

Abstract
3,7-Diazabicyclo[3.3.1]nonane (bispidine) based nicotinic acetylcholine receptor (nAChR) ligands have been synthesized and evaluated for nAChRs interaction. Diverse spacer motifs were incorporated between the hydrogen bond acceptor (HBA) part and a variety of substituted (hetero)aryl moieties. Bispidine carboxamides bearing spacer motifs often showed high affinity in the low nanomolar range and selectivity for the α4β2* nAChR. Compounds 15, 25, and 47 with Ki values of about 1 nM displayed the highest affinities for α4β2* nAChR. All evaluated compounds are partial agonists or antagonists at α4β2*, with reduced or no effects on α3β4* with the exception of compound 15 (agonist), and reduced or no effect at α7 and muscle subtypes.
Keywords: 3,7-Diazabicyclo[3.3.1]nonane; Bispidine; Nicotinic acetylcholine receptor; nAChR; Structure-activity relationship; Cytisine
Graphical Abstract
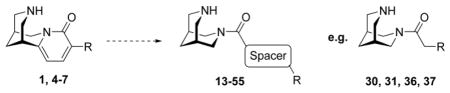
1. Introduction
(Di)azabicyclic templates dominate compound libraries for nicotinic acetylcholine receptors (nAChRs).1 NAChRs are pentameric cation channels found in the central and peripheral nervous systems, as well as in non-neuronal cells and serve as interesting targets especially for various brain diseases.2–5 These (di)azabicyclic templates display cationic/HB cores important for the interaction with the receptors. An additional pharmacophoric point, a hydrogen bond acceptor (HBA) motif, is often introduced as a pyridine moiety and less often as carbonyl functionality or as heteroaryls.6,7 The bulkiness of ring structures containing the potentially charged nitrogen and moieties attached to the HBA system influence both affinity and functionality.8 The diazabicyclic scaffold 3,7-diazabicyclo[3.3.1]nonane (bispidine) originated from the natural product and nAChR ligand cytisine 1 (Fig. 1). It is a privileged scaffold and has been used for the development of nAChR compounds.6,7,8–15 In our previous 3,7-diazabicyclo[3.3.1]nonane project, we explored the influence of different non-heteroaryl based HBA systems.7 We also reported that 3,7-diazabicyclo[3.3.1]nonane is active on nAChRs and that some 3,7-diazabicyclo[3.3.1]nonane carboxamides showed selectivity for the α4β2* nAChR. Herein, we extend the previous study by introducing spacer motifs like methylene, ethylene, ethenylene, ethynylene or (hetero)aryls attached to the HBA system to explore the chemical space for nAChRs further (Fig. 1). Diverse substituents were connected to the spacer motifs.
Figure 1.
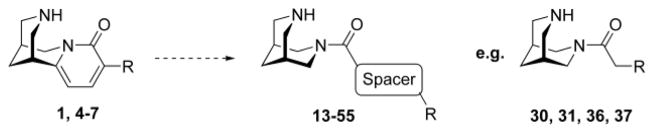
Structures of (-)-cytisine 1, cytisine derivatives 4–7 and the development of the 3,7-diazabicyclo[3.3.1]nonane based compounds 13–55 incorporating a spacer motif, e.g. compounds 30, 31, 36, and 37.
Firstly, we synthesized and tested four cytisine derivatives including the in vivo active 3-(pyridine-3-yl)-cytisine (3PC) 5 displaying at least one rotatable bond and compared them with their analogously substituted 3,7-diazabicyclo[3.3.1]nonane carboxamides to get insight into the influence of a spacer motif.16–18 We have recently shown that 3-(pyridine-3-yl)-cytisine (3PC) 5 serves as an interesting lead for the development of antidepressants.16–18 Now, we want to “open” the “chemical space” of cytisine to have the possibility to obtain compounds with better subtype selectivity pattern than cytisine derivatives along with a much simpler synthetic approach. Secondly, we prepared and tested a series of carboxamides bearing different spacer motifs providing additional possible interaction points with nAChRs and their consequences on affinity and functionality for further profiling projects.
2. Results and discussion
2.1. Chemistry
Like previously described by us, (-)-cytisine was isolated from seeds and pods of Laburnum anagyroides.16,17 The dried and milled plant material was extracted with PE to remove lipids. Then (-)-cytisine was extracted with a mixture of CH2Cl2:MeOH:NH3 (90:10:5) for 8 hours. After filtration and concentration of this solution, it was extracted with HCl (1N) and, subsequently, rendered alkaline, and extracted with CH2Cl2 until completion. The alkaloid 1 was finally purified by column chromatography on silica gel with a mixture of CHCl3 and MeOH (6:1) or by preparative HPLC using a C18-RP stationary phase and a MeOH-H2O gradient.
The N-tboc protection group was introduced by adding di-tert-butyl dicarbonate ((Boc)2O) to a solution of (-)-cytisine 1 and Na2CO3 in a mixture of CH2Cl2 and H2O. The solution was heated to reflux for 2 h and N-tboc-cytisine 2 was obtained in good yields after extraction and recrystallization in PE. Alternatively, N-tboc-cytisine 2 was synthesized by adding portions of (Boc)2O to a solution of 1 and Na2CO3 in a mixture of CH2Cl2 and H2O until the (-)-cytisine spot 1 has disappeared on TLC. After extracting the product 2 it was purified by preparative HPLC using a C18-RP column and a MeOH-H2O gradient.
The reaction of N-tboc-cytisine 2 with N-bromosuccinimide (NBS) for 2 h under reflux in CH2Cl2 afforded a mixture of isomeric bromo-N-tboc-cytisine derivatives bearing the bromo substituents in the 3 or the 5 position of the pyridone moiety. These isomers could be separated by preparative HPLC using a C18-RP column and a MeOH-H2O gradient. Only 3-bromo-N-tboc-cytisine 3 was used for this project.
The cytisine derivatives 4 – 7 were synthesized in a two-step process incorporating a cross-coupling reaction and the N-tboc-deprotection step. The coupling of aromatic or heteroaromatic moieties to the cytisine backbone was achieved by using the Suzuki-Miyaura reaction in a microwave synthesizer. The palladium catalyst Pd(PPh3)4 was added to a solution of 3-bromo-N-tboc-cytisine 3, the appropriate boronic acid and an inorganic base in a mixture of DME or DMF and H2O. Microwave irradiation (30 W) at 80 °C was used for 30 to 90 minutes to synthesize the according N-tboc protected cytisine derivatives Boc-4 – Boc-7. These products were purified on preparative HPLC using a C18-RP column and a MeOH-H2O gradient.
The N-tboc protection group of Boc-4 – Boc-7 was directly removed by using microwave irradiation (150 W) at 150 °C for 30 minutes in pure H2O. The solvent of the final cytisine products 4 – 7 was removed by lyophilisation for at least 24 h.
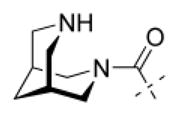
The 3,7-diazabicyclo[3.3.1]nonane (bispidine) scaffold was synthesized using the method described previously, where N-benzyl-N′-tboc-bispidinone 9 was obtained by a double Mannich reaction from commercially available tert-butyl 4-oxopiperidine carboxylate 8, benzylamine, and paraformaldehyde and reduced to the key intermediate N-benzyl-N′-tboc-bispidine 10.7,13 The cleavage of the N-benzyl protecting group from N-benzyl-N′-tboc-bispidine 10 was achieved by using palladium on activated charcoal (Pd/C) 5 % under a hydrogen atmosphere. The resulting N-tboc-bispidine 12 was now used as the starting material for all carboxamides described.
In general, all N-tboc protected intermediates Boc-13 – Boc-55 were purified by flash chromatography on silica gel and analyzed by LC-MS. When the purity was above 90–95 % these intermediates were N-tboc deprotected without further analysis. The N-tboc deprotected final compounds 13 – 55 were purified by flash chromatography and have been analyzed in more details.
Using the method previously described, intermediates Boc-13 – Boc-28 were synthesized by using carbonyldiimidazole (CDI) as a coupling reagent or via carboxylic acid chlorides/aminolysis or via DCC (N,N′-dicyclohexylcarbodiimide) coupling reaction.7,13
After removal of the N-tboc protecting group final compounds were often obtained as oils. For purification purposes and to improve stability, compounds 13–55 were transferred into their corresponding fumaric acid salts 13F–55F. Products 13F–55F have been obtained in excellent purity (mostly > 99 %).
2.2. Biological activity and SAR
Radioligand binding assays as previously described were performed to determine the affinities (Ki values) of cytisine 1 (standard ligand and starting material), cytisine derivatives 4–7 (Table 1) and the bispidine derivatives 13–55 (Table 2).19–21
Table 1.
Affinities (Ki values ± SEM) and calculated physicochemical parameters (logP, TPSA, logBB) of the (-)-cytisine 1 and cytisine derivatives 4–7
| Structure | α4β2* Ki [nM] | α3β4* Ki [nM] | α7* Ki [nM] | (α1)2β1γδ Ki [nM] | Mol. Weight | ClogPa | TPSA | logBBb | |
|---|---|---|---|---|---|---|---|---|---|
|
1c | 0.122 | 18 | 261 | 1,300 | 190.24 | 0.17 | 32.34 | −0.25 |
|
4c | 128 | > 10,000 | > 10,000 | > 10,000 | 266.34 | 2.27 | 32.34 | −0.01 |
|
5c | 0.91 | 119 | 1,100 | > 5,000 | 267.14 | 1.03 | 45.23 | −0.23 |
|
6c | 3.9 | 436 | > 10,000 | > 10,000 | 267.14 | 0.78 | 45.23 | −0.28 |
|
7c | 110 | > 10,000 | > 10,000 | > 10,000 | 310.35 | 2.31 | 50.80 | −0.03 |
Table 2.
Affinities (Ki values ± SEM) and calculated physicochemical parameters (logP, TPSA, logBB) of the bispidine carboxamide derivatives 13–55
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd. | R = | α4β2* Ki [nM] | α3β4* Ki [nM] | α7* Ki [nM] | (α1)2β1γδ Ki [nM] | Mol. Weight | ClogPa | TPSA | logBBb |
| 29 |
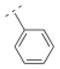
|
453.6 ± 14.8 | > 5,000 | > 10,000 | > 10,000 | 230.31 | 0.56 | 32.34 | −0.09 |
| 30 |
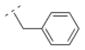
|
57.6 ± 15.4 | n. d. | > 10,000 | n. d. | 244.33 | 1.57 | 32.34 | 0.18 |
| 34 |
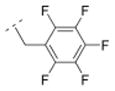
|
> 5,000 | n.d. | n.d. | n.d. | 334.28 | 1.75 | 32.34 | 0.25 |
| 35 |
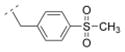
|
22.9 ± 0.9 | > 5,000 | > 5,000 | > 5,000 | 322.42 | −0.15 | 74.86 | −0.10 |
| 13 |
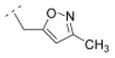
|
46.9 ± 7.2 | > 1,000 | > 1,000 | n. d. | 249.31 | −0.11 | 58.37 | −0.03 |
| 31 |
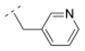
|
11.2 ± 1.5 | > 1,000 | > 1,000 | > 5,000 | 245.32 | 0.08 | 45.23 | −0.01 |
| 36 |
|
14.4 ± 2.0 | > 1,000 | > 1,000 | > 5,000 | 245.32 | 0.08 | 45.23 | −0.01 |
| 37 |
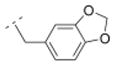
|
13.2 ± 1.2 | > 1,000 | > 1,000 | > 5,000 | 288.34 | 1.43 | 50.80 | −0.13 |
| 38 |
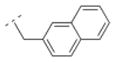
|
10.2 ± 2.1 | > 1,000 | > 1,000 | > 5,000 | 294.39 | 2.80 | 32.34 | −0.19 |
| 32 |
|
52.5 ± 0.7 | 395.8 | > 10,000 | n. d. | 258.36 | 2.08 | 32.34 | 0.04 |
| 39 |
|
39 ± 3.4 | > 5,000 | > 5,000 | > 5,000 | 259.35 | 0.59 | 45.23 | −0.10 |
| 14 |
|
232.6 | > 5,000 | > 10,000 | n. d. | 348.44 | 1.52 | 60.03 | −0.08 |
| 33 |
|
548.0 ± 66.0 | > 5,000 | > 5,000 | > 5,000 | 294.78 | 1.75 | 41.57 | −0.23 |
| 15 |
|
1.2 ± 0.4 | 81.1 | > 10,000 | n. d. | 270.37 | 1.73 | 32.34 | −0.13 |
| 16 |
|
55.1 ± 12.5 | > 5,000 | > 5,000 | > 10,000 | 256.34 | 1.87 | 32.34 | −0.24 |
| 17 |
|
171.8 ± 39.7 | n. d. | n. d. | > 10,000 | 286.37 | 1.87 | 41.57 | −0.21 |
| 18 |
|
23.7 ± 3.2 | n. d. | > 10,000 | n. d. | 290.79 | 2.47 | 32.34 | 0.06 |
| 19 |
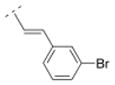
|
24.6 ± 4.2 | n. d. | > 10,000 | n. d. | 335.24 | 2.70 | 32.34 | 0.01 |
| 20 |
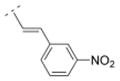
|
38.9 ± 4.4 | > 1,000 | n. d. | > 10,000 | 301.34 | 1.77 | 81.17 | −0.51 |
| 40 |
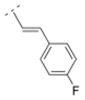
|
20.7 ± 2.4 | > 5,000 | > 5,000 | > 10,000 | 274.33 | 1.87 | 32.34 | −0.03 |
| 21 |
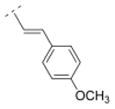
|
39.6 ± 5.4 | n. d. | n. d. | n. d. | 286.37 | 1.81 | 41.57 | −0.23 |
| 22 |
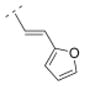
|
287.5 ± 25.7 | > 1,000 | > 1,000 | > 5,000 | 246.30 | 1.49 | 45.48 | −0.07 |
| 23 |
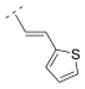
|
96.9 ± 20.5 | 2,000 | > 10,000 | n.d. | 262.37 | 1.68 | 60.58 | −0.32 |
| 24 |
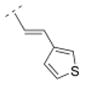
|
57.1 ± 12.3 | n.d. | > 10,000 | n.d. | 262.37 | 1.66 | 60.58 | 0.33 |
| 41 |
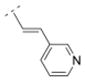
|
32.59 | > 1,000 | > 1,000 | > 5,000 | 257.33 | 0.62 | 45.23 | −0.23 |
| 25 |
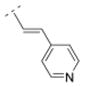
|
1.03 | > 1,000 | > 10,000 | > 5,000 | 257.33 | 0.37 | 45.23 | −0.27 |
| 26 |
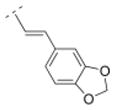
|
36.9 ± 8.0 | > 1,000 | > 1,000 | > 5,000 | 300.35 | 1.99 | 50.80 | −0.15 |
| 27 |
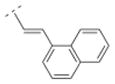
|
296.6 ± 50.7 | > 5,000 | > 1,000 | > 5,000 | 306.40 | 3.10 | 32.34 | −0.11 |
| 42 |

|
10.9 ± 3.4 | > 1,000 | > 5,000 | > 1,000 | 254.33 | 2.41 | 32.34 | −0.27 |
| 43 |
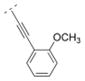
|
241.9 | > 1,000 | > 1,000 | > 5,000 | 284.35 | 2.33 | 41.57 | −0.23 |
| 44 |
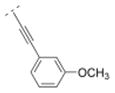
|
6.3 ± 1.1 | > 1,000 | > 1,000 | > 5,000 | 284.35 | 2.33 | 41.57 | −0.23 |
| 45 |
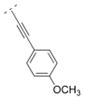
|
7.8 ± 2.0 | > 1,000 | > 1,000 | > 5,000 | 284.35 | 2.33 | 41.57 | −0.23 |
| 46 |
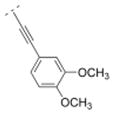
|
2.1 ± 0.8 | > 1,000 | > 1,000 | > 5,000 | 314.38 | 2.15 | 50.80 | −0.24 |
| 47 |
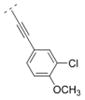
|
0.991 ± 0.02 | > 1,000 | > 1,000 | > 5,000 | 318.80 | 2.87 | 41.57 | −0.19 |
| 48 |
|
10.9 ± 2.1 | > 1,000 | > 1,000 | > 5,000 | 268.35 | 2.87 | 32.34 | −0.03 |
| 49 |
|
7.1 ± 2.4 | > 1,000 | > 1,000 | > 5,000 | 272.32 | 2.46 | 32.34 | −0.17 |
| 50 |
|
3.1 ± 1.0 | > 1,000 | > 1,000 | > 5,000 | 298.34 | 2.27 | 50.80 | −0.14 |
| 51 |
|
46.0 ± 7.5 | > 1,000 | > 1,000 | > 5,000 | 304.39 | 3.64 | 32.34 | −0.09 |
| 28 |
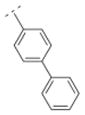
|
39.9 ± 7.1 | 1089.5 ± 290.5 | *c | *c | 306.40 | 2.21 | 32.34 | −0.65 |
| 52 |
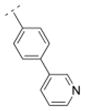
|
25.0 ± 5.5 | > 1,000 | > 1,000 | > 5,000 | 307.39 | 0.90 | 45.23 | −0.85 |
| 53 |
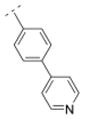
|
7.1 ± 1.3 | > 1,000 | > 1,000 | > 5,000 | 307.39 | 0.83 | 45.23 | −0.86 |
| 54 |
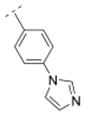
|
5.6 ± 0.9 | > 1,000 | > 1,000 | > 5,000 | 296.37 | 0.33 | 49.64 | −0.29 |
| 55 |
|
> 1,000 | n.d. | n.d. | n.d. | 314.41 | 1.58 | 73.47 | −0.34 |
Values are generated from 2–10 independent experiments; n. d. = not determined;
logP values have been calculated using the ACD/Labs Algorithm;
logBB was calculated from logP derived from using the ACD/Labs Algorithm (ACD);
radioligand binding was increased.
Cytisine derivatives in comparison with their analogously substituted 3,7-diazabicyclo[3.3.1]nonane based carboxamides
In the past, we and several other research groups studied structure-activity relationships (SAR) around the intact cytisine template to gain insights into nAChR affinity and functional selectivity.1,16,17,22
3-Phenyl-cytisine 4 exhibited an affinity for the α4β2* subtype (Ki = 128 nM) what is about 1000 times lower than cytisine 1, but with enhanced subtype selectivity. The introduction of a heteroaryl substitutent provided compounds with high affinity for α4β2* (5 (3PC): Ki = 0.91 nM; 6: Ki = 3.9 nM). The pyridyl ring can function as an additional HBA system enhancing the affinity for α4β2* when comparing with ligand 4, but decreasing the subtype selectivity for e.g. α3β4 (5 (3PC): Ki = 119 nM; 6: Ki = 436 nM). Compound 7, bearing an additional 3,4-methylendioxy substituent at the phenyl ring, displayed similar affinity for α4β2* (Ki = 110 nM) like 3-phenyl-cytisine 4 (Ki = 128 nM) along with high subtype selectivity.
Compounds 30, 31, 36, and 37 (Table 2) can be directly compared to cytisine derivatives 4 – 7. They can be seen as simplified and more flexible cytisine analogs, but lacking the pyridone ring system of 1 (table 1). Compounds 30 and 37 (Ki = 57.6 and 13.2 nM, respectively) showed higher affinity for the α4β2* nAChR than their more rigid cytisine analogs 4 and 7 (Ki = 128 and 110 nM, respectively). In contrast, cytisine derivatives 5 and 6 (Ki = 0.91 and 3.9 nM) bearing a pyridine substituent displayed slightly higher affinity for the α4β2* nAChR than their more flexible bispidine analogs 31 and 36 (Ki = 11.2 and 14.4 nM, respectively). 3,7-Diazabicyclo[3.3.1]nonane based carboxamides can therefore provide easier synthetically accessible nAChR ligands with high α4β2* affinity and enhanced subtype selectivity.
3,7-Diazabicyclo[3.3.1]nonane based carboxamides with methylene spacer
The introduction of the methylene spacer into compound 29 (Ki = 454 nM for α4β2*) increased the affinity for the α4β2* subtype by about 8-fold (30; Ki = 58 nM). Compound 30 showed also high subtype selectivity. The perfluorinated compound 34 failed to interact with nAChRs tested. Other compounds bearing a methylene spacer (compounds 13, 31 and 35 – 38) between the amide bond and the (hetero)aryl substituents also exhibited high affinity in the low nanomolar range for the α4β2* nAChR. Compound 35 substituted with a methylsulfonyl moiety in the para position of the phenyl ring showed a Ki value of 22.9 nM for the α4β2* subtype, and no affinity for e.g. α3β4* nAChR. The 3-methyl-5-isoxazolyl derivative 13 (Ki = 46.9 nM) bearing a substituent which can be considered as a bioisosteric replacement of the phenyl ring displayed similar Ki values for α4β2*. For compounds 31 (3-pyridyl), 36 (4-pyridyl) and 37 (3,4-methylenedioxyphenyl) Ki values of 11.2 nM (31), 14.4 nM (36) and 13.2 nM (37), respectively, were obtained. These moieties have additional hydrogen bond acceptors which might be beneficial for increasing the affinity for α4β2* nAChR further. Compound 38 with the bulkier 2-naphthyl moiety exhibited a Ki value in the low nanomolar range (Ki = 10.2 nM) for this receptor subtype. These results indicate that the introduction of a methylene spacer is tolerated by the α4β2* subtype and can increase the affinity for this subtype.
3,7-Diazabicyclo[3.3.1]nonane based carboxamides with an ethylene, oxymethylene, or cyclopropyl spacer
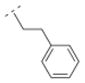
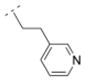
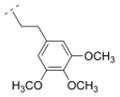
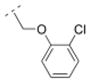
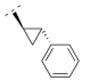
The homolog ethylene spacer in compound 32 did not change the affinity for α4β2* nAChR (Ki = 52.5 nM) comparing with the methylene spacer compound 30 (Table 2). Compound 32 also displayed an affinity for the α3β4* subtype (Ki = 395.8 nM), but no affinity for the α7* nAChR. The introduction of a 3-pyridyl ring (compound 39) increased the affinity for the α4β2* subtype only slightly (Ki = 39 nM), whereas the introduction of three methoxy groups in the meta- and the para positions (compound 14) resulted in a drop of affinity for this receptor subtype (Ki = 232.6 nM). Compound 33, bearing a 2-chloro aryl moiety and an oxymethylene spacer displayed lower affinity for the α4β2* subtype (Ki = 548 nM) compared to compound 32. This shows, that either a small electron withdrawing group in the ortho position of the aromatic ring or a heteroatom in the spacer motif can decrease affinity for α4β2* nAChR. In contrast, compound 15, bearing a cyclopropyl ring with a trans substitution pattern as a spacer motif displayed high affinity for the α4β2* nAChR (Ki = 1.2 nM), but also for the α3β4* subtype (Ki = 81.1 nM). The rigidity of the cyclopropyl spacer could be beneficial for high affinity for the α4β2* nAChR subtype, but different trans and cis arrangements needs to be explored in the future to evaluate the possibility to reduce α3β4* interaction and maintain α4β2* activity.
3,7-Diazabicyclo[3.3.1]nonane based carboxamides with a ethenylene spacer
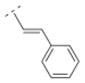
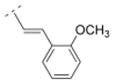
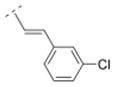
Bispidine derivative 16, with a cinnamic acid coupled to the bispidine backbone, showed affinity in the nanomolar range (Ki = 55.1 nM) for α4β2* nAChR, but lacked affinity for any of the other nAChR subtypes tested. This spacer motif exhibited more rigidity compared to its more flexible ethylene spacer (compound 32). The Ki values of both compounds (32 vs. 16) are almost identical (Ki = 55.1 vs. 52.5 nM, respectively). The introduction of an electron donating methoxy substituent into the ortho position of the phenyl ring (compound 17) showed lower affinity for the α4β2* subtype (Ki = 171.8 nM) compared to its unsubstituted cinnamic acid derivative 16. However, when small electron withdrawing groups were introduced in the meta (chloro: 18, bromo: 19, nitro: 20) or para position (fluoro: 40), compounds with higher affinity for the α4β2* subtype (18: Ki = 23.7 nM; 19: Ki = 24.6 nM; 20: Ki = 38.9 nM; 40: Ki = 20.7 nM) were obtained. Also, compound 21 with an electron donating methoxy group in the para position displayed a slightly higher affinity (Ki = 39.6 nM) for the α4β2* subtype compared to compound 16. So, small electron withdrawing groups in the meta or para position increase the affinity for the α4β2* subtype and it seems that the position of the small electron withdrawing or donating substituent might be more important than its electronic properties.
The affinity for the α4β2* nAChR subtype was decreased when the phenyl ring of compound 16 was replaced by smaller five-membered, aromatic rings systems, such as furanyl (compound 22) or thiophenyl (compounds 23 and 24). Ki values were 287.5 nM for the 2-furanyl (22), 96.9 nM for the 2-thiophenyl (23), and 57.1 nM for the 3-thiophenyl derivative (24). When the phenyl ring of compound 16 was replaced by a 3-pyridyl (41) or a 4-pyridyl moiety (25), the affinity increased for the α4β2* subtype (41: Ki = 32.6 nM; 25: 1.03 nM). The Ki values of the 3-pyridyl compound 41 and its less rigid counterpart (compound 39, Ki = 39 nM) are very similar for α4β2*, indicating that both spacer motifs allow similar orientation in the binding pocket. When the simple phenyl moiety of compound 16 was extended by a fused 3,4-methylendioxy moiety (compound 26), a Ki value of 36.9 nM was obtained for the α4β2* nAChR subtype. The extension to a 1-naphthyl ring system (compound 27), however, decreased the affinity for the α4β2* subtype (Ki = 296.6 nM).
3,7-Diazabicyclo[3.3.1]nonane based carboxamides with an ethynylene spacer
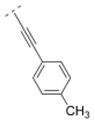
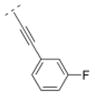
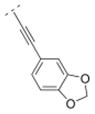
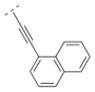
Compound 42 displayed a 5-times higher affinity for the α4β2* nAChR subtype (Ki = 10.9 nM) than its more flexible analogs 32 and 16. The introduction of an electron donating methoxy group (compound 43) caused a drop in affinity for α4β2* (Ki = 241.9 nM). A similar observation was made for compound 17, which means that ortho substitutions are less tolerated by the receptor. Small electron donating or withdrawing groups at meta and/or para positions (compounds 44–50) increased the affinity for the α4β2* subtype (up to about 11 fold). E.g. a methoxy group in the meta or para position increased the affinity slightly (44: Ki = 6.3 nM; 45: Ki = 7.8 nM) and a di-substitution (compound 46) resulted in an even higher affinity for the α4β2* subtype (Ki = 2.1 nM). The combination of a 3-chloro/4-methoxy substitution (compound 47) resulted in a compound with the highest affinity in this compound library (Ki = 0.991 nM). Compound 48, bearing a 4-methyl group displayed the same affinity (Ki = 10.9 nM) for the α4β2* nAChR subtype as the unsubstituted compound 42. 3-Fluoro substitution (compound 49) also provided a ligand with high affinity for α4β2* (Ki value of 7.1 nM). The affinity of compound 50 (3,4-methylendioxy substituent; Ki = 3.13 nM) is similar to its ring open 3,4-dimethoxy analog 46 (Ki = 2.1 nM), and both compounds possess about a 10-fold higher affinity than its analog bearing a vinyl spacer (26). A 1-naphthyl moiety (compound 51) decreased affinity for the α4β2* subtype (K i = 46 nM) compared to compound 42. A similar observation has been made with compounds 27 vs. 16. In summary, bispidine compounds bearing an ethynylene spacer moiety along with substitutions in the meta- and/or para position are well tolerated by the α4β2* nAChR subtype.
3,7-Diazabicyclo[3.3.1]nonane based carboxamides with an (hetero)aryl spacer
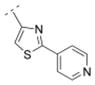
As previously reported, the biphenyl derivative 28 has a Ki value of 39.9 nM for α4β2* nAChR, and about 1,100 nM for the α3β4* subtype.7 In line with the observations for compounds 31 and 36 vs. 30, 39 vs. 32, and 41 and 25 vs. 16, the replacement of the phenyl moiety by 3- or 4-pyridyls increased the affinity for the α4β2* nAChR subtype (3-pyridyl-phenyl derivative 52: Ki = 25 nM; 4-pyridyl-phenyl derivative 53: Ki = 7.1 nM). Additionally, the replacement of the phenyl ring by an imidazolyl ring (compound 54) also increased affinity for the α4β2* subtype (Ki = 5.6 nM). The phenyl group is tolerated as a spacer motif for α4β2* nAChRs. Compound 55, where a 4-pyridyl substituent was connected via a thiazole spacer with the bispidine amide backbone, did not show any affinity for nAChRs. Similar observations have been made on the previous set of bispidine compounds, when heteroatoms in close proximity to the HBA system caused a dramatic drop in affinity.7
In summary, various spacer moieties like methylene, ethylene, ethenylene, ethynylene or phenyl, between the HBA motif and (hetero)aryl moieties are tolerated by the α4β2* nAChR subtype, even with high affinity and subtype selectivity.
Most compounds showed partial agonism/antagonism in initial characterization assays using previously described electrophysiological experiments with diverse nAChRs expressed in Xenopus oocytes (Figure 2).7,23–25 Compounds 35, 15, 46, 47, 50, 52, 53, and 54 produced the strongest antagonistic effects for α4β2* receptors. Compound 35 was further evaluated and recently discovered as an in vivo active, highly selective agent with antidepressive effect in a mouse model.26 Compound 15 was the most potent agonist at α3β4* in this compound series. No strong effects were observed for α7 subtype. Compound 46 had some antagonistic effect at the muscle subtype. In general, spacer motifs lead to compounds with partial agonist/antagonist profiles with the strongest effects observed for α4β2* nAChRs. In contrast, the cyclopropyl spacer (trans form) produced compound 15 with an additional α3β4* agonistic profile.
Figure 2.

Responses of oocytes expressing diverse nAChR subtypes to 1 or 10 μM of selected compounds (numbering are following the sequence in the tables) relative to ACh control responses. Responses of oocytes expressing diverse nAChRs to compounds co-applied at 1 μM with ACh compared to responses to ACh alone. Bars above zero indicate additive effects; bars below zero indicate reduced responses.
2.3. Physicochemical properties and drug-likeness
Physicochemical properties and druglikeness parameters ClogP, TPSA, and logBB were calculated using ACD/ADME Suite 5.0 (ACD/Labs) software. Most compounds are in the range for CNS druglikeness parameter values (Mr: 250 and 350; ClogP: between 1–3; PSA < 75). Additional parameters important for CNS compounds have been calculated (Table 3; supplementary data) for those compounds showing nanomolar affinity for α4β2* (Ki < 1,000 nM).27 Most active compounds showed “good” values (see Table 3; supplementary data). There were no correlations between affinity and ClogP or TPSA values or rotatable bonds. Most compounds have 2–3 rotatable bonds due to their spacer motif which had an influence on their functionality like stated above. For the likelihood of blood brain barrier (BBB) penetration, logBB values were calculated can be from logP and TPSA values where logBB values below −0.5 would reflect very poor or no BBB penetration and > 0.7 very high penetrants. 3,7-diazabicyclo[3.3.1]nonane carboxamides bearing the ethenylene or ethynylene spacer could function as Michael acceptors, properties which should be avoided for potential drug candidates for chronic treatment. Results derived from these compounds serve as a basis for further compound profiling projects and therefore an important part in this early SAR study.
3. Conclusions
In summary, we synthesized and evaluated a small set of cytisine (4–7) derivatives and a series of 3,7-diazabicyclo[3.3.1]nonane carboxamide (13–55) for their affinities at various nAChRs. Selected compounds were screened for agonist and antagonist functionality. Most compounds displayed α4β2* subtype selectivity regarding their affinities measured.
Cytisine derivatives 4–7 were compared with the four analogously substituted bispidine derivatives 30, 31, 36, and 37. Their affinities for the α4β2* nAChR were in the same range. It seems that the impact of flexibility/rigidity on α4β2* affinity is of minor importance. The introduction of an additional hydrogen bond acceptor (HBA) motif, e.g. pyridyl groups, increased the affinity for the α4β2* subtype in both sets of derivatives.
The incorporation of spacer moieties like methylene, ethylene, ethenylene, ethynylene or phenyl at the 3,7-diazabicyclo[3.3.1]nonane carboxamide template increased the affinity for the α4β2* nAChR. Heteroaryl substituents with HBA functionality increased the affinity for α4β2* nAChR further.
Compounds 15, 25, and 47 with Ki values of about 1 nM showed the highest affinities for α4β2* nAChR.
From the electrophysiology point of view, all evaluated compounds displayed partial agonism or antagonism at α4β2*, reduced or no effects on α3β4* with the exception of compound 15 (agonist), and reduced or nor effect at α7 and muscle type.
4. Experimental section
All reagents and solvents were obtained from various suppliers (ABCR, Acros, Aldrich, Alfa Aesar, Fluka, Merck or Sigma) and used without further purification unless otherwise noted. Dichloromethane was freshly distilled from calcium hydride. Methanol was treated with sodium, distilled afterwards and stored under nitrogen. Sodium wires were used to dry diethyl ether, petroleum ether, tetrahydrofuran, and toluene. Water was taken from a water purification system PureLab Plus UV (ELGA Labwater) or Direct-Q™ 5 (Millipore). Amines were purified prior to use with a Kugelrohr distillation apparatus (Büchi). Reactions were monitored by thin-layer chromatography (TLC) using aluminum sheets coated with silica gel 60 F254 (Merck). Compounds were visualized using UV light (254 or 365 nm) and using a KMnO4 (1 %). Column chromatography was carried out on silica gel (0.035–0.060 nm) using different mixtures of CH2Cl2 with MeOH (40:1, 20:1 or 9:1) or of PE with EtOAc (4:1 or 3:1) as mobile phases. 1H NMR spectra (400 or 500 MHz) and 13C NMR spectra (100 or 125 MHz) were recorded on an Avance 400 or on an Avance 500 NMR spectrometer (Bruker). All NMR spectra were recorded at rt. Chemical shifts (δ) are given in parts per million (ppm) relative to the remaining protons of the deuterated solvents used as internal standard. Coupling constants J are given in Hertz (Hz) and spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet) and br (broad). Mass spectra were recorded on an API 2000 mass spectrometer with an electron spray ionization source (Applied Biosystems) coupled to an Agilent 1100 HPLC system (LC/ESI-MS) or on a Varian 500-MS mass spectrometer (ESI-MS). The purity of the compounds was determined by LC/ESI-MS or a Shimadzu Prominence HPLC system at an appropriate wavelength. HRMS runs were performed on Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS. All compounds proved to possess ≥95 % purity. Melting points were determined in open capillary tubes with a melting point apparatus (Weiss-Gallenkamp) or with a Melting Point B-540 (Büchi) and are uncorrected. Infrared spectroscopy was performed with a Tensor-27 FTIR infrared spectrometer (Bruker Optic) using KBr pellet or with a Nicolet iS10 (Thermo Scientific). Elemental microanalyses (C, H, N) were performed with a VarioEL apparatus (Elementar Analysensysteme) or a Costech elemental combustion apparatus and the determined values are generally within ±0.4 % of the theoretical values. Hydrogen for hydrogenations was produced by a Hogen GC hydrogen generator (Proton Energy Systems) or by a 60H hydrogen generator (Parker, domnick hunter). Lyophilizations were performed with an Alpha 1–4 LSC freeze dryer (Martin Christ).
4.1. General procedure A: Suzuki cross-coupling reaction with 3-bromo-N-tboc-cytisine
3-Bromo-N-tboc-cytisine (100 mg, 0.27 mmol), the appropriate boronic acid (0.41 mmol), a base (0.6 mmol), DME (3 mL) and H2O (1 mL) were added into a 10 mL microwave glass tube. The solution was washed with argon for 10 min. After the addition of Pd(PPh3)4 (30 mg, 0.027 mmol) the reaction vessel was sealed with a septum and placed into the microwave cavity. Microwave irradiation of 30 W was used and the temperature ramped from rt to 80 °C. Once 80 °C was reached the reaction mixture was held for 30 to 90 min. before the mixture was allowed to cool to rt. The reaction vessel was opened and the solvents were evaporated under reduced pressure. The brown residue was extracted on a C-18 SPE column eluting with a mixture of MeOH/H2O 70:30 or 60:40 v/v and the aqueous solution was concentrated under reduced pressure, subsequently. The residue was purified on a preparative HPLC system using RP C-18 silica gel and appropriate MeOH/H2O gradients. The chromatograms were scanned at 254 nm and the appropriate fractions were collected. The fractions containing the desired products were concentrated under reduced pressure on a rotary evaporator.
4.2. General procedure B: Cleavage of the N-tboc protecting group from N-tboc protected cytisine derivatives
The concentrated aqueous solution of the N-tboc protected cytisine derivative (approx. 70 mL) was put into a 80 mL microwave glass tube, sealed and placed into the microwave cavity. Microwave irradiation of 150 W was used and the temperature ramped from rt to 150 °C. Once 150 °C was reached the reaction mixture was held for 30 min. before the mixture was allowed to cool to rt. The reaction vessel was opened and the solvent was evaporated by lyophilization for at least 24 h.
4.3. Isolation of (1R,5S)-1,2,3,4,5,6-hexahydro-1,5-methano-8H-pyrido[1,2-a][1,5]diazocin-8-one [(-)-Cytisine] (1)
Seeds and pods of Laburnum anagyroides watereri were collected in the Cologne-Bonn area (Germany) in the months September and October. The plant material was air-dried at least for 3 months and ground to a powder consistence. The plant material was extracted with CH2Cl2/MeOH/aq. NH3 (10:4:1) through homogenization by Ultra-turrax for 8 hours. The evaporated solvents were replaced, exactly the same amounts of each solvent were added to the homogenate during the extraction. The homogenate was centrifuged (2,000 x min, 40 min) and the supernatant collected. The dark green solution was concentrated under reduced pressure to the final volume of 500 ml and extracted with 1M HCl (3 x 100 ml). The aqueous acid solution was rendered alkaline with 26% NH4OH (pH 11–12) and the free base extracted with CH2Cl2 (10 x 100 ml). The organic layers were collected and the solvent evaporated in vacuo. The dark green/brownish residue was purified by column chromatography on silica gel column with CHCl3/MeOH (6:1). The alkaloid 1 was recrystallized from perchloroethylene or directly used in the next step (synthesis of N-tboc-cytisine 2). Isolated yields ranged from 0.11 to 0.48 % of 1 calculated from the dry weight; mp 155–156 °C. 1H NMR (500 MHz, CDCl3) δ 1.65 (br s, 2H), 2.03 (br s, 1H), 2.62 (br s, 1H), 2.70–2.75 (m, 4H), 3.57 (dd, J = 15.7, 6.6 Hz, 1H), 3.77 (d, J = 15.4 Hz, 1H), 5.77 (d, J = 6.9 Hz, 1H), 6.17 (d, J = 9.1 Hz, 1H), 7.05 (dd, J = 9.1, 6.9 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 25.6, 27.0, 34.9, 49.1, 52.3, 53.3, 104.2, 115.7, 138.1, 150.7, 162.8. LC/ESI-MS: positive mode m/z = 190.9 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3316, 3282, 3084, 3032, 1650, 1541, 1443, 1141, 821, 737. Anal. (C11H14N2O*0.25H2O) C, H, N.
4.4. 8-oxo-1,5,6,8-tetrahydro-2H,4H-1,5-methano-pyrido[1,2-a][1,5]diazocine-3-carboxylic acid tert-butyl ester [N-tboc-cytisine] (2)
Cytisine 1 (500 mg, 2.63 mmol), di-tert-butyl dicarbonate (688 mg, 3.15 mmol, 1.2 eq) and Na2CO3 (334 mg, 3.15 mmol, 1.2 eq) were stirred in 25 ml CH2Cl2 and 6 ml H2O at 60 °C for 2 hours. The reaction mixture was allowed to cool to rt and 10 ml of concentrated NaCl solution was added. The organic layer was dried over MgSO4 and the solvent was evaporated. Product 2 was recrystallized from PE and obtained as an off-white crystalline powder (590–690 mg, 77–90 %); mp 149–150 °C. 1H NMR (500 MHz, CDCl3) δ 1.30 (s, 9H), 1.87 (m, 1H), 1.93 (m, 1H), 2.38 (br s, 1H), 2.94–3.05 (m, 3H), 3.79 (dd, J = 15.7, 6.6 Hz, 1H), 4.00–4.19 (m olv, 2H), 4.14 (d, J = 15.4 Hz, 1H), 6.03 (br s, 1H), 6.41 (d, J = 9.1 Hz, 1H), 7.24 (dd, J = 9.1, 6.3 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 26.1, 27.5, 28.0, 34.8, 48.9, 50.5, 51.6, 80.3, 105.8, 117.1, 138.9, 148.7, 154.5, 163.4. LC/ESI-MS: positive mode m/z = 291.3 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3217, 3099, 1687, 1654, 1465, 1445, 1421, 1364, 818, 760, 572. Anal. (C16H22N2O3) C, H, N.
4.5. 9-bromo-8-oxo-1,5,6,8-tetrahydro-2H,4H-1,5-methano-pyrido[1,2-a][1,5]diazocine-3-carboxylic acid tert-butyl ester [3-Bromo-N-tboc-cytisine] (3)
N-tboc-cytisine 2 (1 g, 3.44 mmol) and N-bromosuccinimide (613 mg, 3.44 mmol, 1 eq) were stirred in 30 ml CH2Cl2 at 60°C for 2 hours. The reaction mixture was allowed to cool to rt and the solvent was evaporated in vacuo. The oily residue was dissolved in 150 ml MeOH/H2O (60:40) and the two isomers were separated and purified by preparative HPLC. Product 3 was obtained in 38–52 % yield as a white crystalline powder; mp 131 °C. 1H NMR (500 MHz, CDCl3) δ 1.30 (s, 9H), 1.94 (t, J = 13.2 Hz, 2H), 2.40 (br s, 1H), 2.99–3.06 (m, 3H), 3.85 (dd, J = 15.5, 6.3 Hz, 1H), 4.06–4.35 (m ovl, 2H), 4.23 (d, J = 15.8 Hz, 1H), 5.96 (br s, 1H), 7.64 (dd, J = 7.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 26.0, 27.4, 28.0, 34.7, 49.2, 50.2, 51.4, 80.6, 105.7, 112.5, 140.8, 148.5, 154.4, 159.4. LC/ESI-MS: positive mode m/z = 369.0 and 371.0 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3092, 2974, 1692, 1648, 1587, 1423, 1365, 1237, 1129, 760. Anal. (C16H21BrN2O3*0.25H2O) C, H, N.
4.6. 9-phenyl-1,2,3,4,5,6-hexahydro-1,5-methano-pyridino[1,2-a]diazocin-8-one [3-Phenyl-cytisine] (4)
The Suzuki reaction was performed according to general procedure A with 3-bromo-N-tboc-cytisine 3 (100 mg, 0.27 mmol), phenylboronic acid (50 mg, 0.41 mmol), Na2CO3 (64 mg, 0.6 mmol), Pd(PPh3)4 (30 mg, 0.027 mmol), DME and H2O. The reaction time was 30 min. For the SPE purification a mixture of MeOH/H2O 70:30 v/v (100 mL) was used. After the HPLC purification, general procedure B was used for the N-tboc deprotection. Final product 4 was obtained as a white solid (42 mg, 58 %). mp 139.8–140.6 °C. 1H NMR (500 MHz, CDCl3) δ 1.96 (br s, 2H), 2.34 (br s, 1H), 2.91 (br s, 1H), 3.02 (br d, J = 12.3 Hz, 2H), 3.07 (dd, J = 12.3, 2.2 Hz, 1H), 4.19 (d, J = 15.5 Hz, 1H), 6.09 (d, J = 7.2 Hz, 1H), 7.29 (tt, J = 7.2, 1.3 Hz, 1H), 7.38 (t, J = 7.2 Hz, 2H), 7.46 (d, J = 7.2 Hz, 1H), 7.69 (dt, J = 7.2, 1.3 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 26.3, 27.9, 35.7,50.2, 53.0, 54.0, 105.0, 127.2, 127.4, 128.0, 128.6, 137.0, 137.4, 150.3, 162.1. HRMS (EI) calcd for C17H18N2O 266.1419, found 266.1426.
4.7. 9-pyridin-3-yl-1,2,3,4,5,6-hexahydro-1,5-methano-pyridino[1,2-a]diazocin-8-one [3-(Pyridin-3′-yl)cytisine] (5)
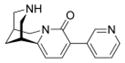
The Suzuki reaction was performed according to general procedure A with 3-bromo-N-tboc-cytisine 3 (100 mg, 0.27 mmol), 3-pyridineboronic acid (49 mg, 0.41 mmol), K3PO4 (126 mg, 0.6 mmol), Pd(PPh3)4 (30 mg, 0.027 mmol), DME and H2O. The reaction time was 60 min. For the SPE purification a mixture of MeOH/H2O 60:40 v/v (100 mL) was used. After the HPLC purification, general procedure B was used for the N-tboc deprotection. Final product 5 was obtained as a yellow solid (48 mg, 66 %). mp 79.8–81.6 °C. 1H NMR (500 MHz, CDCl3) δ 1.96 (br s, 2H), 2.36 (br s, 1H), 2.95 (br s, 1H), 2.99–3.03 (m, 2H), 3.06 (dd, J = 12.0, 2.2 Hz, 1H), 3.11 (d, J = 12 Hz, 1H), 3.94 (dd, J = 15.6, 6.9 Hz, 1H), 4.16 (d, J = 15.6 Hz, 1H), 6.11 (d, J = 7.3 Hz, 1H), 7.29 (ddd, J = 7.9, 4.7, 0.9 Hz, 1H), 7.49 (d, J = 7.3 Hz, 1H), 8.16 (ddd, J = 7.9, 2.2, 1.6 Hz, 1H), 8.49 (dd, J = 4.7, 1.6 Hz, 1H), 8.78 (d, J = 1.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 26.2, 27.8, 35.7, 50.3, 53.0, 53.9, 105.0, 122.8, 123.9, 133.2, 136.1, 137.2, 148.2, 149.1, 151.5, 161.9. HRMS (EI) calcd for C16H17N3O 267.1371, found 267.1376.
4.8. 9-pyridin-4-yl-1,2,3,4,5,6-hexahydro-1,5-methano-pyridino[1,2-a]diazocin-8-one) [3-(Pyridin-4′-yl)cytisine] (6)
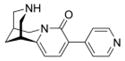
The Suzuki reaction was performed according to general procedure A with 3-bromo-N-tboc-cytisine 3 (100 mg, 0.27 mmol), 4-pyridineboronic acid (49 mg, 0.41 mmol), K3PO4 (126 mg, 0.6 mmol), Pd(PPh3)4 (30 mg, 0.027 mmol), DME and H2O. The reaction time was 90 min. For the SPE purification a mixture of MeOH/H2O 60:40 v/v (100 mL) was used. After the HPLC purification, general procedure B was used for the N-tboc deprotection. Final product 6 was obtained as a yellow solid (45 mg, 62 %). mp n.d. °C. 1H NMR (500 MHz, CDCl3) δ 1.96 (br s, 2H), 2.36 (br s, 1H), 2.94 (br s, 1H), 3.00 (ddd, J = 12.3, 2.5, 1.2 Hz, 1H), 3.04 (d, J = 12.3 Hz, 1H), 3.07 (dd, J = 12.3, 2.5 Hz, 1H), 3.11 (dd, J = 12.3, 2.5 Hz, 1H), 3.95 (dd, J = 15.8, 6.6 Hz, 1H), 4.16 (d, J = 15.8 Hz, 1H), 6.13 (d, J = 7.4 Hz, 1H), 7.57 (d, J = 7.4 Hz, 1H), 7.67 (dd, J = 6.1, 1.6 Hz, 2H), 8.57 (dd, J = 6.1, 1.6 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 26.2, 27.8, 35.8, 50.3, 53.0, 53.9, 104.9, 122.8, 123.9, 137.8, 144.9, 149.6, 152.6, 161.5. HRMS (EI) calcd for C16H17N3O 267.1371, found 267.1379.
4.9. 9-(benzo[1,3]dioxol-5-yl)-1,2,3,4,5,6-hexahydro-1,5-methano-pyridino[1,2-a]diazocin-8-one) [3-(3′,4′-Methylenedioxyphenyl)cytisine] (7)
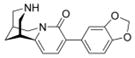
The Suzuki reaction was performed according to general procedure A with 3-bromo-N-tboc-cytisine 3 (100 mg, 0.27 mmol), 3,4-methylenedioxyphenylboronic acid (68 mg, 0.41 mmol), Na2CO3 (64 mg, 0.6 mmol), Pd(PPh3)4 (30 mg, 0.027 mmol), DME and H2O. The reaction time was 30 min. For the SPE purification a mixture of MeOH/H2O 70:30 v/v (100 mL) was used. After the HPLC purification, general procedure B was used for the N-tboc deprotection. Final product 7 was obtained as an off-white solid (30 mg, 36 %). mp 259.1–261.6 °C. 1H NMR (500 MHz, CDCl3) δ 1.95 (br s, 2H), 2.34 (br s, 1H), 2.90 (br s, 1H), 3.00 (br d, J = 12.0 Hz, 2H), 3.06 (dd, J = 12.0, 2.2 Hz, 1H), 3.11 (d, J = 12.0 Hz, 1H), 3.93 (dd, J = 15.7, 6.6 Hz, 1H), 4.16 (d, J = 15.7 Hz, 1H), 5.94 (br s, 2H), 6.05 (d, J = 7.3 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 7.12 (dd, J = 8.2, 1.6 Hz, 1H), 7.26 (d, J = 1.6 Hz, 1H), 7.38 (d, J = 7.3 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 26.4, 27.9, 35.7, 50.2, 53.0, 54.0, 100.9, 104.9, 108.0, 109.4, 122.1, 127.1, 131.4, 136.4, 146.8, 147.3, 149.9, 162.1. HRMS (EI) calcd for C18H18N2O3 310.1317, found 310.1324.
4.10. General procedure C: Synthesis of N-tboc protected bispidinecarboxamides
Methyl iodide (570 mg, 4 mmol) was added to a stirred solution of 12 (320 mg, 1 mmol) dissolved in dry MeCN (2 mL) and dry THF (2 mL) at rt. The volatiles were removed under reduced pressure after 24 h, the residue was dissolved in dry MeCN (4 mL), and Et3N (101 mg, 1 mmol) and the appropriate carboxylic acid (1 mmol) were added. The solution was allowed to stir at rt for 12–120 h before the volatiles were removed under reduced pressure. The residue was purified by flash chromatography (silica gel, mixtures of CH2Cl2 and MeOH - 40:1, 20:1 or 9:1).
4.11. General procedure D: Synthesis of N-tboc protected bispidinecarboxamides
The appropriate carboxyl chloride (1 mmol), either neat or dissolved in dry toluene (1–2 mL), was added dropwise to a stirred solution of N-tboc-bispidine 11 (230 mg, 1 mmol) and Et3N (101 mg, 1 mmol) in dry toluene (5 mL) at rt. The volatiles were removed under reduced pressure after 2 h and the residue was purified by flash chromatography (silica gel, mixtures of CH2Cl2 and MeOH - 40:1, 20:1 or 9:1).
4.12. General procedure E: Synthesis of N-tboc protected bispidinecarboxamides
The appropriate carboxylic acid (1 mmol) and N-tboc-bispidine 11 (248 mg, 1.1 mmol) were dissolved in dry CH2Cl2 (5 mL) and cooled to 0 °C. DMAP (6.1 mg, 0.05 mmol) and DCC (206 mg, 1 mmol) were added and the mixture was allowed to warm up and stir at rt. The precipitate was filtered off after 12 h and washed with cold CH2Cl2 (2 mL) The solvent of the filtrate was evaporated under reduced pressure and the residue was purified by flash chromatography (silica gel, mixtures of CH2Cl2 and MeOH - 40:1. 20:1 or 9:1).
4.13. General procedure F: Cleavage of the N-tboc protecting group from N-tboc protected bispidine derivatives
HCl in 1,4-dioxane (4 M, 4–5 mL) was added to a stirred solution of the N-tboc protected bispidine derivative (0.2 – 1.0 mmol), dissolved in 4–5 mL of 1,4-dioxane, and the mixture was allowed to stir at rt for 2–12 h. The volatiles were removed under reduced pressure and before the residue was dissolved in KOH solution (0.25 M, 20 mL) and extracted with of CH2Cl2 (3–5 x 20 mL). The combined organic layers were washed with saturated NaHCO3 solution (10 mL), water (10 mL), dried with MgSO4 and filtered. The product was obtained after evaporating the solvent under reduced pressure.
4.14. General procedure G: Cleavage of the N-tboc protecting group from N-tboc protected bispidine derivatives
The N-tboc protected bispidine derivative (0.2 – 1.0 mmol) was dissolved in dry CH2Cl2 (5 mL), anhydrous ZnBr2 (2–3 equiv.) was added, and the mixture was allowed to stir at rt for 12–120 h. After the removal of the volatiles under reduced pressure the residue was dissolved in KOH solution (0.25 M, 20 mL) and extracted with CH2Cl2 (5 x 20 mL). The combined organic layers were washed with saturated NaHCO3 solution (10 mL), water (10 mL), dried over MgSO4, and filtered. The product was obtained after evaporating the solvent under reduced pressure.
4.15. General procedure H. Formation of fumaric acid salts of bispidine derivatives
The amine (0.2 – 1.0 mmol) was dissolved in a mixture of Et2O and MeOH (9:1, 2–5 mL). A saturated solution of fumaric acid in the same mixture of solvents was added dropwise to a stirred solution of the amine until no further precipitation was observed. The solution was kept at 4–8 °C overnight before the precipitate was filtered off and washed with the same mixture of solvents (2 x 5 mL) and dry Et2O (5 mL). The solid was dissolved in water (20–30 mL), and freeze-dried.
4.16. General procedure I. Formation of fumaric acid salts of bispidine derivatives
The amine (0.2 – 1.0 mmol) was dissolved in isopropanol (3–5 mL), filtered, and heated to 70–80 °C. The same molar amount of fumaric acid was dissolved in isopropanol (3 mL) and also heated to 70–80 °C. The two solutions were combinedf and allowed to cool to rt. Dry Et2O (10 mL) were added and the mixture was kept at 4–8 °C overnight. The solid was filtered off, washed with dry Et2O (3 x 5 mL), dissolved in water (20–30 mL), and freeze-dried.
4.17. (1R,5S)-tert-butyl 7-benzyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3-carboxylate (9)
Compound 9 was obtained following the synthetic procedures published by Stead et al.{{2 Stead,D. 2005;}} A solution of tert-butyl 4-oxopiperidine-1-carboxylate 8 (10.0 g, 50.2 mmol), acetic acid (2.9 mL, 50.9 mmol), and benzylamine (5.5 mL, 50.4 mmol) in methanol (40 mL) was added dropwise to a stirred suspension of paraformaldehyde (3.32 g, 110.6 mmol). The resulting mixture was heated under reflux for 1 h before another portion of paraformaldehyde (3.32 g, 110.6 mmol) was added. This mixture was heated at reflux for additional 5 h before the reaction was allowed to cool to rt and the solvent was evaporated under reduced pressure. The residue was dissolved in Et2O (150 mL) and washed with aqueous KOH solution (1 M, 2 x 80 mL). The combined aqueous layers were extracted with Et2O (3 x 50 mL). The combined organic layers were dried with MgSO4, filtered, and the solvent was evaporated under reduced pressure. Flash column chromatography (silica gel, PE:EtOAc 3:1) of the residue afforded a white solid 9 (13.0 g, 78 %) after extensive evaporation of the solvents; mp 83°C. 1H NMR (500 MHz, CDCl3) δ 1.53 (s, 9H), 2.42 (br m, 2H), 2.70 (br m, 2H), 3.17 (br m, 2H), 3.27 (br d, J = 12.5 Hz, 1H), 3.35 (br d, J = 12.5 Hz, 1H), 3.52 (br m, 2H), 4.41 (br d, J = 12.9 Hz, 1H), 4.57 (br d, J = 12.9 Hz, 1H), 7.24–7.38 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 28.7, 47.7, 50.0, 50.6, 58.8, 59.1, 62.0, 80.2, 127.4, 128.5, 128.9, 137.5, 154.9, 213.6. LC/ESI-MS: positive mode m/z = 331.3 ([M + H]+). Purity (> 98.5 %). IR (KBr, cm−1) 1731, 1695. Anal. (C19H26N2O3) C, H, N.
4.18. (1R,5S)-tert-butyl 7-benzyl-3,7-diazabicyclo[3.3.1]nonane-3-carboxylate (10)
A solution of 9 (18.9 g, 57.2 mmol), NaOH (10.0 g, 250 mmol), and hydrazine hydrate (80 %, 10.0 mL, 160 mmol) in 150 mL of diethylene glycol was heated at 125 °C under reflux conditions. After 2 h the reflux condenser was exchanged for a Dean-Stark apparatus and the mixture was heated at 140 °C for additional 8 h. After cooling to rt 250 mL of water were added and the resulting mixture was extracted with toluene (4 x 150 mL). The combined organic layers were washed with saturated solution (1 x 30 mL), water (2 x 30 mL), dried with NaHCO3MgSO4 and filtered. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography (silica gel, PE:EtOAc 4:1) to afford 10 as a white solid (13.2 g, 73 %); mp 66 °C. 1H NMR (500 MHz, CDCl3) δ 1.52 (s, 9H), 1.61 (m, 1H), 1.66 (m, 1H), 1.79 (br s, 1H), 1.87 (br s, 1H), 2.16 (br d, J = 10.9 Hz, 1H), 2.22 (br d, J = 10.9 Hz, 1H), 2.89 (br d, J = 10.8 Hz, 1H), 2.99 (br d, J = 10.9 Hz, 1H), 3.05 (ddd, J = 13.1, 3.9, 1.7 Hz, 1H), 3.10 (ddd, J = 13.1, 3.9, 1.7 Hz, 1H), 3.30 (d, J = 13.5 Hz, 1H), 3.44 (d, J = 13.5 Hz, 1H), 3.99 (br d, J = 13.1 Hz, 1H), 4.16 (br d, J = 13.1 Hz, 1H), 7.19–7.34 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 28.9, 29.2, 31.3, 47.7, 48.6, 58.9, 59.2, 63.7, 78.9, 126.8, 128.2, 128.7, 139.1, 155.2. LC/ESI-MS: positive mode m/z = 317.1 ([M + H]+). Purity (> 99.5 %). IR (KBr, cm−1) 1681. Anal. (C19H28N2O2) C, H, N.
4.19. (1R,5S)-tert-butyl 3,7-diazabicyclo[3.3.1]nonane-3-carboxylate fumaric acid salt (11F)
200 mg of Pd/C (5 %) was added to a solution of 10 (1.5 g, 4.74 mmol) in MeOH (7 mL) and the mixture was allowed to react under an atmosphere of hydrogen (10 psi) at rt for 4 h. After the mixture was filtered and washed thoroughly with MeOH the solvent was evaporated under reduced pressure. The product was obtained as a clear oil (1.05 g, 4.64 mmol) in 98 % yield. The free amine 10 was used for further syntheses. General procedure H was used to transfer this clear oil 10 (161 mg, 0.71 mmol) into a white solid, its fumaric acid salt 10F (200 mg, 82 %); mp 170 °C (dec). 1H NMR (500 MHz, D2O) δ 1.47 (s, 9H), 1.86 (br d, J = 13.5 Hz, 1H), 1.96 (br d, J = 13.5 Hz, 1H), 2.25 (br s, 2H), 3.17 (br d, J = 13.2 Hz, 2H), 3.31 (br d, J = 13.2 Hz, 2H), 3.48 (br d, J = 13.2 Hz, 2H), 4.05 (br d, J = 13.2 Hz, 2H), 6.80 (s, 2.0H). 13C NMR (125 MHz, MeOD) δ 28.2, 30.3, 30.5, 50.7, 50.9, 85.3, 137.6, 161.0, 174.4. LC/ESI-MS: positive mode m/z = 226.9 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3438, 1691, 1657, 985. Anal. (C12H22N2O2*1.0C4H4O4*0.1H2O) C, H, N.
4.20. (1R,5S)-tert-butyl 7-(1H-imidazole-1-carbonyl)-3,7-diazabicyclo[3.3.1]nonan-3-carboxylate (12)
N-tBoc-bispidine 11 (1.5 g, 6.63 mmol) was dissolved in dry THF (20 mL) and CDI (1.18 g, 7.29 mmol) was added. The solution was refluxed for 2 h before the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (silica gel, mixture of CH2Cl2 and MeOH - 20:1). Evaporation of the solvents afforded a white solid 12 (1.98 g, 6.2 mmol, 93 %); mp 143 °C (dec). 1H NMR (500 MHz, CDCl3) δ 1.43 (s, 9H), 1.87 (s, 2H), 1.96 (br s, 2H), 3.01 (br m, 1H), 3.09 (br m, 1H), 3.26 (br s, 2H), 3.97 (br s, 1H), 4.25 (br m, 3H), 7.07 (s, 1H), 7.33 (s, 1H), 7.89 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 27.8, 28.5, 31.2, 47.4, 48.8, 50.2, 51.8, 80.3, 118.0, 129.4, 137.0, 151.8, 155.0. LC/ESI-MS: positive mode m/z = 321.1 ([M + H]+), negative mode m/z = 319.9 ([M – H]−). Purity (> 99.8 %). IR (KBr, cm−1) 1675. Anal. (C16H24N4O3) C, H, N.
4.21. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(3-methylisoxazol-5-yl)ethanone fumaric acid salt (13F)
The N-tboc protected compound was obtained by using the general procedure C with 3-methyl-5-isoxazoleacetic acid (141 mg, 1 mmol) for 48 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a yellowish oil (234 mg, 67 %) was obtained. The N-tboc protection group of this oil (180 mg, 0.52 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (348 mg, 1.55 mmol) for 72 h and an off-white solid 13 (126 mg, 98 %) was obtained after extraction. This solid 13 (74 mg, 0.30 mmol) was transferred to its fumaric acid salt 13F by using the general procedure I with fumaric acid (34 mg, 0.30 mmol). Compound 13F (79 mg, 71 %) was obtained as a white solid in 46 % yield over three steps; mp 168–169 °C (dec). 1H NMR (500 MHz, D2O) δ 1.96 (br m, 1H), 2.02 (br m, 1H), 2.29 (s, 3H), 2.35 (br s, 2H), 3.13 (br d, J = 13.9 Hz, 1H), 3.30–3.40 (br m, 2H), 3.46 (br d, J = 13.2 Hz, 1H), 3.53 (br d, J = 13.3 Hz, 2H), 4.05 (br d, J = 17.0 Hz, 1H), 4.13 (br d, J = 16.7 Hz, 2H), 4.39 (br d, J = 13.9 Hz, 1H), 6.26 (s, 1H), 6.69 (s, 2.0H). 13C NMR (125 MHz, D2O) δ 13.3, 28.0, 28.4, 30.1, 35.3, 49.1, 50.2, 50.5, 52.4, 108.1, 137.6, 164.7, 168.7, 174.4, 174.5. LC/ESI-MS: positive mode m/z = 250.4 ([M + H]+). Purity (> 99.5 %). IR (KBr, cm−1) 3474, 3139, 1704, 1657, 970. Anal. (C13H19N3O2*1.0C4H4O4*0.6H2O) C, H, N.
4.22. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3,4,5-trimethoxyphenyl)propan-1-one fumaric acid salt (14F)
The N-tboc protected compound was obtained by using the general procedure C with 3-(3,4,5-trimethoxyphenyl)propionic acid (140 mg, 1 mmol) for 48 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a clear oil (375 mg, 84 %) was obtained. The N-tboc protection group of this oil (270 mg, 0.60 mmol) was cleaved using the general procedure F for 12 h and an off-white solid 14 (192 mg, 92 %) was obtained after extraction. This solid 14 (96 mg, 0.28 mmol) was transferred to its fumaric acid salt 14F by using the general procedure I with fumaric acid (32 mg, 0.28 mmol). Compound 14F (96 mg, 73 %) was obtained as a white solid in 56 % yield over three steps; mp 124–126 °C (dec). 1H NMR (500 MHz, D2O) δ 1.83 (br d, J = 13.5 Hz, 1H), 1.95 (br d, J = 13.4 Hz, 1H), 2.21 (br s, 1H), 2.29 (br s, 1H), 2.71 (br m, 1H), 2.84–3.01 (br m, 5H), 3.29 (br m, 2H), 3.42 (br m, 2H), 3.76 (s, 3H), 3.85 (s, 6H), 4.01 (br d, J = 13.2 Hz, 1H), 4.39 (br d, J = 13.9 Hz, 1H), 6.63 (s, 2H), 6.67 (s, 2.0H). 13C NMR (125 MHz, D2O) δ 28.0, 28.4, 30.2, 34.1, 37.9, 48.8, 50.1, 50.5, 52.2, 58.9, 63.8, 108.9, 137.5, 138.1, 140.7, 155.3, 174.4, 180.0. LC/ESI-MS: positive mode m/z = 349.1 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3440, 1707, 1641, 973. Anal. (C19H28N2O4*1.0C4H4O4*0.75H2O) C, H, N.
4.23. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl((1R,2R)-2-phenylcyclopropyl)methanone fumaric acid salt (15F)
The N-tboc protected compound was obtained by using the general procedure C with trans-2-phenylcyclopropane-1-carboxylic acid (162 mg, 1 mmol) for 48 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a clear oil (326 mg, 88 %) was obtained. The N-tboc protection group of this oil (250 mg, 0.67 mmol) was cleaved using the general procedure F for 12 h and a clear oil 15 (177 mg, 97 %) was obtained after extraction. This oil 15 (67 mg, 0.25 mmol) was transferred to its fumaric acid salt 15F by using the general procedure I with fumaric acid (29 mg, 0.25 mmol). Compound 15F (28 mg, 28 %) was obtained as a white solid in 24 % yield over three steps; mp 101–102 °C (dec). 1H NMR (500 MHz, D2O) δ (rotamers present) 1.41–1.73 (br m, 2H), 1.90–2.03 (br m, 2H), 2.23–2.60 (br m, 4H), 3.11 (br d, J = 14.0 Hz, 1H), 3.28–3.58 (br m, 5H), 4.26–4.43 (br m, 2H), 6.67 (s, 1.6H), 7.19–7.45 (m, 5H). 13C NMR (125 MHz, D2O) δ (rotamers present) 17.6, 27.3, 28.1, 28.4, 28.6, 30.3, 49.5, 50.3, 50.6, 52.3, 128.9, 129.6, 131.7, 137.6, 143.3, 174.5, 179.2. LC/ESI-MS: positive mode m/z = 271.3 ([M + H]+). Purity (> 99.7 %). IR (KBr, cm−1) 3433, 3028, 1705, 1637, 1459, 983, 972. Anal. (C17H22N2O*0.8C4H4O4*1.25H2O) C, H, N.
4.24. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-phenylprop-2-en-1-one fumaric acid salt (16F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-phenylacrylic acid (148 mg, 1 mmol) for 48 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (256 mg, 72 %) was obtained. The N-tboc protection group of this solid (240 mg, 0.67 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (303 mg, 1.35 mmol) for 72 h and a white solid 16 (150 mg, 87 %) was obtained after extraction. This solid 16 (73 mg, 0.28 mmol) was transferred to its fumaric acid salt 16F by using the general procedure I with fumaric acid (33 mg, 0.28 mmol). Compound 16F (40 mg, 37 %) was obtained as a white solid in 23 % yield over three steps; mp 155–159 °C (dec). 1H NMR (500 MHz, D2O) δ 2.02 (br m,2H), 2.38 (br s, 2H), 3.20 (br d, J = 13.7 Hz, 1H), 3.32–3.42 (br m, 2H), 3.46–3.59 (br m, 3H), 4.38 (br d, J = 12.7 Hz, 1H), 4.49 (br d, J = 13.8 Hz, 1H), 6.68 (s, 1.8H), 7.16 (d, J = 15.6 Hz, 1H), 7.49 (m, 3H), 7.59 (d, J = 15.6 Hz, 1H), 7.68 (m, 2H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.3, 50.6, 52.4, 120.5, 130.9, 132.0, 133.3, 137.5, 137.6, 146.5, 173.9, 174.5. LC/ESI-MS: positive mode m/z = 257.4 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3433, 3059, 1650, 1616, 1451, 975. Anal. (C16H20N2O*0.9C4H4O4*1.2H2O) C, H, N.
4.25. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(2-methoxyphenyl)prop-2-en-1-one fumaric acid salt (17F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(2-methoxyphenyl)acrylic acid (178 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a clear oil (335 mg, 87 %) was obtained. The N-tboc protection group of this oil (275 mg, 0.71 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (481 mg, 2.13 mmol) for 48 h and a white solid 17 (192 mg, 94 %) was obtained after extraction. This solid 17 (142 mg, 0.50 mmol) was transferred to its fumaric acid salt 17F by using the general procedure I with fumaric acid (58 mg, 0.50 mmol). Compound 17F (149 mg, 72 %) was obtained as a white solid in 59 % yield over three steps; mp 141–144 °C (dec). 1H NMR (500 MHz, D2O) δ 1.97 (br m, 1H), 2.03 (br m, 1H), 2.36 (br s, 2H), 3.17 (br d, J = 12.9 Hz, 1H), 3.31–3.40 (br m, 2H), 3.47–3.58 (br m, 3H), 3.92 (s, 3H), 4.33 (br d, J = 12.8 Hz, 1H), 4.48 (br d, J = 13.4 Hz, 1H), 6.66 (s, 2.0H), 7.07 (dd, J = 7.7, 7.5 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 7.14 (d, J = 15.7 Hz, 1H), 7.46 (m, 1H), 7.64 (dd, J = 7.7, 1.6 Hz, 1H), 7.83 (d, J = 15.7 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.3, 50.6, 52.4, 58.7, 115.0, 120.9, 124.1, 126.2, 131.5, 134.8, 137.5, 141.5, 160.6, 174.1, 174.4. LC/ESI-MS: positive mode m/z = 287.1 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3432, 3044, 1705, 1645, 1462, 976. Anal. (C17H22N2O2*1.0C4H4O4*0.75H2O) C, H, N.
4.26. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-chlorophenyl)prop-2-en-1-one fumaric acid salt (18F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(3-chlorophenyl)acrylic acid (182 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (287 mg, 73 %) was obtained. The N-tboc protection group of this solid (225 mg, 0.58 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (389 mg, 1.73 mmol) for 48 h and a white solid 18 (160 mg, 96 %) was obtained after extraction. This solid 18 (103 mg, 0.35 mmol) was transferred to its fumaric acid salt 18F by using the general procedure I with fumaric acid (41 mg, 0.35 mmol). Compound 18F (88 mg, 62 %) was obtained as a white solid in 43 % yield over three steps; mp 141–144 °C (dec). 1H NMR (500 MHz, D2O) δ 1.99 (br m, 1H), 2.04 (br m, 1H), 2.37 (br s, 2H), 3.18 (br d, J = 13.7 Hz, 1H), 3.36 (br m, 2H), 3.52 (br m, 3H), 4.33 (br d, J = 12.9 Hz, 1H), 4.47 (br d, J = 13.9 Hz, 1H), 6.65 (s, 1.58H), 7.11 (d, J = 15.6 Hz, 1H), 7.39–7.46 (m, 2H), 7.48 (d, J = 15.6 Hz, 1H), 7.54 (m, 1H), 7.66 (m, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.2, 50.5, 52.4, 121.7, 129.3, 130.4, 132.8, 133.3, 137.1, 137.6, 139.4, 144.9, 173.4, 174.4. LC/ESI-MS: positive mode m/z = 291.4 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3427, 3058, 1702, 1649, 1475, 971. Anal. (C16H19ClN2O*0.79C4H4O4*1.15H2O) C, H, N.
4.27. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-bromophenyl)prop-2-en-1-one fumaric acid salt (19F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(3-bromophenyl)acrylic acid (178 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (337 mg, 77 %) was obtained. The N-tboc protection group of this solid (285 mg, 0.65 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (442 mg, 1.96 mmol) for 48 h and a white solid 19 (206 mg, 94 %) was obtained after extraction. This solid 19 (132 mg, 0.39 mmol) was transferred to its fumaric acid salt 19F by using the general procedure I with fumaric acid (46 mg, 0.39 mmol). Compound 19F (97 mg, 55 %) was obtained as a white solid in 40 % yield over three steps; mp 142–144 °C (dec). 1H NMR (500 MHz, D2O) δ 2.00 (br m, 2H), 2.37 (br s, 2H), 3.18 (br d, J = 12.5 Hz, 1H), 3.30–3.41 (br m, 2H), 3.45–3.58 (br m, 3H), 4.31 (br d, J = 12.3 Hz, 1H), 4.46 (br d, J = 13.1 Hz, 1H), 6.64 (s, 1.7H), 7.09 (d, J = 15.6 Hz, 1H), 7.34 (m, 1H), 7.45 (d, J = 15.6 Hz, 1H), 7.57 (m, 1H), 7.80 (m, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.2, 50.5, 52.4, 121.7, 125.2, 129.7, 133.3, 133.5, 135.7, 137.5, 139.6, 144.8, 173.4, 174.5. IR (KBr, cm−1) 3423, 3054, 1703, 1649, 1474, 971. Anal. (C16H19BrN2O*0.85C4H4O4*0.9H2O) C, H, N.
4.28. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-nitrophenyl)prop-2-en-1-one fumaric acid salt (20F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(3-nitrophenyl)acrylic acid (193 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a yellow solid (294 mg, 73 %) was obtained. The N-tboc protection group of this solid (230 mg, 0.57 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (387 mg, 1.72 mmol) for 48 h and a yellow solid 20 (170 mg, 98 %) was obtained after extraction. This solid 20 (120 mg, 0.40 mmol) was transferred to its fumaric acid salt 20F by using the general procedure I with fumaric acid (46 mg, 0.40 mmol). Compound 20F (97 mg, 57 %) was obtained as a yellow solid in 41 % yield over three steps; mp 184–187 °C (dec). 1H NMR (500 MHz, D2O) δ 2.03 (br m, 2H), 2.40 (br s, 2H), 3.22 (br d, J = 14.0 Hz, 1H), 3.32–3.43 (br m, 2H), 3.47–3.62 (br m, 3H), 4.38 (br d, J = 13.2 Hz, 1H), 4.50 (br d, J = 13.7 Hz, 1H), 6.64 (s, 1.7H), 7.27 (d, J = 15.6 Hz, 1H), 7.58 (d, J = 15.6 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.98 (d, J = 7.8 Hz, 1H), 8.24 (ddd, J = 8.3, 2.3, 0.9 Hz, 1H), 8.45 (t, J = 1.9 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.2, 50.5, 52.4, 123.2, 125.3, 127.4, 133.0, 137.2, 137.6, 139.1, 143.8, 151.1, 173.1, 174.5. LC/ESI-MS: positive mode m/z = 302.0 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3423, 3082, 1702, 1652, 1529, 1443, 983, 974. Anal. (C16H19N3O3*0.85C4H4O4 *1.5H2O) C, H, N.
4.29. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(4-methoxyphenyl)prop-2-en-1-one fumaric acid salt (21F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(4-methoxyphenyl)acrylic acid (178 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (249 mg, 64 %) was obtained. The N-tboc protection group of this solid (240 mg, 0.62 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (418 mg, 1.86 mmol) for 24 h and a white solid 21 (175 mg, 98 %) was obtained after extraction. This solid 21 (175 mg, 0.61 mmol) was transferred to its fumaric acid salt 21F by using the general procedure I with fumaric acid (71 mg, 0.61 mmol). Compound 21F (194 mg, 76 %) was obtained as a white solid in 48 % yield over three steps; mp 169–170 °C (dec). 1H NMR (500 MHz, D2O) δ 2.00 (br m, 2H), 2.36 (br s, 2H), 3.12–3.19 (br m, 1H), 3.31–3.40 (br m, 2H), 3.45–3.58 (br m, 3H), 3.87 (s, 3H), 4.35 (br d, J = 12.0 Hz, 1H), 4.47 (br d, J = 13.5 Hz, 1H), 6.65 (s, 2.0H), 6.99 (d, J = 15.5 Hz, 1H), 7.03 (ddd, J = 8.8, 3.0, 2.0 Hz, 2H), 7.53 (d, J = 15.5 Hz, 1H), 7.62 (ddd, J = 8.7, 2.9, 1.9 Hz, 2H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.3, 50.6, 52.3, 58.3, 117.4, 117.9, 130.5, 132.8, 137.6, 146.3, 163.6, 174.0, 174.5. LC/ESI-MS: positive mode m/z = 287.4 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3427, 3032, 1737, 1650, 1604, 979. Anal. (C17H22N2O2*1.0C4H4O4*0.75H2O) C, H, N.
4.30. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(furan-2-yl)prop-2-en-1-one fumaric acid salt (22F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(furan-2-yl)acrylic acid (138 mg, 1 mmol) for 72 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents an orange solid (176 mg, 51 %) was obtained. The N-tboc protection group of this solid (175 mg, 0.51 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (341 mg, 1.52 mmol) for 12 h and an orange oil 22 (84 mg, 68 %) was obtained after extraction. This oil 22 (84 mg, 0.34 mmol) was transferred to its fumaric acid salt 22F by using the general procedure I with fumaric acid (40 mg, 0.34 mmol). Compound 22F (65 mg, 51 %) was obtained as an orange solid in 18 % yield over three steps; mp 142–144 °C (dec). 1H NMR (500 MHz, D2O) δ 2.00 (br m, 1H), 2.09 (br m, 1H), 2.32 (br s, 2H), 3.30–3.40 (br m, 4H), 3.51 (br s, 2H), 4.48 (br s, 2H), 6.58 (dd, J = 3.4, 1.7 Hz, 1H), 6.72 (s, 2.0H), 6.77 (d, J = 3.4 Hz, 1H), 7.05 (d, J = 15.2 Hz, 1H), 7.45 (d, J = 15.2 Hz, 1H), 7.65 (d, J = 1.7 Hz, 1H). 13C NMR (125 MHz, D2O) δ 27.7, 29.5, 49.0, 113.7, 115.8, 116.5, 131.4, 136.5, 146.3, 153.2, 171.1, 171.7. LC/ESI-MS: positive mode m/z = 247.4 ([M + H]+). Purity (> 99 %). IR (KBr, cm−1) 3426, 3095, 1679, 1648, 1608, 984, 968. Anal. (C14H18N2O2*1.0C4H4O4*0.75H2O) C, H, N.
4.31. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(thiophen-2-yl)prop-2-en-1-one fumaric acid salt (23F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(thiophen-2-yl)acrylic acid (154 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a clear oil (220 mg, 61 %) was obtained. The N-tboc protection group of this oil (170 mg, 0.47 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (317 mg, 1.41 mmol) for 48 h and a clear oil 23 (122 mg, 99 %) was obtained after extraction. This oil 23 (44 mg, 0.17 mmol) was transferred to its fumaric acid salt 23F by using the general procedure I with fumaric acid (19 mg, 0.17 mmol). Compound 23F (24 mg, 37 %) was obtained as an off-white solid in 22 % yield over three steps; mp 173–178 °C (dec). 1H NMR (500 MHz, D2O) δ 1.87–2.06 (br m, 2H), 2.33–2.48 (br m, 2H), 3.13–3.24 (br m, 1H), 3.30–3.58 (br m, 5H), 4.30–4.52 (br m, 2H), 6.67 (s, 2.0H), 6.91 (d, J = 15.2 Hz, 1H), 7.15 (dd, J = 5.1, 3.6 Hz, 1H), 7.42 (d, J = 3.6 Hz, 1H), 7.57 (d, J = 5.1, 1H), 7.75 (d, J = 15.2, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.3, 50.6, 52.3, 118.6, 131.4, 132.0, 134.5, 137.6, 139.5, 142.4, 173.5, 174.5. LC/ESI-MS: positive mode m/z = 263.4 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3441, 3074, 1700, 1638, 1600, 968. Anal. (C14H18N2OS*1.0C4H4O4*0.6H2O) C, H, N.
4.32. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(thiophen-3-yl)prop-2-en-1-one fumaric acid salt (24F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(thiophen-3-yl)acrylic acid (154 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (312 mg, 86 %) was obtained. The N-tboc protection group of this solid (240 mg, 0.66 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (447 mg, 1.98 mmol) for 48 h and a clear oil 24 (168 mg, 97 %) was obtained after extraction. This oil 24 (126 mg, 0.48 mmol) was transferred to its fumaric acid salt 24F by using the general procedure I with fumaric acid (56 mg, 0.48 mmol). Compound 24F (129 mg, 70 %) was obtained as a white solid in 58 % yield over three steps; mp 155–159 °C (dec). 1H NMR (500 MHz, D2O) δ 1.97 (br m, 1H), 2.02 (br m, 1H), 2.36 (br s, 2H), 3.17 (br d, J = 12.2 Hz, 1H), 3.30–3.40 (br m, 2H), 3.45–3.58 (br m, 3H), 4.34 (br d, J = 12.5 Hz, 1H), 4.47 (br d, J = 13.3 Hz, 1H), 6.66 (s, 1.8H), 6.97 (d, J = 15.4 Hz, 1H), 7.47 (dd, J = 5.1, 1.1 Hz, 1H), 7.52 (dd, J = 5.1, 3.0 Hz, 1H), 7.59 (d, J = 15.4 Hz, 1H), 7.74 (dd, J = 2.8, 1.1 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.2, 28.5, 30.3, 49.4, 50.3, 50.6, 52.3, 119.6, 128.0, 130.5, 131.8, 137.5, 140.4, 140.5, 174.1, 174.4. LC/ESI-MS: positive mode m/z = 263.4 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3428, 3087, 1705, 1647, 1594, 973. Anal. (C14H18N2OS*0.9C4H4O4*0.9H2O) C, H, N.
4.33. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(pyridin-4-yl)prop-2-en-1-one fumaric acid salt (25F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(pyridin-4-yl)acrylic acid (149 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (9:1) was used for the chromatographic purification. After removal of the solvents a white solid (243 mg, 68 %) was obtained. The N-tboc protection group of this solid (185 mg, 0.52 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (350 mg, 1.55 mmol) for 48 h and an off-white oil 25 (116 mg, 87 %) was obtained after extraction. This oil 25 (63 mg, 0.24 mmol) was transferred to its fumaric acid salt 25F by using the general procedure I with fumaric acid (28 mg, 0.24 mmol). Compound 25F (70 mg, 72 %) was obtained as a white solid in 43 % yield over three steps; mp 155–159 °C (dec). 1H NMR (500 MHz, D2O) δ 2.02 (br m, 2H), 2.38 (br s, 1H), 2.40 (br s, 1H), 3.24 (br d, J = 14.7 Hz, 1H), 3.33–3.41 (br m, 2H), 3.49 (br d, J = 13.3 Hz, 1H), 3.54–3.62 (br m, 2H), 4.33 (br d, J = 13.2 Hz, 1H), 4.48 (br d, J = 13.9 Hz, 1H), 6.58 (s, 2.0H), 7.56 (s, 2H), 8.02 (d, J = 6.7 Hz, 1H); 8.70 (d, J = 6.2 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.1, 28.4, 30.2, 49.4, 50.1, 50.4, 52.5, 127.3, 130.4, 138.0, 140.4, 146.6, 152.7, 172.2, 176.2. LC/ESI-MS: positive mode m/z = 258.4 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3427, 3038, 1699, 1653, 1602, 982. Anal. (C15H19N3O*1.0C4H4O4*1.25H2O) C, H, N.
4.34. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(benzo[d][1,3]dioxol-5-yl)prop-2-en-1-one fumaric acid salt (26F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(benzo[d][1,3]dioxol-5-yl)acrylic acid (192 mg, 1 mmol) for 72 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (284 mg, 71 %) was obtained. The N-tboc protection group of this solid (235 mg, 0.59 mmol) was cleaved using the general procedure F for 2 h and an off-white solid 26 (128 mg, 73 %) was obtained after extraction. This solid 26 (128 mg, 0.43 mmol) was transferred to its fumaric acid salt 26F by using the general procedure I with fumaric acid (49 mg, 0.43 mmol). Compound 26F (106 mg, 58 %) was obtained as an off-white solid in 30 % yield over three steps; mp 147–148 °C (dec). 1H NMR (500 MHz, D2O) δ 1.97 (br m, 1H), 2.03 (br m, 1H), 2.37 (s, 2H), 3.16 (br d, J = 12.2 Hz, 1H), 3.31–3.41 (br m, 2H), 3.44–3.58 (br m, 3H), 4.34 (br d, J = 11.6 Hz, 1H), 4.46 (br d, J = 13.2, 1H), 6.02 (s, 2H), 6.65 (s, 2.0H), 6.91 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 15.3 Hz, 1H), 7.12 (dd, J = 8.1, 1.6 Hz, 1H), 7.19 (d, J = 1.6 Hz, 1H), 7.47 (d, J = 15.4 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.1, 28.5, 30.3, 49.4, 50.3, 50.6, 52.3, 104.6, 109.3, 111.6, 118.0, 127.6, 131.9, 137.5, 146.5, 150.8, 152.0, 173.9, 174.3. LC/ESI-MS: positive mode m/z = 301.4 ([M + H]+). Purity (> 99.5 %). IR (KBr, cm−1) 3434, 3054, 1706, 1646, 1607, 1448, 976. Anal. (C17H20N2O3*1.0C4H4O4*0.75H2O) C, H, N.
4.35. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(naphthalen-1-yl)prop-2-en-1-one fumaric acid salt (27F)
The N-tboc protected compound was obtained by using the general procedure C with trans-3-(naphthalene-1-yl)acrylic acid (198 mg, 1 mmol) for 48 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (344 mg, 85 %) was obtained. The N-tboc protection group of this solid (270 mg, 0.66 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (449 mg, 1.99 mmol) for 72 h and an off-white solid 27 (196 mg, 96 %) was obtained after extraction. This solid 27 (119 mg, 0.39 mmol) was transferred to its fumaric acid salt 27F by using the general procedure I with fumaric acid (45 mg, 0.39 mmol). Compound 27F (103 mg, 61 %) was obtained as a white solid in 50 % yield over three steps; mp 171–174 °C (dec). 1H NMR (500 MHz, D2O) δ 1.86 (br d, J = 13.3 Hz, 1H), 1.97 (br d, J = 13.4 Hz, 1H), 2.21 (br s, 1H), 2.32 (br s, 1H), 3.03 (br d, J = 13.0 Hz, 1H), 3.20 (br d, J = 12.3 Hz, 1H), 3.30 (br d, J = 13.0 Hz, 2H), 3.44 (br d, J = 13.4 Hz, 2H), 4.15 (br d, J = 12.3 Hz, 1H), 4.41 (br d, J = 13.4 Hz, 1H), 6.61 (s, 1.9H), 6.96 (d, J = 15.3 Hz, 1H), 7.50 (dd, J = 7.8, 7.7 Hz, 1H), 7.56 (m, 2H), 7.74 (d, J = 7.2 Hz, 1H), 7.91 (dd, J = 7.7, 6.7 Hz, 2H), 8.13 (d, J = 8.2 Hz, 1H), 8.21 (d, J = 15.3 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.1, 28.4, 30.2, 49.3, 50.2, 50.5, 52.2, 122.8, 126.0, 127.9, 128.6, 129.3, 130.0, 131.6, 133.3, 133.8, 134.5, 136.2, 137.5, 143.0, 173.4, 174.3. LC/ESI-MS: positive mode m/z = 307.5 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3433, 3048, 1701, 1643, 1601, 1458, 970. Anal. (C20H22N2O*0.95C4H4O4*1.1H2O) C, H, N.
4.36. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl(biphenyl-4-yl)methanone fumaric acid salt (28F)
The N-tboc protected compound was obtained by using the general procedure C with 4-biphenylcarboxylic acid (198 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a clear oil (293 mg, 72 %) was obtained. The N-tboc protection group of this oil (250 mg, 0.61 mmol) was cleaved using the general procedure F for 12 h and a clear oil 28 (141 mg, 75 %) was obtained after extraction. This oil 28 (60 mg, 0.20 mmol) was transferred to its fumaric acid salt 28F by using the general procedure I with fumaric acid (23 mg, 0.20 mmol). Compound 28F (54 mg, 63 %) was obtained as a white solid in 34 % yield over three steps; mp 148–150 °C (dec). 1H NMR (500 MHz, D2O) δ 1.99 (br m, 2H), 2.20 (br s, 1H), 2.43 (br s, 1H), 3.30–3.41 (br m, 3H), 3.42–3.54 (br m, 3H), 3.91 (br m, 1H), 4.53 (br m, 1H), 6.67 (s, 2.0H), 7.48 (m, 1H), 7.56 (m, 4H), 7.74 (d, J = 7.9 Hz, 2H), 7.79 (d, J = 8.3 Hz, 2H). 13C NMR (125 MHz, D2O) δ 28.0, 28.5, 30.3, 49.4, 49.6, 50.6, 54.8, 129.9, 130.0, 130.5, 131.2, 132.1, 136.2, 137.6, 142.4, 145.5, 174.6, 177.8. LC/ESI-MS: positive mode m/z = 307.5 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3427, 1705, 1624, 982, 970. Anal. (C20H22N2O*1.0C4H4O4*0.9H2O) C, H, N.
4.37. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl(phenyl)methanone fumaric acid salt (29F)
The N-tboc protected compound was obtained by using the general procedure D with benzoyl chloride (141 mg, 1 mmol). A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (325 mg, 97 %) was obtained. The N-tboc protection group of this solid (200 mg, 0.61 mmol) was cleaved using the general procedure F for 12 h and an off white solid 29 (138 mg, 99 %) was obtained after extraction. This solid 29 (67 mg, 0.29 mmol) was transferred to its fumaric acid salt 29F by using the general procedure I with fumaric acid (34 mg, 0.29 mmol). Compound 29F (46 mg, 44 %) was obtained as a white solid in 42 % yield over three steps; mp 162–165 °C (dec). 1H NMR (500 MHz, D2O) δ 1.99 (br m, 1H), 2.07 (br m, 1H), 2.27 (br s, 2H), 3.35–3.37 (br m, 2H), 3.37–3.40 (br m, 2H), 3.46 (br d, J = 13.1 Hz, 1H), 3.98 (br m, 1H), 4.56 (br s, 1H), 6.73 (s, 1.8H), 7.53 (m, 5H). 13C NMR (125 MHz, D2O) δ 27.3, 29.1, 48.3, 48.5, 128.3, 129.7, 131.2, 136.2, 136.8, 171.3, 175.4. LC/ESI-MS: positive mode m/z = 231.4 ([M + H]+). Purity (> 99.9 %). IR (KBr, cm−1) 3420, 1701, 1613, 987, 969. Anal. (C14H18N2O*0.9C4H4O4*1.5H2O) C, H, N.
4.38. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-phenylethanone fumaric acid salt (30F)
The N-tboc protected compound was obtained by using the general procedure D with phenylacetyl chloride (155 mg, 1 mmol). A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (343 mg, 98 %) was obtained. The N-tboc protection group of this solid (310 mg, 0.90 mmol) was cleaved using the general procedure F for 12 h and an off white solid 30 (210 mg, 97 %) was obtained after extraction. This solid 30 (180 mg, 0.74 mmol) was transferred to its fumaric acid salt 30F by using the general procedure H. Compound 30F (158 mg, 66 %) was obtained as a white solid in 62 % yield over three steps; mp 145–147 °C (dec). 1H NMR (500 MHz, D2O) δ 1.91 (br m, 1H), 1.97 (br m, 1H), 2.27 (br s, 1H), 2.32 (br s, 1H), 2.78 (br d, J = 13.4 Hz, 1H), 3.27–3.33 (br m, 3H), 3.39 (br d, J = 12.9 Hz, 1H), 3.45 (br d, J = 13.2 Hz, 1H), 3.85–3.97 (br m, 2H), 4.17 (br d, J = 13.2 Hz, 1H), 4.40 (br d, J = 13.7 Hz, 1H), 6.69 (s, 0.9H), 7.27–7.45 (m, 5H). 13C NMR (125 MHz, D2O) δ 28.0, 28.3, 30.2, 43.5, 49.1, 50.2, 50.6, 52.5, 130.2, 131.9, 132.1, 137.3, 138.0, 176.5, 178.8. LC/ESI-MS: positive mode m/z = 245.4 ([M + H] +). Purity (> 99 %). IR (KBr, cm−1) 3443, 3031, 1696, 1640, 1457, 984, 969. Anal. (C15H20N2O*0.45C4H4O4*1.5H2O) C, H, N.
4.39. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(pyridine-3-yl)ethanone fumaric acid salt (31F)
The N-tboc protected compound was obtained by using the general procedure E with 2-(pyridine-3-yl)acetic acid (137 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (262 mg, 76 %) was obtained. The N-tboc protection group of this solid (222 mg, 0.64 mmol) was cleaved using the general procedure F for 2 h and a clear oil 31 (143 mg, 90 %) was obtained after extraction. This oil 31 (143 mg, 0.58 mmol) was transferred to its fumaric acid salt 31F by using the general procedure I with fumaric acid (67 mg, 0.58 mmol). Compound 31F (96 mg, 47 %) was obtained as a white solid in 32 % yield over three steps; mp 166–168 °C (dec). 1H NMR (400 MHz, D2O) δ 1.97 (br d, J = 13.6 Hz, 1H), 2.03 (br d, J = 13.3 Hz, 1H), 2.36 (br s, 2H), 3.14 (br d, J = 13.5 Hz, 1H), 3.30–3.42 (br m, 2H), 3.46 (br d, J = 13.0 Hz, 1H), 3.53–3.64 (br m, 2H), 4.04 (br d, J = 18.1 Hz, 1H), 4.19 (br d, J = 12.2 Hz, 1H), 4.22 (br d, J = 17.1 Hz, 1H), 4.38 (br d, J = 13.9 Hz, 1H), 6.55 (s, 1.84H), 7.91 (dd, J = 7.9, 5.8 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.61 (s, 1H), 8.67 (d, J = 5.3 Hz, 1H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 27.9, 37.9, 47.0, 48.0, 48.2, 49.8, 126.9, 135.3, 142.1, 144.4, 147.0, 174.2, 174.3. ESI-MS: positive mode m/z = 246.3 ([M + H]+). Purity (> 99.9 %). IR (cm−1) 2926, 2859, 1699, 1640, 1219, 1106, 970, 639. Anal. (C14H19N3O *1.0C4H4O4*0.4H2O) C, H, N.
4.40. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-phenylpropan-1-one fumaric acid salt (32F)
The N-tboc protected compound was obtained by using the general procedure D with 3-phenylpropionic acid chloride (169 mg, 1 mmol). A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (355 mg, 97 %) was obtained. The N-tboc protection group of this solid (340 mg, 0.95 mmol) was cleaved using the general procedure F for 12 h and a clear oil 32 (235 mg, 96 %) was obtained after extraction. This oil 32 (103 mg, 0.40 mmol) was transferred to its fumaric acid salt 32F by using the general procedure I with fumaric acid (46 mg, 0.40 mmol). Compound 32F (115 mg, 75 %) was obtained as a white solid in 70 % yield over three steps; mp 153–156 °C (dec). 1H NMR (500 MHz, D2O) δ 1.86 (br m, 1H), 1.94 (br m, 1H), 2.22 (br s, 1H), 2.29 (br s, 1H), 2.78 (br m, 1H), 2.88 (br m, 1H), 2.90–2.96 (br m, 1H), 2.99 (br d, J = 14.0 Hz, 1H), 3.14 (br d, J = 13.7 Hz, 1H), 3.27 (br m, 2H), 3.39 (br m, 2H), 4.02 (br d, J = 13.5 Hz, 1H), 4.37 (br d, J = 14.1 Hz, 1H), 6.69 (s, 2.0H), 7.30 (m, 3H), 7.38 (m, 2H). 13C NMR (125 MHz, D2O) δ 28.0, 28.3, 30.1, 33.6, 37.9, 48.8, 50.2, 50.6, 52.2, 129.4, 131.4, 131.7, 137.5, 143.7, 174.4, 180.2. LC/ESI-MS: positive mode m/z = 259.5 ([M + H]+). Purity (> 99.8 %). IR (KBr, cm−1) 3433, 3054, 1701, 1645, 1454, 984. Anal. (C16H22N2O*1.0C4H4O4*0.5H2O) C, H, N.
4.41. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(2-chlorophenoxy)ethanone fumaric acid salt (33F)
The N-tboc protected compound was obtained by using the general procedure C with (2-chlorophenoxy)acetic acid (187 mg, 1 mmol) for 24 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a clear oil (186 mg, 47 %) was obtained. The N-tboc protection group of this oil (130 mg, 0.33 mmol) was cleaved using the general procedure F for 12 h and a white solid 33 (95 mg, 98 %) was obtained after extraction. This solid 33 (46 mg, 0.16 mmol) was transferred to its fumaric acid salt 33F by using the general procedure I with fumaric acid (18 mg, 0.16 mmol). Compound 33F (40 mg, 62 %) was obtained as a white solid in 29 % yield over three steps; mp 145–149 °C (dec). 1H NMR (500 MHz, D2O) δ 2.02 (br m, 2H), 2.34 (br s, 1H), 2.37 (br s, 1H), 3.18 (br d, J = 13.4 Hz, 1H), 3.35 (br m, 2H), 3.49 (br d, J = 13.1 Hz, 1H), 3.59 (br m, 2H), 4.00 (br d, J = 13.2 Hz, 1H), 4.38 (br d, J = 13.9 Hz, 1H), 4.85 (br d, J = 14.3 Hz, 1H), 5.15 (br d, J = 14.6 Hz, 1H), 6.68 (s, 1.8H), 7.07 (m, 2H), 7.33 (m, 1H), 7.50 (dd, J = 8.2, 1.3 Hz, 1H). 13C NMR (125 MHz, D2O) δ 28.0, 28.2, 30.2, 49.0, 50.2, 50.5, 51.2, 70.1, 117.2, 124.7, 125.8, 131.2, 133.3, 137.5, 155.5, 174.2, 176.4. LC/ESI-MS: positive mode m/z = 295.1 ([M + H]+). Purity (>97 %). IR (KBr, cm−1) 3441, 3065, 1662, 1457, 970. Anal. (C15H19ClN2O2*0.9C4H4O4*0.8H2O) C, H, N.
4.42. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(2,3,4,5,6-pentafluorophenyl)ethanone fumaric acid salt (34F)
The N-tboc protected compound was obtained by using the general procedure E with 2,3,4,5,6-pentafluorophenylacetic acid (226 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (370 mg, 85 %) was obtained. The N-tboc protection group of this solid (200 mg, 0.46 mmol) was cleaved using the general procedure F for 4 h and a white solid 34 (149 mg, 97 %) was obtained after extraction. This solid 34 (145 mg, 0.43 mmol) was transferred to its fumaric acid salt 34F by using the general procedure I with fumaric acid (50 mg, 0.43 mmol). Compound 34F (149 mg, 79 %) was obtained as a white solid in 65 % yield over three steps; mp 176–180 °C (dec.). 1H NMR (400 MHz, DMSO-d6) δ 1.78 (br d, J = 13.6 Hz, 1H), 1.84 (br d, J = 13.8 Hz, 1H), 2.17 (br s, 1H), 2.20 (br s, 1H), 2.96 (br d, J = 13.2 Hz, 1H), 3.15 (br m, 2H), 3.26 (br d, J = 13.1 Hz, 1H), 3.39 (br d, J = 13.0 Hz, 1H), 3.46 (br d, J = 12.7 Hz, 1H), 3.74 (br d, J = 17.3 Hz, 1H), 3.96 (br d, J = 16.4 Hz, 1H), 4.00 (br d, J = 11.7 Hz, 1H), 4.17 (br d, J = 13.9 Hz, 1H), 6.48 (s, 1.80H). 13C NMR (100 MHz, DMSO-d6) δ 26.5, 27.0, 28.6, 29.5, 47.9, 48.8, 49.1, 50.7, 136.4, 172.5, 173.7. LC/ESI-MS: positive mode m/z = 335.2 ([M + H]+). HRMS (EI) calcd for C15H15F5N2O 335.1177, found 335.1186. Purity (> 99 %). IR (cm−1) 1669, 1657, 1506, 1410, 1222, 1004, 974, 642. Anal. (C15H15F5N2O*1.0C4H4O4*0.4H2O) C, H, N.
4.43. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(4-(methylsulfonyl)phenyl)ethanone fumaric acid salt (35F)
The N-tboc protected compound was obtained by using the general procedure E with 4-(methylsulfonyl)phenylacetic acid (214 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (296 mg, 70 %) was obtained. The N-tboc protection group of this solid (246 mg, 0.58 mmol) was cleaved using the general procedure F for 2 h and a white solid 35 (147 mg, 78 %) was obtained after extraction. This solid 35 (147 mg, 0.46 mmol) was transferred to its fumaric acid salt 35F by using the general procedure I with fumaric acid (53 mg, 0.46 mmol). Compound 35F (121 mg, 61 %) was obtained as a white solid in 33 % yield over three steps; mp 188–191 °C (dec). 1H NMR (400 MHz, D2O) δ 1.93 (br d, J = 13.5 Hz, 1H), 2.01 (br d, J = 13.6 Hz, 1H), 2.33 (br s, 2H), 3.11 (br d, J = 13.2 Hz, 1H), 3.27 (s, 3H), 3.29–3.39 (br m, 2H), 3.42–3.57 (br m, 3H), 4.03 (s, 2H), 4.16 (br d, J = 13.2 Hz, 1H), 4.39 (br d, J = 13.9 Hz, 1H), 6.68 (s, 1.90H), 7.51 (d, J = 8.3 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 27.9, 41.0, 44.0, 46.9, 48.0, 48.3, 50.1, 128.0, 131.5, 135.4, 138.1, 142.4, 172.1, 175.5. ESI-MS: positive mode m/z = 323.3 ([M + H]+). HRMS (EI) calcd for C16H22N2O3S 323.1424, found 323.1416. Purity (> 99.9 %). IR (cm−1) 2930, 2857, 2720, 2626, 1652, 1582, 1253, 1144, 962, 751, 634. Anal. (C16H22N2O3S *1.0C4H4O4*0.7H2O) C, H, N.
4.44. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(pyridine-4-yl)ethanone fumaric acid salt (36F)
The N-tboc protected compound was obtained by using the general procedure E with 2-(pyridine-4-yl)acetic acid (137 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (276 mg, 80 %) was obtained. The N-tboc protection group of this solid (220 mg, 0.64 mmol) was cleaved using the general procedure F for 2 h and a clear oil 36 (128 mg, 82 %) was obtained after extraction. This oil 36 (128 mg, 0.52 mmol) was transferred to its fumaric acid salt 36F by using the general procedure I with fumaric acid (61 mg, 0.52 mmol). Compound 36F (157 mg, 74 %) was obtained as a white solid in 48 % yield over three steps; mp 174–175 °C (dec). 1H NMR (400 MHz, D2O) δ 1.97 (br d, J = 13.6 Hz, 1H), 2.03 (br d, J = 13.4 Hz, 1H), 2.37 (br s, 2H), 3.14 (br d, J = 13.5 Hz, 1H), 3.30–3.42 (br m, 2H), 3.47 (br d, J = 13.2 Hz, 1H), 3.54–3.63 (br m, 2H), 4.15 (br d, J = 12.6 Hz, 1H), 4.19–4.32 (m, 2H, exchangeable), 4.38 (br d, J = 13.9 Hz, 1H), 6.62 (s, 2.80H), 7.93 (d, J = 6.3 Hz, 2H), 8.72 (d, J = 6.3 Hz, 2H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 27.8, 40.8, 46.9, 48.0, 48.2, 49.9, 129.5, 135.6, 141.5, 157.0, 173.08, 173.14. ESI-MS: positive mode m/z = 246.3 ([M + H]+). HRMS (EI) calcd for C14H19N3O 246.1601, found 246.1591. Purity (> 99.9 %). IR (cm−1) 2971, 2848, 1671, 1568, 1404, 1271, 976, 784, 631, 590. Anal. (C14H19N3O *1.0C4H4O4*1.3H2O) C, H, N.
4.45. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(benzo[d][1,3]dioxol-5-yl)ethanone fumaric acid salt (37F)
The N-tboc protected compound was obtained by using the general procedure E with 2-benzo[d][1,3]dioxol-5-yl)acetic acid (180 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (248 mg, 64 %) was obtained. The N-tboc protection group of this solid (207 mg, 0.53 mmol) was cleaved using the general procedure F for 2 h and a white solid 37 (152 mg, 99 %) was obtained after extraction. This solid 37 (152 mg, 0.53 mmol) was transferred to its fumaric acid salt 37F by using the general procedure I with fumaric acid (61 mg, 0.53 mmol). Compound 37F (76 mg, 37 %) was obtained as a white solid in 23 % yield over three steps; mp 179–185 °C (dec). 1H NMR (400 MHz, D2O) δ 1.91 (br d, J = 13.2 Hz, 1H), 1.98 (br d, J = 13.4 Hz, 1H), 2.29 (br s, 1H), 2.31 (br s, 1H), 3.08 (br d, J = 13.5 Hz, 1H), 3.26–3.50 (br m, 5H), 3.81 (m, 2H), 4.15 (br d, J = 13.0 Hz, 1H), 4.39 (br d, J = 13.7 Hz, 1H), 5.97 (s, 2H), 6.67 (s, 1.75H), 6.74 (d, J = 8.0 Hz, 1H), 6.80 (s, 1H), 6.88 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, D2O) δ 25.8, 26.3, 27.9, 40.8, 46.9, 48.0, 48.4, 50.2, 101.8, 109.3, 110.4, 123.2, 128.7, 135.5, 146.8, 148.1, 172.2, 176.6. ESI-MS: positive mode m/z = 289.3 ([M + H]+). HRMS (EI) calcd for C16H20N2O3 289.1547, found 289.1550. Purity (> 97 %). IR (cm−1) 2869, 1698, 1659, 1596, 1443, 1398, 1253, 1213, 1192, 1034, 787. Anal. (C16H20N2O3*1.1C4H4O4*0.4H2O) C, H, N.
4.46. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(naphthalene-2-yl)ethanone fumaric acid salt (38F)
The N-tboc protected compound was obtained by using the general procedure E with 2-naphthylacetic acid (186 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (366 mg, 93 %) was obtained. The N-tboc protection group of this solid (309 mg, 0.78 mmol) was cleaved using the general procedure F for 2 h and a white solid 38 (228 mg, 99 %) was obtained after extraction. This solid 38 (228 mg, 0.78 mmol) was transferred to its fumaric acid salt 38F by using the general procedure I with fumaric acid (90 mg, 0.78 mmol). Compound 38F (122 mg, 32 %) was obtained as a white solid in 30 % yield over three steps; mp 181–185 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.71 (br d, J = 11.8 Hz, 1H), 1.79 (br d, J = 12.3 Hz, 1H), 2.10 (br s, 1H), 2.13 (br s, 1H), 2.88 (br d, J = 11.9 Hz, 1H), 3.04–3.34 (br m, 5H), 3.83 (br d, J = 14.2 Hz, 1H), 3.95 (br d, J = 14.5 Hz, 1H), 4.06 (br d, J = 10.9 Hz, 1H), 4.25 (br d, J = 12.2 Hz, 1H), 6.48 (s, 1.36H), 7.29 (d, J = 7.8 Hz, 1H), 7.42 (m, 2H), 7.61 (s, 1H), 7.72–7.86 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 25.4, 25.7, 28.1, 40.6, 45.9, 46.7, 46.8, 49.0, 125.4, 125.9, 127.33, 127.34, 127.4, 127.5, 128.3, 131.7, 133.0, 133.6, 135.1, 167.9, 171.6. ESI-MS: positive mode m/z = 295.4 ([M + H]+). HRMS (EI) calcd for C19H22N2O 295.1805, found 295.1808. Purity (> 98 %). IR (cm−1) 2952, 2852, 1656, 1402, 1218, 1104, 980, 796. Anal. (C19H22N2O*0.8C4H4O4*1.0H2O) C, H, N.
4.47. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-(pyridine-4-yl)propanone fumaric acid salt (39F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(pyridine-3-yl)propionic acid (151 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (232 mg, 64 %) was obtained. The N-tboc protection group of this solid (190 mg, 0.53 mmol) was cleaved using the general procedure F for 2 h and a clear oil 39 (120 mg, 88 %) was obtained after extraction. This oil 39 (120 mg, 0.46 mmol) was transferred to its fumaric acid salt 39F by using the general procedure I with fumaric acid (54 mg, 0.46 mmol). Compound 39F (127 mg, 65 %) was obtained as a white solid in 37 % yield over three steps; mp 149–155 °C (dec). 1H NMR (400 MHz, D2O) δ 1.92 (br d, J = 13.6 Hz, 1H), 1.99 (br d, J = 13.6 Hz, 1H), 2.31 (br s, 2H), 2.86–3.08 (br m, 3H), 3.15 (q, J = 7.0 Hz, 2H), 3.39–3.54 (br m, 3H), 4.06 (br d, J = 13.0 Hz, 1H), 4.34 (br d, J = 13.8 Hz, 1H), 6.62 (s, 2.78H), 7.98 (dd, J = 7.7, 6.2 Hz, 1H), 8.50 (d, J = 8.1 Hz, 1H), 8.63 (d, J = 5.6 Hz, 1H), 8.70 (s, 1H). 13C NMR (100 MHz, D2O) δ 25.8, 26.1, 27.7, 34.2, 46.6, 48.0, 48.3, 49.6, 127.5, 135.6, 139.7, 141.7, 142.3, 147.5, 173.1, 176.3. ESI-MS: positive mode m/z = 260.3 ([M + H]+). HRMS (EI) calcd for C15H21N3O 260.1757, found 260.1751. Purity (> 99.9 %). IR (cm−1) 2949, 2852, 1731, 1642, 1562, 1312, 1180, 985, 787, 641. Anal. (C15H21N3O*1.4C4H4O4*1.0H2O) C, H, N.
4.48. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(4-fluorophenyl)prop-2-en-1-one fumaric acid salt (40F)
The N-tboc protected compound was obtained by using the general procedure E with trans-3-(4-fluorophenyl)acrylic acid (166 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (242 mg, 65 %) was obtained. The N-tboc protection group of this solid (210 mg, 0.56 mmol) was cleaved using the general procedure F for 2 h and a clear oil 40 (149 mg, 97 %) was obtained after extraction. This oil 40 (149 mg, 0.54 mmol) was transferred to its fumaric acid salt 40F by using the general procedure I with fumaric acid (63 mg, 0.54 mmol). Compound 40F (151 mg, 71 %) was obtained as a white solid in 45 % yield over three steps; mp 199–201 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.76 (br d, J = 12.5 Hz, 1H), 1.87 (br d, J = 12.7 Hz, 1H), 2.07 (br s, 2H), 2.88–3.42 (br m, 6H), 4.34 (br d, J = 12.9 Hz, 2H), 6.46 (s, 2.0H), 7.25 (d, J = 15.3 Hz, 1H), 7.24–7.28 (m, 2H), 7.41 (d, J = 15.5 Hz, 1H), 7.77 (dd, J = 8.2, 5.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 26.6, 28.4, 46.7, 115.7 (d, JC,F = 21.6 Hz), 120.0 (d, JC,F = 2.1 Hz), 130.0 (d, JC,F = 8.3 Hz), 132.0 (d, JC,F = 3.1 Hz), 135.1 (br d, JC,F = 1.9 Hz), 139.0, 162.7 (d, JC,F = 247.1), 167.5, 167.9. ESI-MS: positive mode m/z = 275.1 ([M + H]+). HRMS (EI) calcd for C16H19FN2O 275.1554, found 275.1547. Purity (> 99.9 %). IR (cm-1) 2952, 2851, 2717, 2640, 1732, 1652, 1620, 1558, 1507, 1316, 1217, 972, 826, 787, 645. Anal. (C16H19FN2O *1.5C4H4O4) C, H, N.
4.49. (E)-1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(pyridin-3-yl)prop-2-en-1-one fumaric acid salt (41F)
The N-tboc protected compound was obtained by using the general procedure E with trans-3-(pyridine-3-yl)acrylic acid (149 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. An additional HPLC purification (RP18 silica gel, MeOH-H2O gradient) was necessary. After removal of the solvents a white solid (238 mg, 67 %) was obtained. The N-tboc protection group of this solid (160 mg, 0.45 mmol) was cleaved using the general procedure F for 4 h and a clear oil 41 (110 mg, 95 %) was obtained after extraction. This oil 41 (92 mg, 0.36 mmol) was transferred to its fumaric acid salt 41F by using the general procedure I with fumaric acid (41 mg, 0.36 mmol). Compound 41F (115 mg, 86 %) was obtained as a white solid in 55 % yield over three steps; mp 82–85 °C (dec). 1H NMR (400 MHz, D2O) δ 2.03 (br m, 2H), 2.39 (br s, 2H), 3.23 (br d, J = 13.5 Hz, 1H), 3.37 (br t, J = 12.1 Hz, 2H), 3.46–3.62 (br m, 3H), 4.35 (br d, J = 13.4 Hz, 1H), 4.49 (br d, J = 14.0 Hz, 1H), 6.58 (s, 1.96H), 7.37 (d, J = 15.7 Hz, 1H), 7.59 (d, J = 15.9 Hz, 1H), 7.83 (dd, J = 8.1, 5.5 Hz, 1H), 8.49 (d, J = 8.2 Hz, 1H), 8.65 (d, J = 5.3 Hz, 1H), 8.86 (s, 1H). 13C NMR (100 MHz, D2O) δ 26.0, 26.3, 28.0, 47.2, 48.0, 48.3, 50.2, 123.4, 126.7, 133.8, 135.7, 137.9, 141.0, 144.9, 145.7, 170.5, 173.5. ESI-MS: positive mode m/z = 258.3 ([M + H]+). HRMS (EI) calcd for C15H19N3O 258.1601, found 258.1597. Purity (> 99 %). IR (cm−1) 2858, 1699, 1651, 1574, 1418, 1222, 1107, 971, 545. Anal. (C15H19N3O*0.4C4H4O4*7.0H2O) C, H, N.
4.50. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-2-phenylprop-2-yn-1-one fumaric acid salt (42F)
The N-tboc protected compound was obtained by using the general procedure E with phenylpropiolic acid (146 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (303 mg, 86 %) was obtained. The N-tboc protection group of this solid (250 mg, 0.71 mmol) was cleaved using the general procedure G with anhydrous ZnBr2 (318 mg, 1.41 mmol) for 48 h and an off-white solid 42 (148 mg, 82 %) was obtained after extraction. This solid 42 (148 mg, 0.58 mmol) was transferred to its fumaric acid salt 42F by using the general procedure I with fumaric acid (67 mg, 0.58 mmol). Compound 42F (110 mg, 53 %) was obtained as a white solid in 37 % yield over three steps; mp 151–154 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.78 (br d, J = 13.0 Hz, 1H), 1.87 (br d, J = 12.7 Hz, 1H), 2.07 (br s, 2H), 2.96–3.14 (br m, 4H), 3.24 (br d, J = 12.8 Hz, 1H), 3.46 (br d, J = 13.0 Hz, 1H), 4.26 (br d, J = 13.5 Hz, 1H), 4.37 (br d, J = 12.0 Hz, 1H), 6.48 (s, 1.80H), 7.44–7.55 (m, 3H), 7.60–7.65 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 25.5, 26.0, 28.6, 45.6, 47.3, 47.4, 50.3, 82.5, 89.4, 120.0, 128.9, 130.4, 132.2, 135.0, 154.3, 167.7. ESI-MS: positive mode m/z = 255.3 ([M + H]+). HRMS (EI) calcd for C16H18N2O 255.1492, found 255.1507. Purity (> 99.5 %). IR (cm−1) 3536, 3390, 2218, 1658, 1607, 1425, 980. Anal. (C16H18N2O*1.0C4H4O4*3.0H2O) C, H, N.
4.51. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(2-methoxyphenyl)prop-2-yn-1-one fumaric acid salt (43F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(2-methoxyphenyl)propiolic acid (176 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (370 mg, 96 %) was obtained. The N-tboc protection group of this solid (370 mg, 0.96 mmol) was cleaved using the general procedure F for 4 h and a white solid 43 (260 mg, 95 %) was obtained after extraction. This solid 43 (260 mg, 0.91 mmol) was transferred to its fumaric acid salt 43F by using the general procedure I with fumaric acid (106 mg, 0.91 mmol). Compound 43F (284 mg, 78 %) was obtained as a white solid in 71 % yield over three steps; mp 155–159 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.77–1.90 (br m, 2H), 2.20 (br s, 2H), 2.96–3.04 (br m, 1H), 3.07–3.23 (br m, 3H), 3.28 (br d, J = 13.2 Hz, 1H), 3.43 (br d, J = 13.0 Hz, 1H), 3.78 (s, 3H), 4.21 (br d, J = 13.8 Hz, 1H), 4.41 (br d, J = 13.3 Hz, 1H), 6.46 (s, 2.00H), 6.97 (t, J = 7.5 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 7.45 (m, 1H), 7.50 (dd, J = 7.6, 1.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 25.8, 26.0, 28.2, 46.4, 47.4, 47.5, 51.1, 56.8, 86.7, 89.1, 109.3, 112.5, 121.9, 133.7, 134.9, 136.0, 156.7, 161.8, 169.7. ESI-MS: positive mode m/z = 285.3 ([M + H]+). HRMS (EI) calcd for C17H20N2O2 285.1598, found 285.1603. Purity (> 99.5 %). IR (cm−1) 2939, 2200, 1712, 1609, 1421, 1254, 968, 762, 728. Anal. (C17H20N2O2*1.0C4H4O4*1.6H2O) C, H, N.
4.52. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-methoxyphenyl)prop-2-yn-1-one fumaric acid salt (44F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(3-methoxyphenyl)propiolic acid (176 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a clear oil (349 mg, 91 %) was obtained. The N-tboc protection group of this oil (310 mg, 0.81 mmol) was cleaved using the general procedure F for 4 h and a white solid 44 (228 mg, 99 %) was obtained after extraction. This solid 44 (228 mg, 0.80 mmol) was transferred to its fumaric acid salt 44F by using the general procedure G with fumaric acid (93 mg, 0.80 mmol). Compound 44F (202 mg, 67 %) was obtained as a white solid in 60 % yield over three steps; mp 123–126 °C (dec). 1H NMR (400 MHz, D2O) δ 1.97 (br d, J = 13.5 Hz, 1H), 2.04 (br d, J = 13.3 Hz, 1H), 2.36 (br s, 2H), 3.16 (br d, J = 14.0 Hz, 1H), 3.29–3.41 (br m, 2H), 3.45 (br d, J = 13.0 Hz, 1H), 3.52–3.59 (br m, 2H), 3.81 (s, 3H), 4.37 (br d, J = 14.1 Hz, 1H), 4.55 (br d, J = 13.5 Hz, 1H), 6.63 (s, 1.60H), 7.10 (dd, J = 8.3, 2.5 Hz, 1H), 7.15 (br s, 1H), 7.23 (d, J = 7.5 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H). 13C NMR (100 MHz, D2O) δ 25.6, 26.1, 28.1, 46.5, 47.7, 48.1, 51.5, 56.1, 80.7, 94.2, 118.0, 118.1, 120.9, 126.2, 130.8, 135.3, 157.9, 159.4, 172.1. ESI-MS: positive mode m/z = 285.4 ([M + H]+). HRMS (EI) calcd for C17H20N2O2 285.1598, found 285.1605. Purity (> 99.5 %). IR (cm−1) 2935, 2863, 2207, 1718, 1703, 1611, 1417, 1231, 1106, 989, 682. Anal. (C17H20N2O2*0.84C4H4O4*1.4H2O) C, H, N.
4.53. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(4-methoxyphenyl)prop-2-yn-1-one fumaric acid salt (45F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(4-methoxyphenyl)propiolic acid (176 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a clear oil (377 mg, 98 %) was obtained. The N-tboc protection group of this oil (320 mg, 0.83 mmol) was cleaved using the general procedure F for 4 h and a white solid 45 (236 mg, 99 %) was obtained after extraction. This solid 45 (236 mg, 0.83 mmol) was transferred to its fumaric acid salt 45F by using the general procedure I with fumaric acid (96 mg, 0.83 mmol). Compound 45F (272 mg, 83 %) was obtained as a white solid in 81 % yield over three steps; mp 169–174 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.77 (br d, J = 12.5 Hz, 1H), 1.86 (br d, J = 12.8 Hz, 1H), 2.07 (br s, 2H), 2.98 (br dd, J = 13.7, 2.8 Hz, 1H), 3.01–3.12 (br m, 3H), 3.23 (br d, J = 12.8 Hz, 1H), 3.41–3.48 (br m, 1H), 3.81 (s, 3H), 4.25 (br d, J = 13.6 Hz, 1H), 4.37 (br d, J = 13.0 Hz, 1H), 6.47 (s, 1.92H), 7.02 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 25.5, 25.9, 28.5, 45.6, 47.1, 47.3, 50.3, 55.4, 81.7, 90.1, 111.7, 114.6, 134.1, 135.0, 154.6, 160.8, 167.7. ESI-MS: positive mode m/z = 285.3 ([M + H]+). HRMS (EI) calcd for C17H20N2O2 285.1598, found 285.1594. Purity (> 99 %). IR (cm−1) 2868, 2198, 1717, 1600, 1511, 1254, 1172. Anal. (C17H20N2O2*0.95C4H4O4*1.6H2O) C, H, N.
4.54. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3,4-dimethoxyphenyl)prop-2-yn-1-one fumaric acid salt (46F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(3,4-dimethoxyphenyl)propiolic acid (206 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (299 mg, 72 %) was obtained. The N-tboc protection group of this solid (260 mg, 0.63 mmol) was cleaved using the general procedure F for 4 h and a white solid 46 (192 mg, 97 %) was obtained after extraction. This solid 46 (192 mg, 0.61 mmol) was transferred to its fumaric acid salt 46F by using the general procedure I with fumaric acid (71 mg, 0.61 mmol). Compound 46F (158 mg, 60 %) was obtained as a white solid in 42 % yield over three steps; mp 152–153 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.78 (br d, J = 12.5 Hz, 1H), 1.87 (br d, J = 12.9 Hz, 1H), 2.08 (br s, 2H), 2.98 (br dd, J = 13.7, 3.1 Hz, 1H), 3.01–3.15 (br m, 3H), 3.24 (br d, J = 12.4 Hz, 1H), 3.44 (br dd, J = 12.9 Hz, 2.1 Hz, 1H), 3.79 (s, 3H), 3.80 (s, 3H), 4.25 (br d, J = 13.4 Hz, 1H), 4.37 (br d, J = 12.9 Hz, 1H), 6.46 (s, 2.11H), 7.02 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 1.9 Hz, 1H), 7.22 (dd, J = 8.3, 1.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 25.5, 25.9, 28.5, 45.6, 47.0, 47.2, 50.3, 55.6, 55.7, 81.4, 90.4, 111.6, 111.9, 114.9, 126.2, 135.0, 148.6, 150.9, 154.6, 167.8. ESI-MS: positive mode m/z =315.4 ([M + H]+). HRMS (EI) calcd for C18H22N2O3 315.1703, found 315.1702. Purity (> 99 %). IR (cm−1) 3081, 2202, 1695, 1603, 1514, 1252, 1107. Anal. (C18H22N2O3*1.5C4H4O4*0.5H2O) C, H, N.
4.55. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-chloro-4-methoxyphenyl)prop-2-yn-1-one fumaric acid salt (47F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(3-chloro-4-methoxyphenyl)propiolic acid (211 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents an off-white solid (412 mg, 98 %) was obtained. The N-tboc protection group of this solid (360 mg, 0.86 mmol) was cleaved using the general procedure F for 4 h and an off-white solid 47 (270 mg, 99 %) was obtained after extraction. This solid 47 (270 mg, 0.85 mmol) was transferred to its fumaric acid salt 47F by using the general procedure I with fumaric acid (98 mg, 0.85 mmol). Compound 47F (270 mg, 74 %) was obtained as an off-white solid in 72 % yield over three steps; mp 169–174 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.76–1.89 (br m, 2H), 2.19 (br s, 2H), 3.00 (br d, J = 13.4 Hz, 1H), 3.06–3.17 (br m, 2H), 3.21 (br d, J = 13.4 Hz, 1H), 3.33 (br d, J = 13.2 Hz, 1H), 3.43 (br d, J = 13.1 Hz, 1H), 3.82 (s, 3H), 4.20 (br d, J = 13.4 Hz, 1H), 4.34 (br d, J = 13.3 Hz, 1H), 6.46 (s, 1.96H), 7.11 (d, J = 8.8 Hz, 1H), 7.54 (dd, J = 8.6, 1.9 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 25.6, 26.3, 28.5, 46.8, 47.8, 48.1, 51.6, 57.3, 82.4, 92.3, 113.5, 114.3, 122.8, 134.7, 134.9, 136.3, 157.7, 157.7, 170.9. ESI-MS: positive mode m/z = 319.3 ([M + H]+). HRMS (EI) calcd for C17H19ClN2O2 319.1208, found 319.1212. Purity (> 99.5 %). IR (cm−1) 3088, 2205, 1720, 1609, 1426, 1296, 1255, 969. Anal. (C17H19ClN2O2*0.8C4H4O4*2.0H2O) C, H, N.
4.56. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(4-methylphenyl)prop-2-yn-1-one fumaric acid salt (48F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(4-methylphenyl)propiolic acid (160 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (350 mg, 95 %) was obtained. The N-tboc protection group of this solid (305 mg, 0.83 mmol) was cleaved using the general procedure F for 4 h and a white solid 48 (217 mg, 98 %) was obtained after extraction. This solid 48 (217 mg, 0.81 mmol) was transferred to its fumaric acid salt 48F by using the general procedure I with fumaric acid (94 mg, 0.81 mmol). Compound 48F (244 mg, 78 %) was obtained as a white solid in 75 % yield over three steps; mp 202–207 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.77 (br d, J = 12.7 Hz, 1H), 1.87 (br d, J = 12.7 Hz, 1H), 2.07 (br s, 2H), 2.35 (s, 3H), 2.98 (br dd, J = 13.6 Hz, 1H), 3.01–3.13 (br m, 3H), 3.24 (br d, J = 12.8 Hz, 1H), 3.45 (br dd, J = 13.1, 1.9 Hz, 1H), 4.25 (br d, J = 13.6 Hz, 1H), 4.36 (br d, J = 12.9 Hz, 1H), 6.47 (s, 2.00H), 7.28 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 21.2, 25.5, 25.9, 28.6, 45.6, 47.0, 47.2, 50.3, 82.2, 89.8, 116.9, 129.6, 132.2, 135.0, 140.5, 154.5, 167.8. ESI-MS: positive mode m/z = 269.4 ([M + H]+). HRMS (EI) calcd for C17H20N2O 269.1648, found 269.1650. Purity (> 99.5 %). IR (cm−1) 3408, 2963, 2867, 2202, 1720, 1605, 1431, 1270, 977. Anal. (C17H20N2O*1.0C4H4O4*1.7H2O) C, H, N.
4.57. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(3-fluorophenyl)prop-2-yn-1-one fumaric acid salt (49F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(3-fluorophenyl)propiolic acid (164 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a white solid (327 mg, 88 %) was obtained. The N-tboc protection group of this solid (290 mg, 0.78 mmol) was cleaved using the general procedure F for 4 h and a white solid 49 (210 mg, 99 %) was obtained after extraction. This solid 49 (210 mg, 0.77 mmol) was transferred to its fumaric acid salt 49F by using the general procedure I with fumaric acid (90 mg, 0.77 mmol). Compound 49F (199 mg, 68 %) was obtained as a white solid in 60 % yield over three steps; mp 154–158 °C (dec). 1H NMR (400 MHz, D2O+DMSO-d6) δ 1.84 (br m, 2H), 2.20 (br s, 2H), 3.03 (br d, J = 13.9 Hz, 1H), 3.09–3.21 (br m, 2H), 3.25 (br d, J = 13.1 Hz, 1H), 3.36 (br d, J = 14.0 Hz, 1H), 3.45 (br d, J = 13.2 Hz, 1H), 4.21 (br d, J = 13.9 Hz, 1H), 4.38 (br d, J = 13.4 Hz, 1H), 6.47 (s, 1.80H), 7.14–7.25 (m, 1H), 7.25–7.41 (m, 3H). 13C NMR (100 MHz, D2O+ DMSO-d6) δ 26.2, 26.7, 28.8, 47.2, 48.3, 48.6, 52.1, 82.6, 92.9 (d, JC,F = 3.4 Hz), 119.8 (d, JC,F = 20.8 Hz), 120.5 (d, JC,F = 23.6 Hz), 122.5 (d, JC,F = 9.4 Hz), 130.3 (d, JC,F = 2.7 Hz), 132.4 (d, JC,F = 8.7 Hz), 136.4, 158.0, 163.4 (d, JC,F = 246.0 Hz), 172.0. ESI-MS: positive mode m/z = 273.3 ([M + H]+). HRMS (EI) calcd for C16H17N2O 273.1398, found 273.1401. Purity (> 98 %). IR (cm−1) 3523, 3082, 2867, 2213, 1704, 1610, 1421, 1229, 969, 788. Anal. (C16H17N2O *0.9C4H4O4*1.9H2O) C, H, N.
4.58. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(benzo[d][1,3]dioxol-5-yl)prop-2-yn-1-one fumaric acid salt (50F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(benzo[d][1,3]dioxol-5-yl)propiolic acid (190 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a yellow oil (347 mg, 87 %) was obtained. The N-tboc protection group of this oil (290 mg, 0.73 mmol) was cleaved using the general procedure F for 4 h and an off-white solid 50 (214 mg, 99 %) was obtained after extraction. This solid 50 (214 mg, 0.72 mmol) was transferred to its fumaric acid salt 50F by using the general procedure I with fumaric acid (83 mg, 0.72 mmol). Compound 50F (254 mg, 77 %) was obtained as an off-white solid in 66 % yield over three steps; mp 182–186 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.77 (br d, J = 13.0 Hz, 1H), 1.88 (br d, J = 13.1 Hz, 1H), 2.10 (br s, 2H), 2.98 (br dd, J = 13.7, 2.8 Hz, 1H), 3.04–3.16 (br m, 3H), 3.27 (br d, J = 12.8 Hz, 1H), 3.43 (br d, J = 13.0 Hz, 1H), 4.24 (br d, J = 13.4 Hz, 1H), 4.36 (br d, J = 13.0 Hz, 1H), 6.11 (s, 2H), 6.51 (s, 2.80H), 7.00 (d, J = 7.8 Hz, 1H), 7.18 (dd, J = 9.7, 1.6 Hz, 1H), 7.19 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 25.2, 25.6, 28.1, 45.5, 46.6, 46.8, 50.2, 81.2, 89.9, 101.9, 109.0, 111.7, 112.9, 127.9, 134.8, 147.5, 149.3, 154.7, 167.3. ESI-MS: positive mode m/z = 299.3 ([M + H]+). HRMS (EI) calcd for C17H18N2O3 299.1390, found 299.1393. Purity (> 99 %). IR (cm−1) 3495, 3382, 2196, 1646, 1602, 1425, 1251, 1235, 1038, 977. Anal. (C17H18N2O3*1.2C4H4O4*2.5H2O) C, H, N.
4.59. 1-((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)-3-(naphthalene-1-yl)prop-2-yn-1-one fumaric acid salt (51F)
The N-tboc protected compound was obtained by using the general procedure E with 3-(naphthalene-1-yl)propiolic acid (196 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (40:1) was used for the chromatographic purification. After removal of the solvents a clear oil (108 mg, 27 %) was obtained. The N-tboc protection group of this oil (108 mg, 0.27 mmol) was cleaved using the general procedure F for 4 h and a white solid 51 (74 mg, 91 %) was obtained after extraction. This solid 51 (74 mg, 0.24 mmol) was transferred to its fumaric acid salt 51F by using the general procedure I with fumaric acid (28 mg, 0.24 mmol). Compound 51F (60 mg, 58 %) was obtained as a white solid in 14 % yield over three steps; mp 171–173 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 1.81 (br d, J = 12.5 Hz, 1H), 1.89 (br d, J = 12.7 Hz, 1H), 2.11 (br s, 1H), 2.14 (br s, 1H), 3.02–3.18 (br m, 4H), 3.28 (br d, J = 12.8 Hz, 1H), 3.58 (br d, J = 12.7 Hz, 1H), 4.32 (br d, J = 13.5 Hz, 1H), 4.48 (br d, J = 12.6 Hz, 1H), 6.48 (s, 2.10H), 7.59 (t, J = 7.7 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.91 (d, J = 7.0 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 8.25 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 25.5, 25.9, 28.5, 45.7, 47.0, 47.1, 50.5, 87.4, 87.5, 117.4, 125.1, 125.6, 127.1, 127.9, 128.8, 130.8, 132.1, 132.6, 132.8, 135.1, 154.5, 167.9. ESI-MS: positive mode m/z = 305.4 ([M + H]+). HRMS (EI) calcd for C20H20N2O 305.1648, found 305.1644. Purity (> 99 %). IR (cm−1) 3498, 2840, 2199, 1704, 1635, 1214, 970, 768, 644. Anal. (C20H20N2O *0.75C4H4O4*1.6H2O) C, H, N.
4.60. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl(4-(pyridin-3-yl)phenyl)methanone fumaric acid salt (52F)
The N-tboc protected compound was obtained by using the general procedure E with 4-(3-pyridyl)benzoic acid (199 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (403 mg, 99 %) was obtained. The N-tboc protection group of this solid (380 mg, 0.93 mmol) was cleaved using the general procedure F for 2 h and a white solid 52 (257 mg, 90 %) was obtained after extraction. This solid 52 (257 mg, 0.84 mmol) was transferred to its fumaric acid salt 52F by using the general procedure I with fumaric acid (97 mg, 0.84 mmol). Compound 52F (202 mg, 56 %) was obtained as a white solid in 50 % yield over three steps; mp 149–151 °C (dec). 1H NMR (400 MHz, D2O) δ 2.02 (br m, 2H), 2.24 (br s, 1H), 2.45 (br s, 1H), 3.33–3.42 (br m, 3H), 3.46–3.58 (br m, 3H), 3.91 (br d, J = 12.3 Hz, 1H), 4.55 (br d, J = 12.8 Hz, 1H), 6.56 (s, 2.1H), 7.65 (d, J = 8.2 Hz, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.98 (dd, J = 8.1, 5.6 Hz, 1H), 8.65 (d, J = 8.2 Hz, 1H), 8.72 (d, J = 5.3 Hz, 1H), 9.00 (s, 1H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 28.0, 47.2, 47.4, 48.4, 52.5, 127.3, 128.3, 128.6, 135.7, 136.1, 137.2, 138.9, 142.4, 142.7, 143.0, 173.6, 175.0. ESI-MS: positive mode m/z = 308.4 ([M + H]+). HRMS (EI) calcd for C19H21N3O 308.1757, found 308.1743. Purity (> 99 %). IR (cm−1) 2855, 2579, 1718, 1604, 1435, 1261, 1099, 970, 633. Anal. (C19H21N3O *1.0C4H4O4*1.5H2O) C, H, N.
4.61. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl(4-(pyridin-4-yl)phenyl)methanone fumaric acid salt (53F)
The N-tboc protected compound was obtained by using the general procedure E with 4-(4-pyridyl)benzoic acid (199 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (246 mg, 60 %) was obtained. The N-tboc protection group of this solid (180 mg, 0.44 mmol) was cleaved using the general procedure F for 4 h and a white solid 53 (132 mg, 97 %) was obtained after extraction. This solid 53 (132 mg, 0.43 mmol) was transferred to its fumaric acid salt 53F by using the general procedure I with fumaric acid (50 mg, 0.43 mmol). Compound 53F (120 mg, 58 %) was obtained as a white solid in 34 % yield over three steps; mp 175–180 °C (dec). 1H NMR (400 MHz, D2O) δ 2.02 (br m, 2H), 2.25 (br s, 1H), 2.46 (br s, 1H), 3.34–3.60 (br m, 6H), 3.89 (br d, J = 13.2 Hz, 1H), 4.56 (br d, J = 14.0 Hz, 1H), 6.58 (s, 3.00H), 7.70 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 8.32 (d, J = 6.7 Hz, 2H), 8.81 (d, J = 6.8 Hz, 2H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 28.0, 47.1, 47.7, 48.4, 52.5, 125.3, 128.6, 129.1, 135.5, 137.1, 138.0, 142.4, 157.2, 173.0, 174.7. ESI-MS: positive mode m/z = 308.4 ([M + H]+). HRMS (EI) calcd for C19H21N3O 308.1757, found 308.1749. Purity (> 99.5 %). IR (cm−1) 2906, 2694, 2601, 1698, 1639, 1255, 1171, 969, 834, 636. Anal. (C19H21N3O *1.4C4H4O4*0.75H2O) C, H, N.
4.62. (4-(1H-imidazol-1-yl)phenyl)((1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl)methanone fumaric acid salt (54F)
The N-tboc protected compound was obtained by using the general procedure E with 4-(1H-imidazol-1-yl)benzoic acid (188 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a white solid (366 mg, 92 %) was obtained. The N-tboc protection group of this solid (317 mg, 0.80 mmol) was cleaved using the general procedure F for 2 h and a white solid 54 (209 mg, 88 %) was obtained after extraction. This solid 54 (209 mg, 0.71 mmol) was transferred to its fumaric acid salt 54F by using the general procedure I with fumaric acid (82 mg, 0.71 mmol). Compound 54F (149 mg, 36 %) was obtained as a white solid in 29 % yield over three steps; mp 178–181 °C (dec). 1H NMR (400 MHz, D2O) δ 2.01 (br m, 2H), 2.25 (br s, 1H), 2.45 (br s, 1H), 3.34–3.64 (br m, 6H), 3.87 (br d, J = 12.8 Hz, 1H), 4.54 (br d, J = 13.8 Hz, 1H), 6.52 (s, 2.0H), 7.61 (s, 1H), 7.72 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 7.90 (s, 1H), 9.09 (s, 1H). 13C NMR (100 MHz, D2O) δ 25.8, 26.2, 27.9, 47.2, 47.4, 48.4, 52.5, 121.6, 122.6, 123.3, 129.5, 135.0, 135.9, 136.4, 137.1, 174.2, 174.5. ESI-MS: positive mode m/z = 297.4 ([M + H]+). HRMS (EI) calcd for C17H20N4O 297.1710, found 297.1700. Purity (> 99 %). IR (cm−1) 3104, 2910, 2699, 1704, 1635, 1523, 1257, 969, 851, 636. Anal. (C17H20N4O*0.9C4H4O4*1.0H2O) C, H, N.
4.63. (1R,5S)-3,7-diazabicyclo[3.3.1]nonan-3-yl(2-(pyridine-4-yl)thiazol-4-yl)methanone fumaric acid salt (55F)
The N-tboc protected compound was obtained by using the general procedure E with 2-(4-pyridyl)thiazole-4-carboxylic acid (206 mg, 1 mmol) for 12 h. A mixture of CH2Cl2 and MeOH (20:1) was used for the chromatographic purification. After removal of the solvents a yellow oil (401 mg, 97 %) was obtained. The N-tboc protection group of this oil (401 mg, 0.97 mmol) was cleaved using the general procedure F for 2 h and a yellow oil 55 (252 mg, 83 %) was obtained after extraction. This oil 55 (252 mg, 0.80 mmol) was transferred to its fumaric acid salt 55F by using the general procedure I with fumaric acid (93 mg, 0.80 mmol). Compound 55F (119 mg, 35 %) was obtained as an off-white solid in 28 % yield over three steps; mp 124–128 °C (dec). 1H NMR (400 MHz, D2O) δ 2.06 (br m, 2H), 2.27 (br s, 1H), 2.45 (br s, 1H), 3.34–3.72 (br m, 6H), 4.33 (br s, 1H), 4.51 (br s, 1H), 6.60 (s, 1.58H), 8.19 (d, J = 5.6 Hz, 2H), 8.31 (s, 1H), 8.78 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, D2O) δ 26.3, 28.6, 47.8, 47.9, 48.0, 52.0, 122.9, 128.9, 135.5, 144.3, 147.1, 150.8, 164.8, 167.9, 173.1. ESI-MS: positive mode m/z = 315.4 ([M + H]+). HRMS (EI) calcd for C16H18N4OS 315.1274, found 315.1261. Purity (> 99.5 %). IR (cm−1) 3354, 3201, 2860, 2600, 1704, 1630, 1458, 1406, 1247, 1015, 972, 641. Anal. (C16H18N4OS*1.0C4H4O4*1.0H2O) C, H, N.
4.64. Calculation of physicochemical properties
The physicochemical properties have been calculated using ACD/ADME Suite 5.0 and ACD/PhysChem (ACD/Labs).
Supplementary Material
Scheme 1.
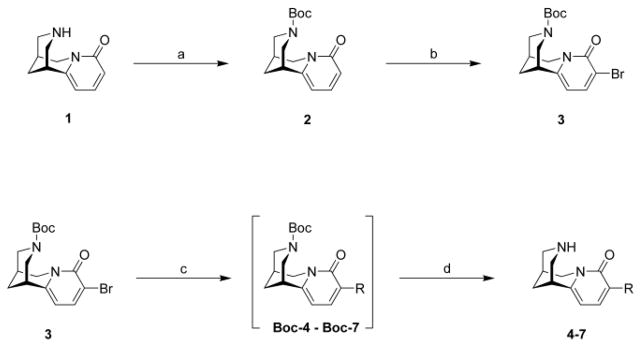
Synthesis of cytisine derivatives. Reagents and conditions: (a) (Boc) 2O, Na2CO3, CH2Cl2, H2O, reflux, 2 h; (b) NBS, CH2Cl2, reflux, 2 h; (c) R-B(OH)2, Pd(PPh3)4, base, DME or DMF, H2O [Ar], MW (30 W), 80 aC, 30 to 60 min; (d) H2O, MW (150 W), 150 °C, 30 min.
Scheme 2.

Synthesis of bispidine derivatives. Reagents and conditions: (a) (CH2O)n, BnNH2, AcOH, MeOH, [Ar] reflux, 6 h; (b) N2H4 (80%), NaOH, diethylene glycol, 125 °C, 2 h, then Dean Stark trap, 140 °C, 8 h; (c) Pd/C (5%), H2 (2–4 bar), MeOH, rt, 4–24 h; (d) CDI, THF, reflux, 2 h; (e) (i) MeI, MeCN, THF, rt, 24 h; (ii) R-COOH, Et3N, MeCN, rt, 12–120 h; (f) HCl/1,4-dioxane (4M), rt, 4 h; (g) anhydr. ZnBr2, CH2Cl2, rt, 12–120 h; (h) R-COCl, Et3N, toluene, rt, 2h; (j) R-COOH, DCC, DMAP, CH2Cl2, 0 °C to rt, 12 h.
Acknowledgments
The authors thank Tim Deinet, Julia Thomas, Florian Schorr, Franziska Krumbiegel, and Alia Abdelraman for their technical assistance. The authors gratefully thank Dr. Jörg Hockemeyer for providing the propioloic acid derivatives. We thank Dr. Marina R. Picciotto for many helpful discussions. This work was financially supported by the German Research Council (DFG; GRK 677) (CE, DG) and the National Institutes of Health P20RR016467 (CE, IT, DG). CS and RLP were supported by the James and Esther King Biomedical Research Grant 1KG12 (CS, RLP). Elemental analyses for some compounds were conducted by the UH Hilo Analytical Laboratory for this project supported in part by the National Science Foundation award number EPS-0903833. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Abbreviations
- BBB
blood-brain barrier
- CD3OD
tetradeuteromethanol
- CDCl3
deuterochloroform
- CH2Cl2
dichloromethane
- CNS
central nervous system
- D2O
deuterium oxide
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-(Dimethylamino)pyridine
- DME
1,2-dimethoxyethane
- DMF
N,N-dimethylformamide
- Et2O
diethyl ether
- Et3N
triethylamine
- EtOAc
ethyl acetate
- HBA
hydrogen bond acceptor
- HCl
hydrogen chloride
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- HSS
HEPES-buffered salt solution
- K2CO3
potassium carbonate
- KBr
potassium bromide
- KMnO4
potassium permanganate
- KOH
potassium hydroxide
- MeCN
acetonitrile
- MeI
iodomethane
- MeOH
methanol
- MgSO4
magnesium sulfate
- nAChR
nicotinic acetylcholine receptor
- NaHCO3
sodium hydrogen carbonate
- NaOH
sodium hydroxide
- Pd/C
palladium on activated charcoal
- PE
petroleum ether
- PEI
poly(ethyleneimine)
- Ro5
rule of five
- THF
tetrahydrofuran
- TPSA
topological polar surface area
- TRIS
Tri(hydroxymethyl)aminomethane
- ZnBr2
zinc bromide
Footnotes
Supplementary data associated with this article can be found in the online version at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Gündisch D, Eibl C. Expert Opin Ther Patents. 2011;21:1867. doi: 10.1517/13543776.2011.637919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Changeux JP. JBC. 2012;287:40207. doi: 10.1074/jbc.R112.407668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Changeux JP, Taly A. Trends Mol Med. 2008;14:93. doi: 10.1016/j.molmed.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Lukas RJ. Biochem Pharmacol. 2011;82:800. doi: 10.1016/j.bcp.2011.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gotti C, Riganti L, Vailati S, Clementi F. Curr Pharm Des. 2006;12:407. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- 6.Mazurov AA, Kombo DC, Akireddy S, Murthy S, Hauser TA, Jordan KG, Gatto GJ, Yohannes D. Bioorg Med Chem Lett. 2013;23:3927. doi: 10.1016/j.bmcl.2013.04.058. [DOI] [PubMed] [Google Scholar]
- 7.Eibl C, Tomassoli I, Munoz L, Stokes C, Papke RL, Gündisch D. BMC (Part 1) doi: 10.1016/j.bmc.2013.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tosco P, Ahring PK, Dyhring T, Peters D, Harpsoe K, Liljefors T, Balle T. J Med Chem. 2009;52:2311. doi: 10.1021/jm801060h. [DOI] [PubMed] [Google Scholar]
- 9.Bunnelle WH, Barlocco CD, Jerome D, Dart MJ, Meyer MD, Ryther KB, Schrimpf MR, Sippy KB, Toupence RB. 2000044755. PCT Int Appl, WO. 2000
- 10.Peters D, Olsen GM, Nielsen E, Nielsen S, Ahring PK, Dyhring T. 2001044243. PCT Int Appl, WO. 2000
- 11.Mazurov A, Miao L, Xiao T-D, Hammond PS, Miller CH, Akireddy SR, Murthy VS, Whitaker RC, Breining SR, Melvin MS. 2008057938. PCT Int Appl, WO. 2007
- 12.Akireddy SR, Bhatti BS, Breining SR, Hammond PS, Heemstra RJ, Mazurov A, Melvin MS, Miao L, Murthy VS, Strachan J-P, Xiao Y-D. 2009111550. PCT Int Appl, WO. 2009
- 13.Eibl C. PhD Thesis. University of Bonn; Germany: Jul, 2009. [Google Scholar]
- 14.Gündisch D, Eibl C. Biochem Pharmacol. 2009;78:905. [Google Scholar]
- 15.Tomassoli I, Eibl C, Wulf M, Papke RL, Picciotto MR, Gündisch D. Biochem Pharmacol. 2011;82:1023. [Google Scholar]
- 16.Munoz L. PhD Thesis. University of Bonn; Germany: Nov, 2005. [Google Scholar]
- 17.Picciotto M, Gündisch D, Munoz L, Andrä M, Mineur Y. 2007100430. PCT Int Appl, WO. 2007
- 18.Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gundisch D, Picciotto MR. J Pharmacol Exp Ther. 2009;329:377. doi: 10.1124/jpet.108.149609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gündisch D, London ED, Terry P, Hill GR, Mukhin AG. Neuroreport. 1999;10:1631. doi: 10.1097/00001756-199906030-00002. [DOI] [PubMed] [Google Scholar]
- 20.Imming P, Klaperski P, Stubbs MT, Seitz G, Gündisch D. Eur J Med Chem. 2001;36:375. doi: 10.1016/s0223-5234(01)01222-3. [DOI] [PubMed] [Google Scholar]
- 21.Mukhin AG, Gündisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, Chambers J, Vaupel DB, King S, Picciotto MR, Innis R, London ED. Mol Pharmacol. 2000;57:642. doi: 10.1124/mol.57.3.642. [DOI] [PubMed] [Google Scholar]
- 22.Perez EG, Mendez-Galvez C, Cassels BK. Nat Prod Reports. 2012;29:555. doi: 10.1039/c2np00100d. [DOI] [PubMed] [Google Scholar]
- 23.Halevi S, Yassin L, Eshel M, Sala F, Sala S, Criado M, Treinin M. JBC. 2003;278:34411. doi: 10.1074/jbc.M300170200. [DOI] [PubMed] [Google Scholar]
- 24.Papke RL, Papke JKP. Br J of Pharm. 2002;137:49. [Google Scholar]
- 25.Papke RL, Stokes C. Methods. 2010;51:121. doi: 10.1016/j.ymeth.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng C, Stokes C, Mineur YS, Picciotto MR, Tian C, Eibl C, Tomassoli I, Guendisch D, Papke RL. J Pharmacol Exp Ther. 2013 Aug 22; doi: 10.1124/jpet.113.206904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghose AK, Herbetz T, Hudkins RL, Dorsey BD, Mallamo JP. ACS Chem Neurosci. 2012;3:50. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.