

SUMMARY
Myelin is essential in vertebrates for the rapid propagation of action potentials, but the molecular mechanisms driving its formation remain largely unknown. Here we show that the initial stage of process extension and axon ensheathment by oligodendrocytes requires dynamic actin filament assembly by the Arp2/3 complex. Unexpectedly, subsequent myelin wrapping coincides with the upregulation of actin disassembly proteins, rapid disassembly of the oligodendrocyte actin cytoskeleton, and does not require Arp2/3. Inducing loss of actin filaments drives oligodendrocyte membrane spreading and myelin wrapping in vivo, and the actin disassembly factor gelsolin is required for normal wrapping. We show that myelin basic protein, a protein essential for CNS myelin wrapping whose role has been unclear, is required for actin disassembly, and its loss phenocopies loss of actin disassembly proteins. Together these findings provide insight into the molecular mechanism of myelin wrapping and identify it as an actin-independent form of mammalian cell motility.
Graphical Abstract
INTRODUCTION
Myelination of axons in the CNS is essential for the rapid propagation of action potentials, and loss of myelin in demyelinating diseases leads to severe disabilities (Bercury and Macklin, 2015). During myelination, oligodendrocyte (OL) precursor cells (OPCs) differentiate and undergo a series of morphological changes: they (1) extend numerous cellular processes to (2) ensheath axons, (3) spirally wrap around the axon underneath previous wraps, (4) simultaneously extend longitudinally along the axon, (5) and compact their cytoplasm to form mature myelin. The geometry of this complex cellular process was recently defined (Snaidero et al., 2014), but its molecular mechanism remains unknown.
Eukaryotic cells control their shape and move by means of a dynamic actin cytoskeleton. “Classical” cell motility is driven by the lamellipodium, an actin-based structure constructed by coordinated activities of actin filament nucleation—by the Arp2/3 complex—and filament capping, crosslinking, and severing by multiple other proteins (Fletcher and Mullins, 2010). The Arp2/3 complex (hereafter Arp2/3) builds branched networks of actin filaments, and is essential for cell migration on a surface (Wu et al., 2012). However, work in the past decade reveals that cells moving in confined, 3-dimensional spaces often utilize actin-independent mechanisms to propel themselves forward (Paluch and Raz, 2013). The current model of CNS myelin wrapping suggests that the OL inner tongue is a modified lamellipodium, using the force of Arp2/3-dependent actin assembly to spirally wrap around axons (Bauer et al., 2009). To date there is little direct experimental evidence to support this hypothesis, and the precise role of the actin cytoskeleton in CNS myelination remains to be elucidated.
Previous work in vitro has suggested that actin dynamics are required for early stages of OL process outgrowth and branching (Wilson and Brophy, 1989; Song et al., 2001). In vivo, signaling pathways that regulate the actin cytoskeleton in other cell types have roles in myelination in both the CNS and PNS, including phosphoinositide signaling (Goebbels et al., 2010; Snaidero et al., 2014), the Rho family GTPases (Thurnherr et al., 2006), and the Arp2/3 activators WAVE1 in the CNS (Kim et al., 2006) and N-Wasp in the PNS (Novak et al, 2011; Jin et al., 2011). Gene profiling studies from our lab and others have revealed that the mRNA of many proteins that regulate the actin cytoskeleton are highly induced when OPCs differentiate into myelinating OLs, and that, surprisingly, a number of well-characterized proteins that cause disassembly of actin filaments (e.g. gelsolin and cofilin family members) are among the most abundant transcripts in myelinating OLs (Liu et al., 2003; Zhang et al., 2014).
Myelin basic protein (MBP) is the major structural element of CNS myelin, where it is essential for both myelin wrapping and compaction (Readhead et al., 1987). MBP binds to PI(4,5)P2 on the cytoplasmic faces of the OL plasma membrane, and self-assembles to promote compaction and membrane polarization (Nawaz et al., 2009; Aggarwal et al., 2011). In addition to compaction, MBP is also essential for myelin wrapping (Rosenbluth, 1980; Shine et al., 1992), but its cellular mechanism for promoting wrapping is unknown. MBP has also been shown to interact with actin in vitro (Baryłko and Dobrowolski, 1984; Boggs and Rangaraj, 2000), but the functional significance of this and whether MBP interacts with actin in vivo are also unknown.
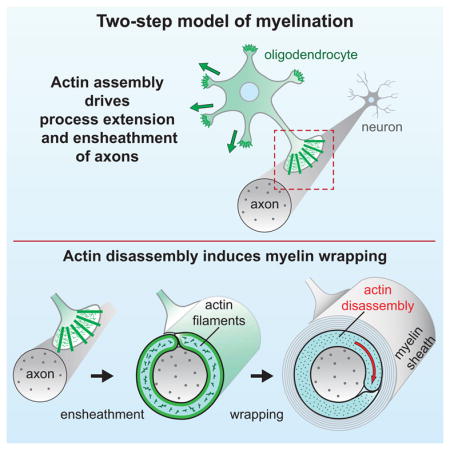
Here we show that the actin cytoskeleton controls CNS myelination in two distinct steps. First, actin assembly by the Arp2/3 complex drives OL process outgrowth and branching; consistent with this, ArpC3 conditional knockout mice fail to ensheath axons. Second, as OLs differentiate they massively disassemble their actin cytoskeleton, which induces myelin wrapping. Mice lacking the actin disassembly factor gelsolin have wrapping defects, and inducing global actin disassembly accelerates the spreading of OL myelin membranes in vitro and myelin wrapping in vivo. In contrast, Arp2/3 is not required for myelin wrapping. Finally, we provide evidence that MBP is required for actin disassembly, which it may regulate by binding to membrane PI(4,5)P2, and releasing actin disassembly proteins to induce myelin wrapping.
RESULTS
Actin dynamics during developmental myelination in vivo
Despite the importance of myelination for the normal function of the CNS, the cellular mechanisms that allow OL membranes to spirally wrap and compact around axons remain poorly understood. Since eukaryotic cells change their shape and move by means of a dynamic actin cytoskeleton, we investigated how the OL cytoskeleton is organized and regulated during myelination. We first examined the actin cytoskeleton in mature myelin in vivo, using phalloidin staining of actin filaments in P45 spinal cord sections. We validated the specificity of phalloidin for actin filaments in tissue sections with the actin filament binding drug, jasplakinolide (Jasp), which blocks phalloidin staining in a dose-dependent manner (Figure S1). Whereas actin filaments were highly enriched in gray matter, white matter tracts containing myelinated axons had comparatively low levels of actin filaments (Figure 1A). Higher magnification confocal micrographs showed that the bulk of actin filaments in white matter was excluded from compact myelin (Figure 1B and C), similar to the PNS (Trapp et al., 1989). In agreement with this observation, purified myelin fractions appeared devoid of actin protein by immunoblotting (Figure 1D).
Figure 1. Loss of actin filaments during myelination in vivo.

(A–C) P45 mouse spinal cord transverse sections stained for compact myelin with FluoroMyelin Red (FMred, red), actin filaments with Alexa 488-phalloidin (phalloidin, green), and nuclei (DAPI, blue). (A) Wide field epifluorescence showing low levels of actin filaments in mature white matter. (B) Confocal microscopy revealed lowest levels of actin filaments (phalloidin, green) in compact myelin (FMred, red). Arrows point to compact myelin.
(C) Representative line scan through two myelinated axons from (B); a.u., arbitrary units.
(D) Immunoblotting of brain lysate and fractionated compact myelin from adult mice. n = 3 mice fractionated on two separate days.
(E) Wide field quantification of phalloidin and FMred staining intensities in mouse dorsal white matter regions of interest (ROI) during postnatal development. Error bars: SEM from n = 5–6 animals per time point; multiplicity adjusted P values shown for P10 (phalloidin) or P5 (FMred) compared to other days, calculated using Tukey’s multiple comparisons test, ***p < 0.001, ****p < 0.0001.
(F and G) Confocal microscopy of developing P5 mouse spinal cord transverse sections, stained for MBP (red) and phalloidin (green). Arrows, early sheaths with low MBP intensity and high phalloidin intensity. Arrowheads, more mature sheaths with high MBP intensity and low phalloidin. (G) Quantification of individual P5 myelin sheaths showed that actin filament levels (phalloidin) decreased with increasing MBP intensity.
See also Figure S1.
We next quantified actin filaments and compact myelin in the developing dorsal white matter of the spinal cord during active myelination. White matter actin filament levels peaked at the start of myelination (P10), but actin filament levels rapidly declined as myelin wrapping and compaction began (Figure 1E). To look more closely at actin in nascent myelin sheaths, we co-stained spinal cord sections for actin filaments and MBP at the onset of myelination (P5). MBP protein level is proportional to the degree of CNS myelin wrapping (Shine et al., 1992), so immunostaining with a validated MBP antibody (Figure S1) indicates the maturity of individual myelin sheaths in tissue sections. Immature myelin sheaths with low levels of MBP had high levels of actin filaments, whereas actin filament levels were dramatically reduced in more mature sheaths (Figure 1, F and G), suggesting that actin filaments are disassembled in OLs during the latter stages of myelination.
Two phases of actin cytoskeletal dynamics during OL differentiation
We next asked whether purified OLs in culture could be used to study the changes in the actin cytoskeleton during myelination that we observed in vivo. We prospectively isolated OPCs from neonatal rats and induced their differentiation into OLs, fixed them at different stages of differentiation over 6 days of culture, and stained their actin cytoskeletons with phalloidin. OLs undergo stereotyped morphological changes as they differentiate (Figure 2A), extending numerous cellular processes to form an arborized morphology, then flattening out into myelin membrane sheets that are similar to “unrolled” myelin (Movie S1 and Figure S2; Aggarwal et al., 2011). Consistent with previous work (Wilson and Brophy, 1989; Song et al., 2001), we found that these early OL processes were highly enriched in actin filaments prior to MBP expression (Figure 2, B–D). Using structured illumination superresolution microscopy we observed that these OL processes were composed of ordered arrays of actin filaments, and were morphologically akin to neuronal growth cones (Figure 2G; Fox et al., 2006). Accordingly, inhibiting actin filament dynamics with latrunculin A (LatA) or Jasp during these early stages arrested OL process extension and arborization (Movies S2 and S3).
Figure 2. Two phases of actin dynamics during OL differentiation.

(A–F) OL differentiation time course. (A), Cartoon depicting stereotyped stages of OL morphology changes during differentiation in culture in the absence (top) or presence (bottom) of neurons. (B–F), Primary rat OPCs were fixed before differentiation (B) or after differentiating for 3 (C), 5 (D), or 6 (E and F) days. OLs were immunostained for MBP (red) and stained for actin filaments (phalloidin, green and on right, D′–F′) and nuclei (DAPI, blue).
(G) Structured illumination microscopy of phalloidin staining in a single OL process.
(H) Quantification of actin filament levels in OLs at each stage of differentiation. Error bars: SEM from n > 50 OLs per day.
(I) Immunoblot of total OL actin protein (top, 42 kDa) and GAPDH (bottom, 37 kDa) at 1, 2, 3, and 5 d differentiation.
As OLs matured further and began to express MBP, however, their actin cytoskeleton was almost completely disassembled, first in MBP+ compact myelin membrane regions (Figure 2E) and then throughout the entire OL (Figure 2, F and H). Immunoblotting revealed that total cellular actin persisted throughout differentiation, so loss of actin filaments corresponded to their disassembly (Figure 2I). Thus, OLs in culture recapitulated the disassembly of actin filaments that we observed in developing myelin in vivo, even in the absence of neurons or a native CNS microenvironment.
Arp2/3-mediated actin assembly is required for myelin initiation in the CNS
Our characterization of the OL actin cytoskeleton in vivo and in culture suggests that myelination proceeds in two phases: first, dynamic actin assembly powers early steps of myelination, and second, the OL cytoskeleton is completely disassembled during the formation of mature, compact myelin. To test this hypothesis, we first explored the role of actin in the initial stage of axon ensheathment using OPC-neuron coculture assays. When plated on dorsal root ganglion neuron (DRG) axons, differentiating OLs initially extended processes enriched in actin filaments, but as they matured and developed MBP+ myelin internodes, the actin filaments disappeared, consistent with isolated OLs above (Figure 3A). The actin assembly inhibiting drugs LatA and Jasp blocked axon ensheathment in a dose-dependent manner, and did so at concentrations that did not affect OL differentiation or viability (Figure S2).
Figure 3. Actin assembly by Arp2/3 is required for myelin initiation.

(A) Cocultures of DRG neurons and OPCs grown for 7 (left) or 9 (right) days, then fixed and stained for MBP, phalloidin, and axons (neurofilament, NF).
(B) OPCs were differentiated for 1d and immunostained for the Arp2/3 subunit ArpC1 (red), and counterstained with phalloidin (green).
(C and D) OLs required Arp2/3 for normal levels of actin filaments and to ensheath axons. (C) A dilution series of Arp2/3 inhibitor CK-666 was applied to OLs for 24 hr, then OLs were fixed and stained for actin filaments with phalloidin (see also Figure S3). (D) Axon ensheathment assay of OPCs on RGC neurons, treated with a dilution series of CK-666 or the inactive control compound CK-689. Graph shows quantification of axonal ensheathment per Olig2+ OPC/OL, normalized to DMSO-treated controls. n = 2 experiments. *p < 0.05, **p < 0.01, Dunnett’s multiple comparison test.
(E and F) Recombination (E) and reduction of ArpC3 protein (F) in OLs purified from P18–20 ArpC3Flox/Flox (Flox), ArpC3Flox/+; CNPCre/+ (heterozygote) and ArpC3Flox/Flox; CNPCre/+ (CNPCre-CKO) mice by immunopanning. Flox-R, recombined; Flox-NR, non-recombined; bands were confirmed by sequencing. n = 4 CNPCre-CKO, 3 Flox, 1 heterozygote. (F) Immunoblot showing reduction of ArpC3 protein in CNPCre-CKO OLs. n = 2 animals per genotype.
(G and H) Transmission electron microscopy of optic nerves from Flox (left), CNPCre-CKO (right), and heterozygote (not shown) mouse littermates. Quantification in (H) shows percent axons myelinated at P18, P45, and P90. Each data point is average from a single animal. n = 5–7 animals per age, per genotype. **p < 0.01, ***p < 0.001, ***p < 0.0001, Dunnett’s multiple comparison test (P18 and P45) or Student’s t-test (P90).
(I) Percent axons myelinated in optic nerves from P72 Flox and ArpC3Flox/Flox; Olig2Cre/+ (Olig2Cre-CKO) mouse littermates. n = 5 animals per genotype; **p < 0.01, Student’s t-test.
(J) EM micrograph shows an example of a myelin outfolding in a P90 CNPCre-CKO mouse.
(K) Quantification of outfoldings in Flox versus CNPCre-CKO littermates at P90. n = 6 animals per genotype; **p < 0.01, Student’s t-test.
Error bars: SEM.
See also Figures S2, S3, S4, S5, and Movies S2 and S3.
To determine the contribution of actin assembly to myelination in vivo, we next addressed the role of Arp2/3, the major actin nucleation factor that assembles branched arrays of actin filaments in the lamellipodia of motile cells (Fletcher and Mullins, 2010; Wu et al., 2012). Our recent RNAseq analysis of CNS glia indicates that all seven subunits of Arp2/3 are expressed by myelinating OLs (Figure S3; Zhang et al., 2014). Both endogenous ArpC1 and GFP-Arp3 localized to actin filament-rich cell edge as well as the OL cell body (Figure 3B and S3). Pharmacological inhibition of Arp2/3 by CK-666 (Nolen et al., 2009) induced a dose-dependent loss of OL actin filaments, suggesting that Arp2/3 plays a major role in constructing the OL cytoskeleton during the early phase of myelination (Figure 3C and S3). Consistent with this finding, CK-666 significantly inhibited OL ensheathment of retinal ganglion cell (RGC) axons in myelinating cocultures, whereas the inactive control compound CK-689 had only a mild effect at the highest concentration (Figure 3D). In isolated OLs CK-666 strongly inhibited the formation of actin-filament rich growth cones, process outgrowth, and branching (Figure S3). CK-666 also mildly inhibited proliferation of OPCs (Figure S3). These results suggested that Arp2/3-dependent actin assembly is required in early stages of OL differentiation and the initiation of myelination.
To determine if Arp2/3 is required for myelination in vivo, we conditionally deleted the essential subunit ArpC3 (Yae et al., 2006) using a floxed allele (Kim et al., 2013) and several orthogonal Cre lines that function in the OL lineage. We first used CNP-Cre (Lappe-Siefke et al., 2003), which is expressed by early OLs at the start of myelination. ArpC3Flox/Flox; CNPCre/+ conditional knockouts (hereafter, CNPCre-CKOs) were born in normal Mendelian frequencies, and developed severe motor defects including tremor and ataxia in the second to third postnatal weeks. In contrast, ArpC3Flox/Flox; CNP+/+ (Flox) and ArpC3Flox/+; CNPCre/+ (heterozygote) littermates appeared normal. As CNP-Cre is also expressed by Schwann cells, motor defects are likely due, at least in part, to myelination defects in the PNS (Zuchero and Barres, unpublished observations). We confirmed recombination and loss of ArpC3 protein in OLs by purifying mature OLs from the brains of Flox, heterozygote, and CNPCre-CKO littermates by immunopanning (Figures 3, E and F, and S4). CNPCre-CKO OLs had a 70.5±3.6% reduction of ArpC3 protein by immunoblotting, and the residual ~30% was likely due to the presence of “escaper” OLs that do not recombine fully (Flox, non-recombined band, Figure 3E) and have a proliferative advantage over CKO OLs.
The optic nerve is an ideal location to quantify CNS myelination, since the axons are aligned, myelination is relatively uniform, and the geometry and timing of myelin wrapping is well described (Dangata and Kaufman, 1997; Snaidero et al., 2014). We observed striking hypomyelination in optic nerves of CNPCre-CKO mice, but not in Flox littermates (Figure 3G). CNPCre-CKOs had fewer than half as many axons myelinated than Flox littermates at all ages analyzed (Figure 3H), suggesting that OLs lacking functional Arp2/3 are unable to initiate axon ensheathment. This phenotype persisted from P18 through P90, so it is not due to a developmental delay. In contrast, heterozygotes displayed small but significant hypomyelination at P18, but appeared grossly normal by P45. Axon caliber and axonal mitochondria size were unaffected in CNPCre-CKOs, and there was only a small increase in the number of degenerated axons at P90 (an expected result of hypomyelination), arguing that hypomyelination is largely due to cell autonomous loss of ArpC3 in OLs (Figure S4).
Since CNP-Cre expression is not completely restricted to OLs, we also utilized Olig2-Cre (Schüller et al., 2008) to conditionally delete ArpC3 in OPCs. ArpC3Flox/Flox; Olig2Cre/+ mice (Olig2Cre-CKOs) were severely hypomyelinated (Figure 3I), similar to CNPCre-CKOs. At P8 there was no gross difference in the number of OPCs in the optic nerves of Olig2Cre-CKO mice compared to Flox littermates, suggesting that hypomyelination in Olig2Cre-CKOs was unlikely due to impaired OPC migration (Figure S4). OLs purified from Olig2Cre-CKO mice had fewer branches and dim phalloidin staining (Figure S4), similar to treating with the Arp2/3 inhibitor CK-666 (Figure S3).
Despite the hypomyelination we observed by EM, immunohistochemical staining of MBP and FluoroMyelin Red revealed no significant differences in staining intensity in the optic nerves between CNPCre-CKO mice and Flox littermates (Figure S5). This suggests that gross levels of myelin membrane are not affected in CNPCre-CKO mice, or that these staining techniques are only sensitive to all-or-none levels of myelin (see Figure S1). Indeed, CNPCre-CKOs displayed a 10-fold higher frequency of redundant myelin membranes, or outfoldings, than Flox littermates (Figures 3, J and K, and S5), similar to those observed in hypermyelinating mutants of phosphoinositide signaling and some myelin pathologies (Goebbels et al., 2010). Outfoldings were often several microns in length and occasionally appeared to aberrantly ensheath multiple axons. An average optic nerve transverse section displayed outfoldings in 2.2 ± 0.5% of CNPCre-CKO myelin sheaths (0.2 ± 0.1% in Flox littermates), but this may underestimate the number of internodes with outfoldings somewhere along their length (Snaidero et al., 2014). We therefore used serial block face scanning EM to reconstruct nerves of single Flox and CNPCre-CKO littermates. In the CKO nerve, outfoldings typically originated focally along an internode, and appeared more common than estimated from single transverse sections (Figure S5). We observed similar myelin outfoldings when ArpC3 was deleted by Olig2-Cre (Figure S5). Together, these data indicate that Arp2/3-mediated actin assembly is essential for normal myelin initiation and axon ensheathment, and provides evidence that actin filaments play a “steering” role during myelin wrapping, to limit aberrant myelin membrane growth and outfoldings.
Myelin wrapping is independent of Arp2/3-mediated lamellipodial growth
Myelin wrapping has been proposed to be driven by dynamic actin assembly by Arp2/3, like the lamellipodium of a motile cell. However, this has been difficult to test, in part because actin plays an essential role during myelin initiation as shown above. Is Arp2/3-mediated actin assembly also required to power myelin wrapping? We used tamoxifen-inducible Plp1-CreERT mice (Doerflinger et al., 2003) to ablate ArpC3 in OLs after ensheathment, but during wrapping (Figure 4A). The mouse optic nerve progresses from entirely unmyelinated at birth to almost completely ensheathed in the second postnatal week; subsequently the majority of myelin wrapping occurs between the second and fifth postnatal week (Dangata and Kaufman, 1997). Thus, we injected mice with five daily injections of tamoxifen from P10–P14 to induce loss of ArpC3 after ensheathment, and measured myelin wrapping at P30.
Figure 4. Arp2/3 is dispensable for myelin wrapping.

(A) Experimental paradigm for tamoxifen injections to ablate ArpC3 during myelin wrapping.
(B) Reduction of ArpC3 mRNA and protein in OLs purified at P25 from ArpC3Flox/Flox (Flox) and ArpC3Flox/Flox; Plp1-CreERT (induced CKO, Plp1Cre-iCKO) mice that were injected with tamoxifen from P10–14. Top left, immunoblotting of ArpC3 from immunopanned OLs. Top right, densitometry of ArpC3 protein in OLs purified from n = 3 animals per genotype; *p < 0.05, Student’s t-test. Bottom, single cell RT-PCR analysis shows ArpC3 knockout OLs (GAPDH but no ArpC3, *), WT OLs (both GAPDH and ArpC3 bands, ●), or no cells (neither GAPDH nor ArpC3, ø). n > 22 reactions per animal, from one Flox and two Plp1Cre-iCKO mice.
(C–E) Transmission electron microscopy of P30 optic nerves showed no difference in number of myelin wraps in Plp1Cre-iCKOs. (C), example micrographs, (D), average number of myelin wraps per animal, and (E), distribution of number of wraps in all animals. Error bars: SEM from n = 5–6 animals per genotype; >100 myelin sheaths per animal; n.s. = not significant, Student’s t-test.
(F) Experimental paradigm for tamoxifen injections to ablate PTEN and/or ArpC3 at P30, and full genotypes of mice.
(G) Transmission electron microscopy of optic nerves at P90 showing increased myelin wrapping in PTEN-iCKO (middle) and ArpC3+PTEN double iCKO (right) mice, compared to double floxed controls (left).
(H) g-ratio analysis of optic nerves at P90 shows lower g-ratios after deletion of PTEN or both ArpC3+PTEN.
(I) Axon diameter was unaffected.
Error bars: SEM from n = 6 animals per genotype; >100 myelin sheaths measured per animal; n.s. = not significant, *p < 0.05, **p < 0.01, Dunnett’s multiple comparison test.
See also Figure S5.
We first confirmed that our tamoxifen injection paradigm induced recombination in OLs in the optic nerve, using the fluorescent reporter mouse, mT/mG (Muzumdar et al., 2007; Figure S5). We next confirmed loss of ArpC3 protein during active wrapping (P25) by immunoblotting purified OLs (Figure 4B). ArpC3 was reduced to 45.0% ± 12.1% of wild-type levels in OLs purified from tamoxifen-injected ArpC3Flox/Flox; Plp1-CreERT mice (induced CKOs, hereafter Plp1Cre-iCKOs) suggesting that some OLs escaped recombination. Consistent with this, single cell RT-PCR analysis of individual immunopanned OLs revealed two discrete populations of OLs from Plp1Cre-iCKO animals, with either (1) normal or (2) undetectable levels of ArpC3 mRNA (Figure 4B). Approximately half of OLs from Plp1Cre-iCKO mice expressed little or no ArpC3, whereas all OLs purified from Flox littermates had normal levels of ArpC3. In vivo, we noted an increased frequency of myelin outfoldings in Plp1Cre-iCKOs compared to Flox littermates injected with tamoxifen (Figure S5), indicating loss of ArpC3 function in at least a subset of Plp1Cre-iCKO OLs. Together these observations support recombination and loss of ArpC3 protein in ~50% of Plp1Cre-iCKO OLs, rather than reduced ArpC3 protein in all Plp1Cre-iCKO OLs, consistent with previous observations of Plp1-CreERT efficacy in the optic nerve (Doerflinger et al., 2003).
Remarkably, ablation of ArpC3 after ensheathment did not prevent myelin wrapping (Figure 4, C–E). We counted the number of myelin wraps in P30 Flox and Plp1Cre-iCKO littermates and found no difference in the average number of wraps (Figure 4D), nor in the distribution of wrap number (Figure 4E). We also saw no significant decrease in the number of axons myelinated in Plp1Cre-iCKOs, confirming that tamoxifen-induced loss of ArpC3 occurs after myelin initiation (Figure S5). Thus, developmental myelin wrapping does not require Arp2/3.
Next, we utilized a hypermyelinating mouse mutant to further test the role of Arp2/3 in wrapping. Myelin wrapping in the optic nerve normally plateaus after P30, but can be experimentally reinitiated by knocking out the lipid phosphatase PTEN in OLs, leading to hypermyelination and increased wrapping (Goebbels et al., 2010; Snaidero et al., 2014). We therefore tested whether ArpC3 was required for increased wrapping stimulated by loss of PTEN. We injected mice with tamoxifen from P30–34 to induce conditional inactivation of PTEN and/or ArpC3 in OLs, then analyzed optic nerves two months later (Figure 4F). As expected, inducing loss of PTEN in OLs caused a significant increase in myelin thickness compared to ArpC3/PTEN double-flox or double-heterozygote controls (Figure 4, G and H) without affecting axon diameter (Figure 4I). Simultaneous deletion of ArpC3 and PTEN led to a similar increase in myelin wrapping, indicating that ArpC3 was not required for the increased wrapping triggered by PTEN deletion. Taken together, our results showed that Arp2/3-dependent actin assembly was required for myelin initiation, but not for spiral wrapping of myelin.
Actin disassembly drives myelin membrane growth and wrapping
If actin assembly by Arp2/3 is not required for OLs to wrap axons, what powers wrapping? Analysis of RNAseq data from three stages of the OL lineage in vivo (Zhang et al., 2014) revealed that, whereas actin assembly promoting proteins (e.g. Arp2/3 subunits) are statistically enriched in OPCs, actin disassembly-promoting proteins are highly enriched in actively myelinating OLs (Figure S6). Two of these proteins, gelsolin and cofilin-1, are among the top 50 most highly expressed genes by OLs at the start of myelination (Figure S6). We confirmed induction of actin disassembly proteins as OLs differentiate in culture using immunoblotting (Figure 5A). The upregulation of these proteins corresponded temporally with actin disassembly in maturing OLs (Figure 2, A–G).
Figure 5. Actin disassembly drives myelin wrapping.

(A) Induction of actin disassembly proteins as OLs differentiate. Primary rat OPCs differentiated for indicated time prior to immunoblotting.
(B) Primary rat OPCs were differentiated for 2 days until arborized, then treated overnight with DMSO (carrier, top left) or 125 nM LatA to disassemble the actin cytoskeleton (micrographs outlined in red). Micrographs show thresholded MBP immunostaining.
(C) Quantification of morphology after overnight treatment with DMSO or LatA. Cartoon shows examples of each morphology. n = 3 biological replicates.
(D) Live cell imaging of 3d differentiated primary rat OPCs treated with LatA. Quantification of percent area change over 5 min before or after LatA treatment, measured from image stills. n = 7 OLs from 2 experimental days, **p < 0.01, paired Student’s t-test.
(E and F) Quantification of g-ratios of myelinated axons as a function of axon diameter, in adult gelsolin KO mice (green) compared to wild-type littermate controls (gray). n = 6 animals per genotype. **p < 0.01, Student’s t-test.
(G–L) Actin disassembly accelerates myelin wrapping in vivo. Gelfoam pre-loaded with DMSO or LatA was surgically implanted in the dorsal thoracic spinal cord of mice from P12–P18. Representative TEM micrographs show DMSO-treated (G) and LatA-treated (H) mice. LatA treatment induced thicker myelin and occasional redundant myelin outfoldings (right). (I) Myelin in LatA-treated mice was thicker, and (J and K) average g-ratio was lower. (L) Percentage of myelin sheaths with obvious outfoldings. n = 4 animals treated with DMSO, 3 with LatA, on two experimental days; *p < 0.05, Student’s t-test.
Error bars: SEM.
See also Figure S6.
To test whether actin disassembly is a signal for morphological changes during myelination, we allowed OPCs to differentiate normally until they were fully arborized, but had not yet flattened into myelin membrane sheets (i.e. arborized stage, Figure 2E), then induced actin disassembly with LatA. LatA sequesters free actin monomers and prevents them from reassembling into filaments, so prolonged treatment causes net disassembly of all dynamically treadmilling actin. Overnight treatment with nanomolar concentrations of LatA caused near-complete disassembly of the OL actin cytoskeleton (Figures S2 and S6), and induced arborized OLs to flatten and spread their membranes (Figure 5, B and C). Induction of cell spreading is rapid, as we observed an 8.2 ± 2.4% increase in cellular area after only 5 min of treatment with LatA (Figure 5D). This result is surprising given that most cells require actin filaments to maintain their shape. OLs that had already disassembled their actin and spread into myelin membrane sheets (i.e. lamellar stage) are unaffected by LatA (Figure 5C; Aggarwal et al., 2011).
Is actin disassembly required for myelin wrapping in vivo? Of all the actin disassembly proteins expressed by OLs, gelsolin is the highest at the RNA level (Figure S6). We analyzed myelin wrapping in the optic nerves of adult gelsolin knockout mice (Gsn−/−) and found a small but significance decrease in myelin thickness, compared to wild-type littermates (Figure 5, E and F). There was no difference in axon caliber in knockout mice (Figure S6), suggesting that myelin differences are not due to an indirect effect on axon caliber. However, gelsolin is not completely essential for wrapping, which could be explained by the high expression of cofilin family proteins in myelinating OLs, and the observation that gelsolin knockout brains display compensatory increases in cofilin activity (Kronenberg et al., 2010).
Since multiple actin disassembly proteins may collaborate to disassemble the OL cytoskeleton, and based on our observation that pharmacological-induced actin disassembly promoted OL actin disassembly and membrane spreading, we utilized LatA in vivo to assess the role of actin disassembly in myelin wrapping during development. We first confirmed that LatA treatment of intact CNS tissue induced significant disassembly of actin filaments in developing white matter (Figure S6). Next, we surgically implanted LatA- or DMSO (carrier)-loaded gelfoam on the surface of the dorsal spinal cord of P12 mice during active CNS myelination. We treated mice continuously for four days, then perfused for EM at P16. LatA induced a robust increase in myelin wrapping compared to DMSO-treated controls (Figure 5, G–K). We also observed occasional redundant myelin outfoldings in LatA-treated animals, similar to ArpC3 CKO animals (Figure 5L), with no obvious cell death or axonal degeneration. The increase in myelin thickness in LatA-treated animals corresponded to a decreased g-ratio, and axon diameter was unaffected (Figure S6). Since, by P12, myelination has already mostly initiated in the dorsal spinal cord (Matthews and Duncan, 1971), the percent of myelinated axons was not significantly affected (Figure S6). Taken together, these data provide evidence that actin disassembly accelerates myelin wrapping.
MBP is required for OL actin disassembly
What signals actin to disassemble in order to promote myelin wrapping? MBP is an intriguing candidate since its expression by OLs and in myelin occurs simultaneously with actin disassembly (Figures 1 and 2), and previous studies have suggested that MBP and actin physically interact in vitro. We used superresolution microscopy to visualize MBP and actin in mature primary OLs. MBP formed a highly ordered latticework on the OL membrane (Figure 6, A and B) suggestive of a “molecular sieve” (Aggarwal et al., 2011). Domains of MBP lattices were typically bounded by actin filaments. Line scans showed that MBP and actin filaments were completely non-overlapping, suggesting that they do not normally interact in OLs (Figure 6C). Temporally, actin filament and MBP levels were anti-correlated in OLs during their differentiation (Figure 6D). MBP domains began as small, sub-micron regions containing adjacent 200–800 nm diameter rings that appeared to spread and/or fuse as OLs matured (not shown), and were surrounded by actin filaments. Intriguingly, knock-down of MBP by RNAi caused aberrant actin filament accumulation in mature, lamellar OLs, suggesting a defect in actin disassembly (Figure 6E; see also Figure 7, E and F).
Figure 6. Loss of MBP leads to accumulation of actin filaments.

(A–C) Structured illumination microscopy of MBP (red) and actin filaments (phalloidin, green) in a 5d differentiated OL; boxed area shown in more detail in (B). (C) Representative line scan showing no spatial overlap between MBP and actin filaments in mature OLs.
(D) MBP and actin filament levels were anticorrelated in OLs through differentiation. Each data point is one OL.
(E) RNAi knockdown of MBP in OLs caused aberrant accumulation of actin filaments. Rat OPCs were transfected with control (nontargetting) or MBP-specific siRNA, differentiated into OLs, and stained for MBP (red), actin filaments (phalloidin, green), and nuclei (DAPI, blue).
(F–I) Failure of OL actin disassembly in Shiverer mice that lack MBP. (F) Spinal cord transverse sections from P36 wild-type (top) and Shiverer (bottom) littermates, stained for compact myelin with FMred (red) and actin filaments with phalloidin (green and grayscale on right). (G) Confocal microscopy revealed high levels of actin filaments (phalloidin, green) in OL processes that ensheath axons (neurofilament, purple). Quantification of phalloidin staining grossly in dorsal white matter (H), and specifically around axons (myelin ROI, I). Error bars: SEM from n = 3 animals per genotype at both P15 and P20, 6 animals per genotype at P42/44; *p < 0.05, ***p < 0.001, Student’s t-test.
See also Figure S7.
Figure 7. Model of how MBP regulates OL actin disassembly.

(A–C) MBP competes with actin disassembly proteins for binding to PI(4,5)P2. (A and B) 10μM (A) or 1μM (B) recombinant human cofilin-1 protein was incubated with control or PI(4,5)P2 beads with the indicated concentration of native MBP, washed, and bound protein eluted with sample buffer. Immunoblots show bound cofilin (top) and bound MBP (bottom). (C) Densitometry quantification of PI(4,5)P2-bound cofilin as a function of the molar ratio of MBP:cofilin. Inset, bound MBP. n = 3 experimental days. See also Figure S7.
(D–F) Dual RNAi of gelsolin and cofilin phenocopies MBP RNAi. OPCs were transfected with nontargeting siRNA (control, top) or siRNAs targeting gelsolin and cofilin (Gsn+Cfl1, bottom) or MBP (see Figure 6E) then differentiated for 6 days into mature OLs. (D) MBP (red) and actin filaments (phalloidin, green and false colored on right). (E and F) Quantification of width (E) and phalloidin intensity (F) in actin rims at the OL cell edge. *p < 0.05, ***p < 0.001, ***p < 0.0001; Dunnett’s multiple comparisons test; n = 3 experimental days. Error bars: SEM.
(G) Model of how MBP may regulate actin disassembly during myelination. Left, actin disassembly factors cofilin and gelsolin are normally sequestered by PI(4,5)P2 (PIP2), preventing them from disassembling actin filaments. Right, MBP binds to PI(4,5)P2 on the OL membrane, releasing cofilin/gelsolin to disassemble actin.
(H) Two-step model of myelin wrapping. (1) Ensheathment of axons by OL processes requires actin filaments (green), which also limit aberrant myelin membrane growth. (2) Local actin disassembly in the inner tongue induces myelin wrapping.
On the basis of these results, we asked whether MBP controls actin disassembly during myelination in vivo. Remarkably, we found that actin filament levels in white matter tracts of Shiverer mice that lack MBP were nearly indistinguishable from gray matter, whereas wild-type littermates appeared normal (Figure 6F). Developmentally, as white matter actin filament levels dropped in wild-type mice, Shiverer white matter maintained high levels of actin filaments (Figure 6H). This does not appear to be due to increased numbers of OPCs in Shiverer white matter (Figure S7). We used confocal microscopy to look specifically at OL processes around axons, and found that Shiverer OLs had a striking accumulation of actin filaments in their ensheathing processes (Figure 6G). We observed ~3-fold higher levels of actin filaments in Shiverer myelin compared to wild-type littermate controls (Figure 6I). This is consistent with the accumulation of actin in cultured OLs lacking MBP (Figure 6E), and suggests that normal actin disassembly is blocked in the absence of MBP.
How does MBP regulate actin disassembly? MBP may directly disassemble actin filaments, although the absence of colocalization between MBP and actin filaments in OLs suggests otherwise. Indeed, ectopic expression of MBP in HeLa cells had no effect on cellular actin filament levels (Figure S7). The high induction of actin disassembly proteins during myelination and the importance of gelsolin for normal wrapping (Figure 5) also suggest that MBP may instead control OL actin disassembly indirectly. A major cellular mechanism for regulating actin disassembly is the binding of disassembly factors to membrane phospholipids. Cofilin and gelsolin family members are sequestered by membrane PI(4,5)P2 and kept inactive; release from PI(4,5)P2 enables actin filament severing and disassembly (reviewed by Hilpelä et al., 2004). Intriguingly, MBP binds to PI(4,5)P2 on OL membranes, requiring it for stable attachment to the membrane and maintenance of compact myelin (Nawaz et al., 2009; Nawaz et al., 2013). Thus MBP may regulate actin disassembly by competing with cofilin and gelsolin for binding to membrane PI(4,5)P2. In this model, high induction of MBP protein and its recruitment to membrane PI(4,5)P2 triggers the release and activation of actin disassembly proteins.
We used PI(4,5)P2-coated microbeads and purified proteins to test whether MBP competes with actin disassembly proteins for binding to PI(4,5)P2. In the absence of MBP, recombinant cofilin-1 bound to PI(4,5)P2 beads. MBP also bound to PI(4,5)P2 beads, and increasing MBP concentrations completely blocked cofilin-1 binding (Figure 7, A and B). Cofilin was completely competed off of PI(4,5)P2 beads by stoichiometric concentrations of MBP, and this competition was dose-dependent (Figure 7C). We also observed similar but less complete competition with gelsolin (Figure S7). In OLs, dual knockdown of cofilin-1 and gelsolin by RNAi prevented actin disassembly, and closely phenocopied RNAi of MBP (Figure 7, D–F). We propose that MBP competes with actin disassembly factors for binding to membrane PI(4,5)P2, enabling it to locally activate actin disassembly at sites of MBP compaction to drive myelin wrapping (Figure 7G). Future work aims to test this competition model in vivo, and determine the signals that control MBP-dependent actin disassembly during myelination.
DISCUSSION
Myelin wrapping is essential in vertebrates for rapid propagation of action potentials, axonal health, and forms of plasticity (Bercury and Macklin, 2015) but the molecular mechanisms that drive wrapping remain unknown. In this study we have used a combination of in vivo and cellular approaches to elucidate the molecular mechanisms that drive CNS myelin initiation and wrapping. Early stages of myelination—OL process outgrowth and ensheathment of axons—require actin assembly by Arp2/3. Unexpectedly, we find that myelin wrapping is induced by disassembly of the OL actin cytoskeleton, and does not require Arp2/3. Further, we show that MBP is required for actin disassembly during myelination, and we provide evidence that it carries out this function by binding membrane PI(4,5)P2 to release and activate actin disassembly proteins. Together, we propose a two-step model of CNS myelination in which actin assembly by the Arp2/3 complex drives OL process extension to ensheath axons, and then MBP induces actin disassembly to trigger myelin wrapping (Figure 7H).
The Arp2/3 complex is required for extension of neuronal growth cones (Yang et al., 2012) and for fibroblast lamellipodia to respond to surface-attached extracellular matrix proteins (haptotaxis; Wu et al., 2012). These may be analogous to the early steps of myelination that we found to be Arp2/3-dependent: OL processes closely resemble growth cones (Figure 2G), and ensheathment of axons may be a form of haptotaxis, requiring the OL process to adhere and respond to molecules on the axonal membrane. Moreover, a role in polarization or “steering” of this lamellipodial ensheathing process could explain why dysregulated actin assembly (in ArpC3 CKOs and following treatment with LatA) leads to inappropriate growth of myelin membrane outfoldings away from the axon (Figures 3J, S5, and 5H). It will be interesting to elucidate axonal and secreted signals that control Arp2/3-dependent OL process outgrowth and ensheathment, and whether these depend on neuronal activity.
We uncovered actin disassembly as an unexpected but stereotyped step in myelination, that occurs in myelin sheaths in vivo during development, in isolated OLs differentiating in culture, and in myelinating cocultures of OLs and neurons. This transition from actin-rich to near-complete disassembly is one of the most dramatic rearrangements of a cell’s cytoskeleton that we are aware of. We identified actin disassembly factors including cofilin-1 and gelsolin as a gene “cassette” that is highly induced at the start of myelination. Since actin disassembly proteins are also essential in lamellipodia for normal actin turnover or in some cases to initiate actin assembly, our results showing a role for gelsolin in myelin wrapping do not, on their own, prove that actin disassembly itself drives wrapping. Four lines of additional evidence, however, strongly support our conclusion that disassembly promotes wrapping independent of lamellipodial growth. First, inducibly deleting ArpC3 after axon ensheathment has no effect on myelin wrapping (Figure 4, A–E). Second, Arp2/3 is not required for the extensive hypermyelination caused by conditional knockout of PTEN in OLs (Figure 4, F–I). Third, global loss of actin filaments using LatA accelerates myelin wrapping during development, and this is not due to effects of LatA on proliferation or differentiation of OLs (Figure 5). Fourth, OLs highly upregulate a number of actin disassembly factors during differentiation, and completely disassemble their actin cytoskeletons (Figures 1, 2, and 5). Together, our data indicate that actin disassembly is essential for myelin wrapping, which, contrary to expectations, does not require Arp2/3-mediated actin assembly.
How is this dramatic disassembly of the OL cytoskeleton regulated? Actin disassembly factors like gelsolin and cofilin are kept inactive by sequestration on membrane PI(4,5)P2, and are normally released and activated upon PI(4,5)P2 hydrolysis by phospholipase C (Hilpelä et al., 2004). However, since MBP requires PI(4,5)P2 for stable association to the myelin membrane (Nawaz et al., 2009), this mechanism is likely incompatible with myelination. Instead, our findings suggest that OLs have evolved a separate mechanism for releasing and activating actin disassembly proteins: direct displacement by MBP. This simple yet elegant mechanism could enable precise coordination between MBP-driven membrane compaction and actin disassembly. In the future it will be important to test this model in vivo, and identify additional signals that control actin disassembly during myelin wrapping.
A blebbing motility model of myelin wrapping
How does actin disassembly promote myelin wrapping? One possibility is that disassembly itself induces expansion of the OL inner tongue. Arp2/3-mediated actin filament assembly in lamellipodia powers cell motility on surfaces, but cells often utilize actin assembly-independent forces to move in confined spaces. These alternative modes of cell motility, collectively called blebbing motility, operate by a general mechanism (Paluch and Raz, 2013). First, disassembly of a cell’s actin filament cortex initiates the site of membrane growth (blebbing). Second, cellular contraction leads to increased intracellular pressure of cytoplasm to deform the membrane at the site of blebbing, pushing the membrane forward. Third, delivery of new plasma membrane by exocytosis or lateral membrane flow allows for persistent growth of the bleb.
The OL inner tongue represents an ideal candidate for blebbing motility, as it grows by tunneling between previous myelin wraps and the axonal membrane in highly confined (nanometer-scaled) spaces. In support of a blebbing model of myelin wrapping, we have shown that actin disassembly promotes myelin wrapping, rather than Arp2/3-dependent actin assembly like in a lamellipodium. As in blebbing motility, OL actin disassembly needs to be spatially regulated, as unrestricted actin disassembly (by Arp2/3 ablation or LatA treatment) induces aberrant growth of myelin membrane outfoldings. Both cellular mechanisms for membrane delivery that drive blebbing have been demonstrated in myelin: the OL inner tongue is a site of exocytosis (Snaidero et al., 2014), and newly synthesized lipids flow laterally through the membrane from the outside to the inside of developing PNS myelin (Gould and Dawson, 1976).
In contrast to other forms of blebbing motility, myelination appears to not require actomyosin contractility, as myosin II expression drops during OL differentiation (Wang et al., 2008; Zhang et al., 2014) and actin filaments are disassembled. However, membrane compaction by MBP may be sufficient to increase OL intracellular pressure in the absence of actomyosin contraction. Indeed, several lines of evidence suggest that myelin compaction may be required for extensive wrapping: MBP is absolutely required for CNS myelin wrapping (Rosenbluth, 1980; Shine et al., 1992), myelin wrapping beyond the first few wraps does not initiate until after compaction begins (Hildebrand et al., 1993), and spiral growth of the inner tongue occurs simultaneously with compaction of outer layers of myelin by MBP (Snaidero et al., 2014). Taken together with previous studies, our findings suggest a modified blebbing model for myelin wrapping in which MBP triggers actin disassembly and compacts the OL membrane, to increase cytoplasmic pressure and drive membrane extension of the inner tongue.
EXPERIMENTAL PROCEDURES
All animal procedures were approved by Stanford University’s Administrative Panel on Laboratory Animal Care. Full details are provided in Extended Experimental Procedures. Complete protocols available upon request from brad.zuchero@gmail.com.
Mouse lines and experiments
Shiverer and PTEN-floxed mice were from The Jackson Laboratory (stock numbers 001428 and 006440). Previously created ArpC3-floxed (Kim et al., 2013), CNP-Cre (Lappe-Siefke et al., 2003), Olig2-Cre (Schüller et al., 2008), and Plp1-CreERT (Doerflinger et al., 2003) mouse lines were maintained by breeding with C57BL/6 mice. Gelsolin knockout mice (Witke et al., 1995) were maintained as described (Kronenberg et al., 2010). All experiments with mutants were performed blindly without knowledge of their genotype. We observed no sexual dimorphism of myelination defects in ArpC3 CKO mice, so male and females were pooled for analysis. For in vivo application of LatA, we used P12 C57BL/6 mice and harvested tissue for EM 4 days post surgery (see Extended Experimental Procedures).
Immunohistochemistry of tissue was performed by standard methods. Compact myelin was stained with FluoroMyelin Red (FMred, Invitrogen) according to the manufacturer’s protocol, and actin filaments were stained with Alexa Fluor-conjugated phalloidin (Invitrogen). Specificity of anti-MBP (Abcam ab7349) was validated using Shiverer OLs and tissue, which lack MBP expression (Figure S1). Transmission electron microscopy (TEM) of optic nerves was performed in conjunction with the Stanford Cell Sciences Imaging Facility.
Purification, culture, and immunostaining of cells
Unless otherwise indicated, OPCs were purified from enzymatically dissociated P7–P8 Sprague-Dawley (Charles Rivers) rat brains by immunopanning and grown in serum-free defined medium, as described previously (Dugas and Emery, 2013). Mouse OPCs were purified from transgenic mice by immunopanning as described (Emery and Dugas, 2013). LatA, CK-666 and CK-689 were from EMD Millipore, and Jasp was from Invitrogen. OLs were processed for immunostaining by standard methods, and stained with Alexa Fluor 488- or 594-phalloidin (Invitrogen) to visualize actin filaments.
Myelinating cocultures of rat OPCs and dispersed RGC or DRG neurons (Zuchero, 2014) were prepared essentially as described (Watkins et al., 2008). Cultures were analyzed for percent cells expressing MBP and degree of myelin ensheathment per OL using a custom automated imaging workflow (Extended Experimental Procedures).
Fluorescence microscopy
Cells and tissues were visualized by epifluorescence unless noted. Structured illumination imaging was conducted with an OMX V4 Structured Illumination microscope and SoftWoRx reconstruction software (GE Healthcare), with the exception of Figure 2H, which was acquired using an Elyra SR-SIM microscope and reconstruction software (Zeiss Microscopy). Identical illumination and acquisition conditions were used for each experiment.
Biochemistry and molecular biology
For PI(4,5)P2 pull-down competition assays, we used native bovine MBP (EMD Millipore), recombinant human cofilin-1 and gelsolin (gifts of Peter Bieling, UCSF), and PI(4,5)P2-coated or uncoated (control) agarose beads (Echelon) by the method of Gálvez-Santisteban (2012). PCR, RT-PCR, and Western immunoblotting were carried out using standard methods (see Extended Experimental Procedures).
Data Analysis and Statistics
All data acquisition and analysis were performed blinded to the experimental condition. We used nested analysis to first average technical replicates (e.g. 3–5 adjacent cryosections for IHC quantification, 2 replicate immunoblots). In all cases n refers to biological replicates, which is either a single animal, or experimental day for culture and biochemistry experiments. Data shown are from all animals tested; none were treated as outliers. Micrographs and blots were analyzed using NIH ImageJ and linearly contrast adjusted for display using Adobe Photoshop, with identical settings for each experiment. Data were analyzed and plotted using Excel (Microsoft) and Prism (GraphPad Software). Unless otherwise stated, error bars are SEM and p-values were calculated using Student’s t-test for single comparisons, or ANOVA followed by Dunnett’s multiple comparison test.
Supplementary Material
Acknowledgments
We thank Will Talbot and Amanda Brosius Lutz for critical reading of the manuscript; members of the Barres lab, especially Mariko Bennett, Shane Liddelow, Andrew Huang, and Cameron Andrews; and Kaylene Young, Feng Mei, and Bud Swiech for discussions and support. We thank Klaus Nave (CNP-Cre mice), David Rowitch (Olig2-Cre mice), Brian Popko (Plp1-CreERT mice), Lawrence LeClaire (ArpC1 antibody), and Scott Hansen, Peter Bieling, and Dyche Mullins (actin and related proteins). We thank the Stanford Neuroscience Microscopy Service (supported by NIH NS069375), John Perrino and the Stanford Electron Microscopy core (supported by NIH 1S10RR02678001), and Midori Hitomi for assistance with EM. We gratefully acknowledge support from NIH grants to B.A.B. (R01 EY10257), S.H.S. (R01 MH103374, S.H.S.), J.B.Z and M.M.F (T32 HD007249), and S.A.S. (F30 MH106261), and from the National Multiple Sclerosis Society (NMSS) to B.A.B., the Myelin Repair Foundation (B.A.B and R.H.M.), and the Sheldon and Miriam Adelson Medical Foundation (B.A.B). K.G. and G.K. receive grant support from Deutsche Forschungsgemeinschaft and BMBF (Center for Stroke Research Berlin). J.B.Z was a HHMI Fellow of the Life Sciences Research Foundation, and is supported by the NMSS Career Transition Award.
Footnotes
AUTHOR CONTRIBUTIONS
J.B.Z and B.A.B conceived of the project, directed the work, and wrote the manuscript. J.B.Z. designed, performed, and analyzed all experiments except where noted. M.M.F performed live cell imaging experiments in Figure 5D. S.A.S. developed the myelin ROI MATLAB application used in Figure 6I, and analyzed RNAseq data in Figure S6A. A.Z, A.V.C, C.K. and R.H.M. performed the gel foam experiments and acquired EM micrographs in Figure 5. A.O. acquired structured illumination micrographs in Figure 2H and Figure 6A. A.I. provided technical assistance with all aspects of the work. J.C.D performed and analyzed coculture assays in Figures 3 and S2. S.W. performed overexpression studies in Figure S7. G.K. and K.G. obtained tissue from gelsolin knockout mice used in Figures 5 and S6. S.H.S. developed and shared the ArpC3-floxed mice before publication. All authors reviewed and made comments on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal S, Yurlova L, Snaidero N, Reetz C, Frey S, Zimmermann J, Pähler G, Janshoff A, Friedrichs J, Müller DJ, et al. A Size Barrier Limits Protein Diffusion at the Cell Surface to Generate Lipid-Rich Myelin-Membrane Sheets. Dev Cell. 2011;21:445–456. doi: 10.1016/j.devcel.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Baryłko B, Dobrowolski Z. Ca2+-calmodulin-dependent regulation of F-actin-myelin basic protein interaction. Eur J Cell Biol. 1984;35:327–335. [PubMed] [Google Scholar]
- Bauer NG, Richter-Landsberg C, Ffrench-Constant C. Role of the oligodendroglial cytoskeleton in differentiation and myelination. Glia. 2009;57:1691–1705. doi: 10.1002/glia.20885. [DOI] [PubMed] [Google Scholar]
- Bercury KK, Macklin WB. Dynamics and mechanisms of CNS myelination. Dev Cell. 2015;32:447–458. doi: 10.1016/j.devcel.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs JM, Rangaraj G. Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry. 2000;39:7799–7806. doi: 10.1021/bi0002129. [DOI] [PubMed] [Google Scholar]
- Dangata YY, Kaufman MH. Myelinogenesis in the optic nerve of (C57BL x CBA) F1 hybrid mice: a morphometric analysis. Eur J Morphol. 1997;35:3–17. doi: 10.1076/ejom.35.1.3.13057. [DOI] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Emery B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb Protoc. 2013;8:745–758. doi: 10.1101/pdb.prot070862. [DOI] [PubMed] [Google Scholar]
- Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb Protoc. 2013;9:854–868. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Afshari FS, Alexander JK, Colello RJ, Fuss B. Growth conelike sensorimotor structures are characteristic features of postmigratory, premyelinating oligodendrocytes. Glia. 2006;53:563–566. doi: 10.1002/glia.20293. [DOI] [PubMed] [Google Scholar]
- Gálvez-Santisteban M, Rodríguez-Fraticelli AE, Bryant DM, Vergarajauregui S, Yasuda T, Bañón-Rodríguez I, Bernascone I, Datta A, Spivak N, Young K, et al. Synaptotagmin-like proteins control the formation of a single apical membrane domain in epithelial cells. Nat Cell Biol. 2012;14:838–849. doi: 10.1038/ncb2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. Journal of Neuroscience. 2010;30:8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould RM, Dawson RM. Incorporation of newly formed lecithin into peripheral nerve myelin. J Cell Biol. 1976;68:480–496. doi: 10.1083/jcb.68.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand C, Remahl S, Persson H, Bjartmar C. Myelinated nerve fibres in the CNS. Prog Neurobiol. 1993;40:319–384. doi: 10.1016/0301-0082(93)90015-k. [DOI] [PubMed] [Google Scholar]
- Hilpelä P, Vartiainen MK, Lappalainen P. Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr Top Microbiol Immunol. 2004;282:117–163. doi: 10.1007/978-3-642-18805-3_5. [DOI] [PubMed] [Google Scholar]
- Jin F, Dong B, Georgiou J, Jiang Q, Zhang J, Bharioke A, Qiu F, Lommel S, Feltri ML, Wrabetz L, et al. N-WASp is required for Schwann cell cytoskeletal dynamics, normal myelin gene expression and peripheral nerve myelination. Development. 2011;138:1329–1337. doi: 10.1242/dev.058677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, DiBernardo AB, Sloane JA, Rasband MN, Solomon D, Kosaras B, Kwak SP, Vartanian TK. WAVE1 is required for oligodendrocyte morphogenesis and normal CNS myelination. J Neurosci. 2006;26:5849–5859. doi: 10.1523/JNEUROSCI.4921-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, Soderling SH. Disruption of Arp2/3 Results in Asymmetric Structural Plasticity of Dendritic Spines and Progressive Synaptic and Behavioral Abnormalities. J Neurosci. 2013;33:6081–6092. doi: 10.1523/JNEUROSCI.0035-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg G, Gertz K, Baldinger T, Kirste I, Eckart S, Yildirim F, Ji S, Heuser I, Schröck H, Hörtnagl H, et al. Impact of actin filament stabilization on adult hippocampal and olfactory bulb neurogenesis. J Neurosci. 2010;30:3419–3431. doi: 10.1523/JNEUROSCI.4231-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Liu A, Muggironi M, Marin-Husstege M, Casaccia-Bonnefil P. Oligodendrocyte process outgrowth in vitro is modulated by epigenetic regulation of cytoskeletal severing proteins. Glia. 2003;44:264–274. doi: 10.1002/glia.10290. [DOI] [PubMed] [Google Scholar]
- Matthews MA, Duncan D. A quantitative study of morphological changes accompanying the initiation and progress of myelin production in the dorsal funiculus of the rat spinal cord. J Comp Neurol. 1971;142:1–22. doi: 10.1002/cne.901420102. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nawaz S, Kippert A, Saab AS, Werner HB, Lang T, Nave KA, Simons M. Phosphatidylinositol 4,5-bisphosphate-dependent interaction of myelin basic protein with the plasma membrane in oligodendroglial cells and its rapid perturbation by elevated calcium. J Neurosci. 2009;29:4794–4807. doi: 10.1523/JNEUROSCI.3955-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz S, Schweitzer J, Jahn O, Werner HB. Molecular evolution of myelin basic protein, an abundant structural myelin component. Glia. 2013;61:1364–1377. doi: 10.1002/glia.22520. [DOI] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak N, Bar V, Sabanay H, Frechter S, Jaegle M, Snapper SB, Meijer D, Peles E. N-WASP is required for membrane wrapping and myelination by Schwann cells. J Cell Biol. 2011;192:243–250. doi: 10.1083/jcb.201010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch EK, Raz E. The role and regulation of blebs in cell migration. Curr Opin Cell Biol. 2013;25:582–590. doi: 10.1016/j.ceb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead C, Popko B, Takahashi N, Shine HD, Saavedra RA, Sidman RL, Hood L. Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the dysmyelinating phenotype. Cell. 1987;48:703–712. doi: 10.1016/0092-8674(87)90248-0. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J. Central myelin in the mouse mutant shiverer. J Comp Neurol. 1980;194:639–648. doi: 10.1002/cne.901940310. [DOI] [PubMed] [Google Scholar]
- Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine HD, Readhead C, Popko B, Hood L, Sidman RL. Morphometric analysis of normal, mutant, and transgenic CNS: correlation of myelin basic protein expression to myelinogenesis. J Neurochem. 1992;58:342–349. doi: 10.1111/j.1471-4159.1992.tb09316.x. [DOI] [PubMed] [Google Scholar]
- Snaidero N, Möbius W, Czopka T, Hekking LHP, Mathisen C, Verkleij D, Goebbels S, Edgar J, Merkler D, Lyons DA, et al. Myelin Membrane Wrapping of CNS Axons by PI(3,4,5)P3-Dependent Polarized Growth at the Inner Tongue. Cell. 2014;156:277–290. doi: 10.1016/j.cell.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Goetz BD, Baas PW, Duncan ID. Cytoskeletal reorganization during the formation of oligodendrocyte processes and branches. Mol Cell Neurosci. 2001;17:624–636. [Google Scholar]
- Thurnherr T, Benninger Y, Wu X, Chrostek A, Krause SM, Nave KA, Franklin RJM, Brakebusch C, Suter U, Relvas JB. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J Neurosci. 2006;26:10110–10119. doi: 10.1523/JNEUROSCI.2158-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Andrews SB, Wong A, O’Connell M, Griffin JW. Co-localization of the myelin-associated glycoprotein and the microfilament components, F-actin and spectrin, in Schwann cells of myelinated nerve fibres. J Neurocytol. 1989;18:47–60. doi: 10.1007/BF01188423. [DOI] [PubMed] [Google Scholar]
- Wang H, Tewari A, Einheber S, Salzer JL, Melendez-Vasquez CV. Myosin II has distinct functions in PNS and CNS myelin sheath formation. J Cell Biol. 2008;182:1171–1184. doi: 10.1083/jcb.200802091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Emery B, Mulinyawe S, Barres BA. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron. 2008;60:555–569. doi: 10.1016/j.neuron.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Brophy PJ. Role for the oligodendrocyte cytoskeleton in myelination. J Neurosci Res. 1989;22:439–448. doi: 10.1002/jnr.490220409. [DOI] [PubMed] [Google Scholar]
- Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 Is Critical for Lamellipodia and Response to Extracellular Matrix Cues but Is Dispensable for Chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yae K, Keng VW, Koike M, Yusa K, Kouno M, Uno Y, Kondoh G, Gotow T, Uchiyama Y, Horie K, et al. Sleeping beauty transposon-based phenotypic analysis of mice: lack of Arpc3 results in defective trophoblast outgrowth. Mol Cell Biol. 2006;26:6185–6196. doi: 10.1128/MCB.00018-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhang XF, Pollard TD, Forscher P. Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J Cell Biol. 2012;197:939–956. doi: 10.1083/jcb.201111052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. Journal of Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB. Purification of dorsal root ganglion neurons from rat by immunopanning. Cold Spring Harb Protoc. 2014;2014:826–838. doi: 10.1101/pdb.prot074948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.