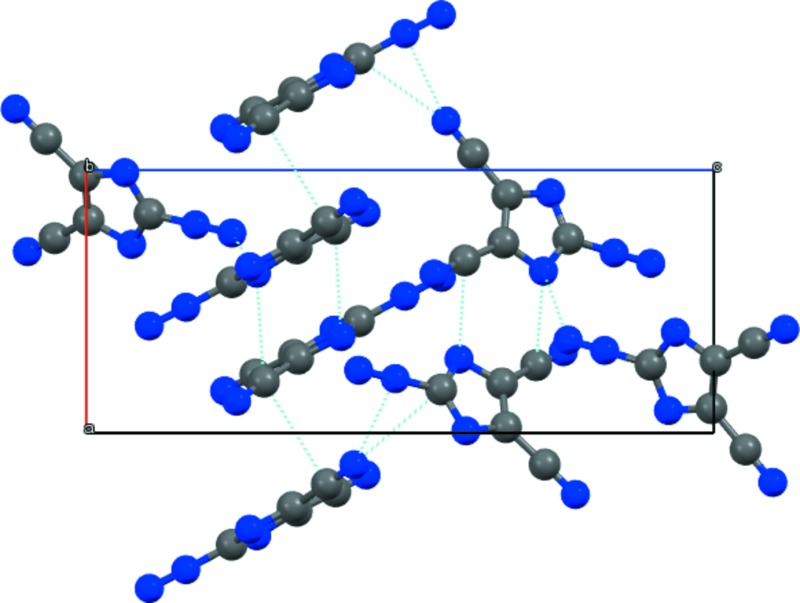
A packing diagram of the title compound viewed along the b axis. Close contacts are represented by dashed lines.
Crystal data
| C5N6 | Dx = 1.520 Mg m−3 |
| Mr = 144.11 | Melting point: 413 K (expl.) K |
| Trigonal, P3221 | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.0746 (3) Å | µ = 0.11 mm−1 |
| c = 16.7315 (6) Å | T = 150 K |
| V = 944.73 (8) Å3 | Plate, purple |
| Z = 6 | 0.37 × 0.30 × 0.08 mm |
| F(000) = 432 |
Data collection
| Bruker SMART APEXII CCD diffractometer | 1269 reflections with I > 2σ(I) |
| Radiation source: fine focus sealed tube | Rint = 0.019 |
| ω scans | θmax = 26.4°, θmin = 3.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −10→10 |
| Tmin = 0.930, Tmax = 0.991 | k = −10→10 |
| 9178 measured reflections | l = −20→20 |
| 1289 independent reflections |
Refinement
| Refinement on F2 | 100 parameters |
| Least-squares matrix: full | 0 restraints |
| R[F2 > 2σ(F2)] = 0.021 | w = 1/[σ2(Fo2) + (0.0333P)2 + 0.1041P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.058 | (Δ/σ)max = 0.001 |
| S = 1.09 | Δρmax = 0.14 e Å−3 |
| 1289 reflections | Δρmin = −0.11 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.61645 (16) | 0.00825 (16) | 0.93835 (6) | 0.0254 (3) | |
| C2 | 0.7396 (2) | 0.16549 (19) | 0.89882 (7) | 0.0226 (3) | |
| N3 | 0.91448 (16) | 0.28157 (17) | 0.92732 (6) | 0.0239 (3) | |
| C4 | 0.90565 (19) | 0.18858 (19) | 0.99570 (7) | 0.0225 (3) | |
| C5 | 0.72502 (19) | 0.02197 (19) | 1.00215 (7) | 0.0240 (3) | |
| N6 | 0.68567 (16) | 0.20682 (16) | 0.82574 (6) | 0.0238 (3) | |
| N7 | 0.6461 (2) | 0.23873 (19) | 0.76756 (7) | 0.0329 (3) | |
| C8 | 1.0629 (2) | 0.2582 (2) | 1.05016 (7) | 0.0258 (3) | |
| N9 | 1.18839 (19) | 0.3094 (2) | 1.09341 (7) | 0.0351 (3) | |
| C10 | 0.6572 (2) | −0.1202 (2) | 1.06366 (8) | 0.0299 (3) | |
| N11 | 0.6055 (2) | −0.2345 (2) | 1.11254 (8) | 0.0441 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0235 (6) | 0.0266 (6) | 0.0250 (5) | 0.0117 (5) | −0.0016 (4) | −0.0005 (5) |
| C2 | 0.0241 (6) | 0.0255 (6) | 0.0200 (5) | 0.0138 (5) | −0.0021 (5) | −0.0011 (5) |
| N3 | 0.0234 (6) | 0.0258 (6) | 0.0226 (5) | 0.0125 (5) | −0.0006 (4) | −0.0012 (4) |
| C4 | 0.0223 (6) | 0.0251 (6) | 0.0223 (6) | 0.0135 (5) | −0.0002 (5) | −0.0022 (5) |
| C5 | 0.0237 (6) | 0.0266 (6) | 0.0237 (6) | 0.0140 (6) | 0.0002 (4) | −0.0005 (5) |
| N6 | 0.0231 (6) | 0.0256 (6) | 0.0244 (5) | 0.0136 (5) | −0.0005 (4) | −0.0020 (4) |
| N7 | 0.0370 (7) | 0.0425 (8) | 0.0282 (6) | 0.0265 (6) | −0.0027 (5) | −0.0003 (5) |
| C8 | 0.0266 (7) | 0.0298 (7) | 0.0239 (6) | 0.0161 (6) | 0.0024 (5) | −0.0005 (5) |
| N9 | 0.0310 (7) | 0.0467 (8) | 0.0306 (6) | 0.0216 (6) | −0.0070 (5) | −0.0064 (5) |
| C10 | 0.0240 (7) | 0.0324 (7) | 0.0314 (7) | 0.0127 (6) | −0.0020 (5) | 0.0025 (6) |
| N11 | 0.0362 (7) | 0.0456 (8) | 0.0451 (8) | 0.0164 (7) | −0.0007 (6) | 0.0179 (6) |
Geometric parameters (Å, º)
| N1—C2 | 1.3327 (18) | C4—C8 | 1.4297 (18) |
| N1—C5 | 1.3502 (16) | C5—C10 | 1.4314 (18) |
| C2—N3 | 1.3325 (18) | N6—N7 | 1.0946 (15) |
| C2—N6 | 1.3936 (15) | C8—N9 | 1.1415 (19) |
| N3—C4 | 1.3507 (17) | C10—N11 | 1.144 (2) |
| C4—C5 | 1.4094 (18) | ||
| C2—N1—C5 | 99.75 (11) | C5—C4—C8 | 128.18 (12) |
| N3—C2—N1 | 121.08 (11) | N1—C5—C4 | 109.72 (11) |
| N3—C2—N6 | 119.59 (12) | N1—C5—C10 | 122.03 (12) |
| N1—C2—N6 | 119.33 (12) | C4—C5—C10 | 128.23 (12) |
| C2—N3—C4 | 99.74 (11) | N7—N6—C2 | 178.50 (12) |
| N3—C4—C5 | 109.70 (11) | N9—C8—C4 | 178.23 (16) |
| N3—C4—C8 | 122.12 (13) | N11—C10—C5 | 178.82 (17) |
| C5—N1—C2—N3 | −0.01 (16) | C2—N1—C5—C4 | 0.36 (14) |
| C5—N1—C2—N6 | 178.72 (11) | C2—N1—C5—C10 | −178.37 (13) |
| N1—C2—N3—C4 | −0.34 (16) | N3—C4—C5—N1 | −0.61 (16) |
| N6—C2—N3—C4 | −179.07 (11) | C8—C4—C5—N1 | 179.57 (12) |
| C2—N3—C4—C5 | 0.53 (14) | N3—C4—C5—C10 | 178.02 (13) |
| C2—N3—C4—C8 | −179.64 (12) | C8—C4—C5—C10 | −1.8 (2) |
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: CQ2016).
References
- Bruker (2008). SADABS and XPREP. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2009). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bugg, C., Lawson, J. & Sass, R. L. (1964). Acta Cryst. 17, 767–768.
- Cambridge Soft (2014). CHEMDRAW Ultra. Cambridge Soft Corporation, Cambridge, Massachusetts, USA.
- Daidone, G., Maggio, B., Raimondi, M. V., Bombieri, G., Marchini, N. & Artali, R. (2005). Heterocycles, 65, 2753–2761.
- Dippold, A. A., Klapötke, T. M., Martin, F. A. & Wiedbrauk, S. (2012). Eur. J. Inorg. Chem. pp. 2429–2443.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Lu, Y. & Just, G. (2001). Tetrahedron, 57, 1677–1687.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheppard, W. A. & Webster, O. W. (1973). J. Am. Chem. Soc. 95, 2695–2697.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015010944/cq2016sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015010944/cq2016Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015010944/cq2016Isup3.cdx
Supporting information file. DOI: 10.1107/S2056989015010944/cq2016Isup4.cml
. DOI: 10.1107/S2056989015010944/cq2016fig1.tif
The molecular structure of the title compound (displacement ellipsoids are drawn at the 50% probability level).
b . DOI: 10.1107/S2056989015010944/cq2016fig2.tif
A packing diagram of the title compound viewed along the b axis. Close contacts are represented by dashed lines.
CCDC reference: 1056377
Additional supporting information: crystallographic information; 3D view; checkCIF report