

Abstract
Epigenetic mechanisms are fundamental in cardiac adaptations, remodeling, reverse remodeling, and disease. A primary goal of translational cardiovascular research is recognizing whether disease related changes in phenotype can be averted by eliminating or reducing the effects of environmental epigenetic risks. There may be significant medical benefits in using gene-by-environment interaction knowledge to prevent or reverse organ abnormalities and disease. This survey proposes that “environmental” forces associated with diastolic RV/LV rotatory flows exert important, albeit still unappreciated, epigenetic actions influencing functional and morphological cardiac adaptations. Mechanisms analogous to Murray's law of hydrodynamic shear-induced endothelial cell modulation of vascular geometry are likely to link diastolic vortex-associated shear, torque and “squeeze” forces to RV/LV adaptations. The time has come to explore a new paradigm in which such forces play a fundamental epigenetic role, and to work out how heart cells react to them. Findings are considered from various disciplines, imaging modalities, computational fluid dynamics, molecular cell biology and cytomechanics. Examined are, among others, structural dynamics of myocardial cells (endocardium, cardiomyocytes, and fibroblasts), cytoskeleton, nucleoskeleton, and extracellular matrix, mechanotransduction and signaling, and mechanical epigenetic influences on genetic expression. To help integrate and focus relevant pluridisciplinary research, rotatory RV/LV filling flow is placed within a working context that has a cytomechanics perspective. This new frontier in contemporary cardiac research should uncover versatile mechanistic insights linking filling vortex patterns and attendant forces to variable expressions of gene regulation in RV/LV myocardium. In due course, it should reveal intrinsic homeostatic arrangements that support ventricular myocardial function and adaptability.
Keywords: intracardiac flow, epigenetic factors, filling vortex forces, cardiac gene regulation, cardiac cytomechanics, flow-imaging modalities, endocardial vortical shear and squeeze, Next Generation Sequencing (NGS) technologies
The main difficulty in defining the concept of phenotype is caused by the fact that animals change in time. … Phenotypic differences between two organisms may be caused by genotypic differences or may be produced by different environments acting on the same genotype. … One might say that the set of organizers and organizing relations to which a certain piece of tissue will be subject during development make up its “epigenetic constitution” or “epigenotype”; then the appearance of a particular organ is the product of the genotype and the epigenotype, reacting with the external environment.
— Professor C. H. Waddington, Sc.D., Fellow of Christ's College, Cambridge. [Ref. 151, pp. 155–156].
1. Introduction and Clinical Perspective
The myocardial cells and the pumping ventricles are continuously challenged to preserve homeostasis notwithstanding a wide-ranging array of stimuli and perturbations, both health-upholding and disease producing. To this end, appropriate morphomechanical/functional modifications must be accomplished that correspond to the changing conditions; they typically entail altered gene expression. This all implies continual adaptive/maladaptive phenotypic changes, and it is this phenotypic flexibility that determines cardiac health or abnormalities. Transcription rates as responses to stimuli, and transcription differences between individuals, are becoming broadly recognized as essential reactions, with important repercussions [1–3].
Our fast expanding capability, especially after the Human Genome Project, to define genetic differences at the DNA base-sequence level is exposing an enormous new domain in the search for gene-by-environment (G×E) interactions. Notwithstanding much information on both genetic and environmental disease-risk factors, there are comparatively few examples of robust gene–environment interactions in the cardiovascular medical literature. Examples of G×E interactions are shown in Table 1 [4–8]. Investigations of G×E interactions seek to define how “environmental” and genetic factors, in parallel, impact manifestation of abnormal organ phenotype and disease. Since the time of publication of Sir Archibald Garrod's classic work of intuition, on the incidence of alkaptonuria, it has been gradually comprehended that the etiology of various common diseases points to not only distinct genetic and/or “environmental” causes, but also to subtle but potent interactions between the two [9].
Table 1. Gene-environment (G × E) interactions in coronary artery disease (CAD).
| Gene | Environment | Interaction | Independently associated with CAD? | Ref |
|---|---|---|---|---|
| ADH1C (also known as ADH3) | Alcohol consumption | Slow-metabolizing γ2 allele homozygotes have higher HDL levels and greatest CAD protection | ADH1C: weak; alcohol: yes | [4] |
| APOE | Smoking | Smoking engenders exaggerated CAD risk in carriers of APOE ε4 allele | APOE: yes; smoking: yes | [5] |
| FGB | Vigorous exercise | Carriers of 455A allele have exaggerated increase in fibrinogen after exercise | FGB: no; exercise: yes | [6] |
| GSTM1, GSTT1 | Smoking | Elevated CAD risk in smokers with null mutations, i.e., gene mutations leading to the genes not being transcribed into RNA and/or translated into a functional protein product | GSTM1, GSTT1: weak; smoking: yes | [7] |
| LDLR | Lifestyle | Mutations have greater effect (early-onset myocardial infarction) in less active people with high-fat diet | LDLR: yes; lifestyle: yes | [8] |
Abbreviations: ADH1C, alcohol dehydrogenase 1C; APOE, apolipoprotein E; FGB, fibrinogen beta chain; GSTM1, glutathione S-transferase mu 1; GSTT1, glutathione S-transferase theta 1; LDLR, low-density lipoprotein receptor.
The present-day progress in genetics and molecular biology is affording us the tools needed to exploit in cardiology such a more sophisticated comprehension. It is broadly recognized currently that genetics and genomics—the study of the genome and of the overall assemblage of expressed and non-expressed genes—are rapidly transforming the face of medicine. Our environment continues to epigenetically influence our genes throughout our lives. And it may be possible to pass down epigenetic modifications to future generations, if the changes occur in sperm or egg cells. The complete set of epigenetic modifications (e.g., DNA methylation, histone acetylation, and chromatin remodeling) [3] on the genome and allied histone proteins of a cell, tissue or organ constitutes the epigenome. The application of molecular genetics and biology can provide us with better ways to approach disease and organ abnormalities, such as hypodynamic ventricular dilatation in failure. Notably, unlike the alterations of gene behavior caused by DNA mutations, epigenetic alterations of gene behavior are generally reversible. Thus, a primary goal of translational cardiovascular research is recognizing whether abnormality/disease related changes in phenotype can be averted by eliminating, or reducing, the effects of environmental “epigenetic” (see Epigraph) risks, with prospective manifold health benefits.
There may be significant medical benefits in using recognition of G×E interactions to prevent or reverse organ abnormalities and disease (see Figure 1). This could allow more effective, rational interventions to achieve therapeutic response in patients, while minimizing complications. It is important to always recognize that genetic factors and environmental factors can interact (the G×E interactions) complicating compound phenotypes, such that any one epigenetic environmental factor may have minimal influence but, acting together, several interacting factors can have a substantial influence on phenotype. An example is phenylketonuria, which occurs only in people with a genetic defect and high dietary intake of phenylalanine. A less straightforward example can be imagined for lung cancer: not everyone who smokes develops it, although smoking is the greatest risk factor known for its development; and some individuals develop it after only a short exposure to tobacco use. One explanation for such discrepancies would be unique G×E interactions, such that the epigenetic/environmental factor is especially harmful in individuals with specific genetic variants, while in others the harm posed by the environmental factor is (partially) offset by other specific genetic/environmental variants. Genetic and environmental factors interact to yield an appreciable influence on the phenotype.
Figure 1. Genotype × Environment = Phenotype.
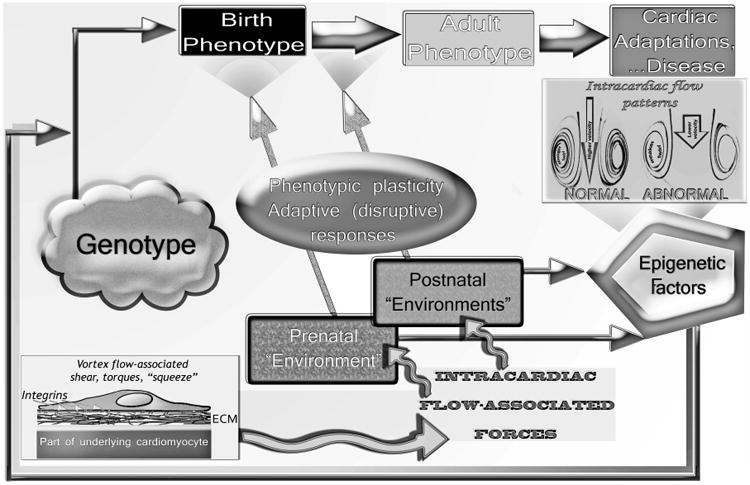
A circular regulatory pathway exists during cardiogenesis and in pre- and postnatal life between: the intracardiac blood flow patterns and associated shear, torque, and “squeeze” (see discussion in text) force transmission to the myocardial walls; epigenetically influenced gene expression; and changes in the morphology of the developing prenatal or in the phenotype of the continuously adapting postnatal and adult heart. Phenotypic plasticity can, in conjunction with normal and anomalous operating “environmental” conditions, lead not only to adaptive but also to disruptive (maladaptive) responses, morphomechanical cardiac abnormalities and disease. Slightly modified with permission of PMPH-USA from Pasipoularides A. Heart's Vortex: Intracardiac Blood Flow Phenomena. Shelton, CT: People's Medical Publishing House, 2010. 960 pp.
Like Janus bifrons, the Roman deity of entryways and exits, of beginnings and endings, genes have two faces or facets. Only one facet, the forward-facing, causal aspect, is acknowledged in the concept of the central dogma of molecular biology. But genes also have another facet, the backward-facing, responsive or regulatory aspect. This aspect is highlighted in epigenetic issues and research, such as the effects of diastolic vortex-associated forces on right and left ventricular (RV/LV) phenotypes [1–3]. At present, the interest in epigenetics, regulation, and complex G×E interactions is witnessing a remarkable increase.
Epigenetic mechanisms are fundamental in cardiac adaptations, remodeling, reverse remodeling, and disease (Figure 1). This two-article series proposes that variable forces associated with diastolic RV/LV rotatory intraventricular flows can exert physiologically and clinically important, albeit still unappreciated, epigenetic actions influencing functional and morphological cardiac adaptations and/or maladaptations. Taken in toto, the two-part survey formulates a new paradigm in which intraventricular diastolic filling vortex-associated forces play a fundamental epigenetic role, and examines how heart cells react to these forces. Albeit still unappreciated, these epigenetic actions influence functional and morphological cardiac adaptations. Part 1 [3] provides a general introduction, focusing on background concepts, on intracardiac vortex imaging methods, and on diastolic filling vortex-associated forces acting epigenetically on RV/LV endocardium and myocardium. Part 2 describes pertinent available pluridisciplinary knowledge/research relating to mechanotransduction mechanisms for intraventricular diastolic vortex forces and myocardial deformations, and to their epigenetic actions on myocardial and ventricular morphomechanics (form–function) and adaptations.
2. Myocardial histoarchitectonic framework for the mechanotransduction of vortical flow forces
Ventricular myocardium encompasses endocardial cells, cardiomyocytes, and fibroblasts, which assimilate multiple signals from their microenvironment, including direct mechanical stimuli associated with vortical flow forces. Mechanical signals may come from the extracellular matrix and neighboring cells (Figure 2), while soluble chemical signals may arise from both adjacent and distant cells [3]. The cardiomyocyte cell membrane, or sarcolemma, invaginates around the myocyte and creates an extensive tubular network (T-tubules) that extends the extracellular space into the interior of the cell. It is at the cell membranes and their attachments to cytoskeletal and extracellular matrix (ECM) components that the necessary mechanotransductive “sensory” systems originate. Understanding how myocardial cells transmit and balance mechanical forces and deformations offers insight into how they can transduce vortical mechanical cues into biological adaptations and activity.
Figure 2.
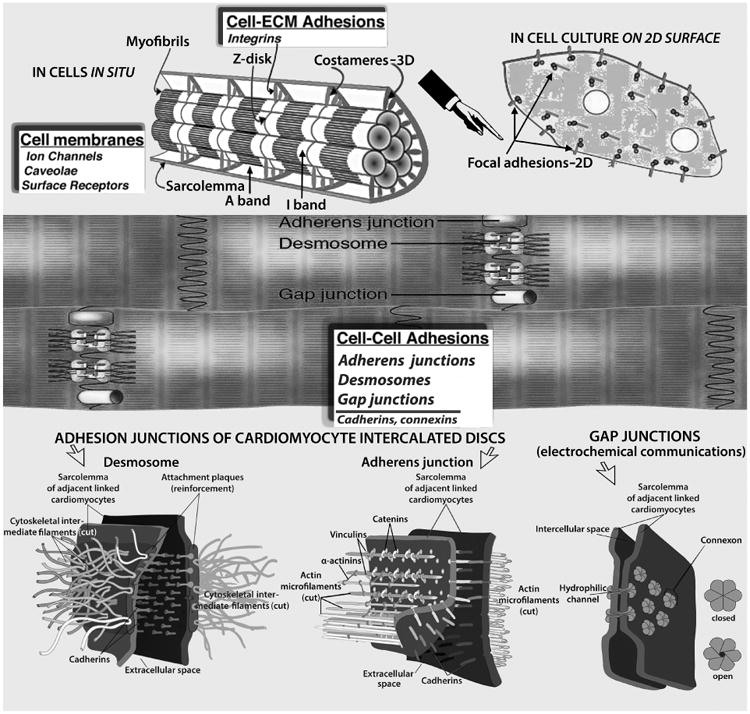
Myocardial cell junctions can be divided into two types: those that link cells to the ECM (costameres and focal adhesions, Top), and intercellular junctions (gap, adherens, and desmosomes, Middle & Bottom) that link cells together directly. Radially arranged integrins, and other specialized proteins, constitute physical links between the Z-disk and sarcolemma; they transmit contractile forces from sarcomeres across the sarcolemma laterally to the extracellular matrix and ultimately to neighboring cardiomyocytes. The desmosome, like the adherens junction, comprises calcium ion-dependent cell adhesion molecules that interact with similar molecules in the adjacent cell. Adherens junctions and focal adhesions not only tether cells together or to the ECM, but they also transduce signals into and out of the cell, influencing a variety of cellular epigenetic responses, notably to flow-associated forces and deformations. The basic building block of the gap junction is the connexin subunit. Six of these in each of the membranes of two adjacent cells come together to form a connexon that interacts with a comparable hexamer in the other cell resulting in formation of a channel, which allows cytoplasmic communication between the cells. Myocardial cell membranes encompass ion channels, surface receptors and caveolae—not shown. (See discussions in text). Costamere diagram adapted, modified, from Ervasti JM [31].
Sensitive to change in its environment, involving dynamic shear (acting parallel to the surface considered) and normal (compressive or tensile) stresses exerted by the vortical intraventricular blood, the myocardium of each ventricle undergoes strains (more or less intense deformations) and can react by adapting accordingly [3]. Mechanical signaling and force transmission within and outside the ventricular myocardium are important factors in the mechanotransduction process. The cytoskeleton is a key component in the structural link between the cell membrane and putative intracellular stress-sensing components, which respond to mechanical signals through multiple pathways, each involving various cascades of internal molecular interactions. Through the intermediation of the ECM, cytoskeletal and nucleoskeletal structures, operating intra- and extracellular forces alter nuclear shape and structure (cf. Figure 4); thus, the nucleus is implicated in the processes of mechanotransduction.
Figure 4.
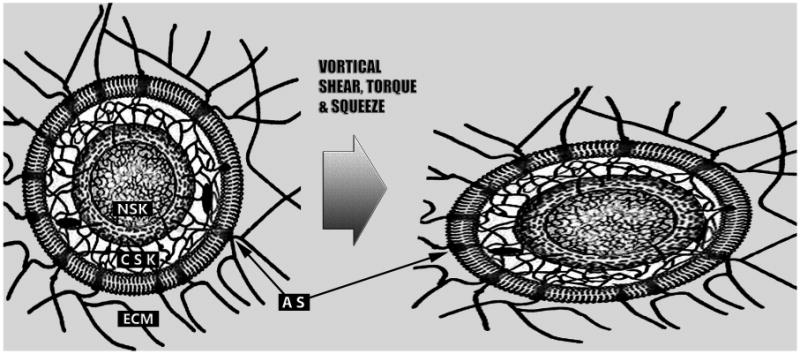
Schematic depiction of the global mechanical interconnectedness of the myocardial extracellular matrix (ECM), cytoskeleton (CSK), and nucleoskeleton (NSK), which has a pivotal role in the epigenetic dynamic actions of the right and left ventricular diastolic toroidal vortex. The inner and outer nuclear membranes and lamins anchored to the nuclear envelope are demarcated. The ECM, CSK, and NSK form a continuous and interconnected communication system, all parts of which work together synergistically. Adhesion structures (AS) are spatially discrete, transmembrane cadherin (cell–cell adhesions) and integrin (cell–ECM adhesions) complexes; they activate many of the same signaling pathways and provoke similar cellular activities, acting as interdependent functional nodes in a larger cytomechanics network. Through the intermediation of ECM, CSK and NSK structures, extra- and intra-cellular forces alter nuclear shape, promote nucleosome disruption and chromatin relaxation, and alter gene activities. (See discussion in text).
The nucleus, which contains almost all of the human genome, is the site of transcriptional regulation [3]. Mitochondrial DNA (mtDNA), distinct from nuclear DNA, is a small genome contained in all mitochondria, which occupy about 25–35% of the volume of ventricular cardiomyocytes. mtDNA is a circular double helix holding genes specific to the manufacture of new mitochondria, as occurs, e.g., in eccentric and concentric myocardial hypertrophy. Cardiomyocytes are either mono- or binucleated (see Section 7.2). Forces applied onto myocardial cell nuclei induce modifications in nuclear shape (cf. Figure 4), which bring about changes in chromatin state and organization; these can, in turn, affect transcription and regulate gene expression [2,3]. Therefore, defects in vortical flow force generation and transmission can be important mechanisms of cardiac morphomechanical phenotypic abnormalities and pathology.
2.1 The cytoskeleton, microfilaments, intermediate filaments and microtubules
Cardiomyocytes have a well-developed cytoskeleton (Figure 2). Microscopically, the cytoskeleton (CSK) is a dynamic, complex 3D filamentous network spanning the space between the nuclear envelope and the cell membrane, and contributing prominently to myocardial cell stability by anchoring subcellular organelles, such as the myofibrils, mitochondria, Golgi apparatus, and nuclei. This network is remarkably dynamic, continuously going through assembly, disassembly, movement and reorganization, even when seemingly stable. It gives cells structural strength besides imparting cell shape (in its place, plant cells and bacteria adopt an extracellular cell wall for structural support), and comprises microfilaments, intermediate filaments and microtubules [10], with auxiliary proteins binding to their sides or endpoints. Besides structural support, CSK affords thoroughfares for the conveyance of subcellular organelles and other structures, facilitates force generation, and can be exploited for the bidirectional transduction of signals: “inside-out,” i.e., force transmission out of cardiomyocytes by traction on their ECM adhesions, to modify the configuration and tension of the ECM and the behavior of adjoining cells and tissues; and “outside-in,” i.e., mechanical distortion of myocardial cells by ECM transmission of, e.g., hydrodynamic vortical shear and “squeeze” forces (discussed in Part 1 [3]), with ensuing readjustments in cytoskeletal organization and nuclear gene expression.
Microfilaments are fine, thread-like fibers of ∼ 3 –7 nm diameter, made of monomeric Gactin (globular) that has polymerized to form F-actin (filamentous, fibrous), which associates with myosin to compose tension-generating cytoskeletal assemblies [11]. By itself, cytoskeletal actin can form a flexible network, or self-assemble into rigid cross-linked struts; they have high compressive strength and act as a major outside-in signal conveyor. The actin cytoskeleton facilitates the transduction of mechanical cues, and can generate the intracellular forces required for cell signaling and many other cellular functions [12]. A high actin filament concentration is commonly found at the cell periphery, near the plasma membrane, and is known as the cell cortex; actin filaments in the cell cortex establish the shape and stiffness of the cell surface. Additionally, actin contributes to the formation and maintenance of cell junctions.
Intermediate filaments are about 10 nm in diameter and form flexible “cables” extending from cell membrane to nucleus [13]; they provide tensile strength to cells, mechanical stability at regions of intercellular contacts, and act as a major cell–cell signal conveyor.
Microtubules are long, hollow cylindrical tubes, 20-25 nm in diameter, composed of α and β subunits of the protein tubulin polymerizing from an organizing center near the nucleus outward to the cell periphery [14]. They provide a set of “tracks” on which molecular motors (cytoskeletal actin-myosin) use chemical energy to create motion and, with a complement of cargo/adaptor proteins [15], can translocate “cargo” (cell organelles and vesicles) from one location to another. When mechanical stresses, such as shear, are applied to cells, microtubules swiftly reorient themselves and actin stress fibers increase in thickness by recruitment and cross-bridging of short actin filaments, in order to reinforce their mechanical strength [16,17].
The microtubules contribute to passive stiffness and additionally oppose compression in myocardial cells, which they cross mostly along their longitudinal axis [18,19]. Importantly, RV/LV pressure overload induces upregulation of the microtubule proteins α-tubulin and β-tubulin, on the mRNA and protein levels, and intensifies microtubule network density [20]. Thus, a pressure overload-induced increased density of the microtubule component of the extramyofilament portion of the cardiomyocyte CSK can physically interfere with the inward-directed shortening of the sarcomeric myofibrils; hence cardiac hypertrophy can usher in a contractile dysfunction [20,21]. In turn, decreased shortening may be important for triggering/exacerbating concentric hypertrophy [22].
2.2 CSK tensegrity/elasticity and cell-to-ECM, cell-to-cell junctions
The solid CSK affords cells “tensegrity” (tensional integrity) [1,12,23]. Cells are prestressed: mechanical stresses applied externally on the cell or generated by the continual internal activity of cytoskeletal actomyosin motors are counterbalanced by the tensional/compressional reaction of the “solid-state” CSK. The existence of prestress in the cytoskeletal bundles was shown by the retraction of a single actin stress fiber bundle in an endothelial cell, ensuing upon severing it with a laser irradiation nanoscissor; as the stress fiber bundle retracted over a period of 15 s, the severed ends splayed apart [24,25].
Cytoskeletal tensions create equilibrium of strut compression and string tension, allowing internal structural dynamic balance. The CSK is active and changes with environmental mechanical cues. Its unconventional (non-sarcomeric) actin and myosin filaments combine and shorten by sliding along each other, generating tension (actin-myosin motors) [26] that gets distributed to intracellular organelles, to the ECM through integrin attachments in the generic class of focal adhesion (FA) complexes and the special to myocytes grouping of costameres, and to contiguous cardiomyocytes through cadherin-mediated adherens junctions and desmosomes (see Figure 2).
Equivalent morphomechanics apply to the CSK of all myocardial cells—including endocardium, myocytes, and fibroblasts. Through actin-myosin motors, cells also apply stresses to fine-tune sensing and responses to external force stimuli [27,28]. In cardiomyocytes, cytoskeletal filaments are overlain spatially on the sarcomeres [29,30]. Besides integrins, costameres [31] contain several of the same CSK adaptor proteins that are present in FA complexes of adherent non-muscle cells. In fact, cardiomyocytes in culture on a 2D surface develop over time typical FA type adhesions, supplanting costameres, as indicated in the top diagrams of Figure 2. Many, or all, of the proteins contained in the costamere in vivo, eventually reassemble within FAs, as isolated cardiomyocytes attach and spread in 2D culture in vitro [32].
Cadherins (indicating calcium-dependent adhesion) are transmembrane proteins important in cell cohesion. They form the adherens junctions that bind cardiomyocytes tightly together during systolic contraction, induced by the sliding of the sarcomeric myofibrils. Cadherins depend on calcium (Ca2+) ions for dynamic regulation of intercellular trans-junctional adhesive interactions [33,34]. Cadherins exert their impact at several different functional levels, ranging from conferring resistance to detachment, controlling the morphogenesis of contacts as cells coalesce into tissue histoarchitectonics, and influencing tissue cellular cohesion and patterning [35]. In particular, they robustly determine tissue cellular alignments both in health and disease, and are involved in cardiac diseases such as dilated and dysplastic cardiomyopathies, in which prominent alterations in tissue architecture occur [36].
Global cell elasticity reflects cytoskeletal dynamics, as conditioned by active and passive tensegrity contrivances [37]. Since it embodies complex regulatory mechanisms, endocardial and cardiomyocyte elasticity pertains to the overall cell rather than specific properties of individual components [38]. Thus, long- and short-term time-resolved elasticity measurements under conditions of impaired RV/LV filling vortex strength, with reduced endocardial shear and myocardial “squeeze” (see companion paper [3]), could provide useful insights into cytoskeletal defects and ventricular remodeling accompanying such abnormalities [1–3]. Diastolic filling vortex formation abnormalities and weakening [39,40] could be an epigenetic factor underlying ventricular dysfunction and remodeling, as is indicated in Figure 1.
2.3 The extracellular matrix (ECM) and its fiber lattice components
Important properties, including cell–cell and cell–ECM adhesiveness and myocardial histoarchitectonics are controlled by the mechanical responses of diverse heart cells (including endocardium, cardiomyocytes, and fibroblasts) to modulating forces of their environment, foreseeably including diastolic vortical shear, torques, and “squeeze” [1–3]. Mural myocardial cells are linked by a dynamic ECM framework composed of different collagen types, large elastin fiber bundles, and adhesive glycoproteins, in an amorphous mixture of easily hydrated, lubricating protein-polysaccharide macromolecules, or proteoglycans [41,42]. Every cardiomyocyte in the myocardium is surrounded by an extensive ECM collagenous meshwork enwrapping it as a thick fibrous lattice, which is essential for development/transmission of diastolic filling and vortical forces and for providing anchoring against which contractile stresses can be generated in systole. It also supports and preserves the 3D arrangement of cardiomyocytes and myocardial blood and lymphatic vessels. Processes of ECM assembly and disassembly by proteases (viz., matrix metalloproteinases, which – after activation – can digest ECM components that encompass glycoproteins, collagen, laminins (see below), and basement membrane proteins), are central to vital cardiac functions, most notably RV/LV remodeling [43–45], in response to applied “environmental” cues.
The collagen-elastin fiber ECM lattice provides elastic support during RV/LV filling and gives dynamic shape to the pumping ventricles and the whole heart [46,47]. Elastin is a highly cross-linked polymer that organizes as fibers or sheets in the ECM. It provides the ventricles with remarkable mechanical properties; most notably, with elastic recoil allowing their myocardial wall laminae to twist under systolic contractile stresses – like torsion-springs under a torque, or twisting force – and then to rebound to their previous state in early diastole, reinforcing rapid filling, especially during hyperdynamic circumstances [47,48]. The elasticity of elastic fibers arises from their hydrophobic regions [49], which are stretched out by tensile forces – such as are exerted by the diastolic vortex – and spontaneously reaggregate when the force is released. Collagen is a group of proteins found in muscle and connective tissues [50]. It is the main constituent of connective tissue, and is the most abundant protein in mammals, representing about 25–35% of the whole-body protein content. Fibroblasts make up the largest cell population of the heart and are the most common cell type that synthesizes collagen and elastin [51]. Besides ECM deposition/assembly and paracrine signaling, cardiac fibroblasts can communicate directly with cardiomyocytes via N-cadherin and connexins, thus influencing electromechanical coupling and cardiac action potential conduction [52].
The laminins are a class of glycoproteins that are an integral part of the structural scaffolding in most tissues, including the myocardium [53]. They are an essential biologically active part of the basal lamina (hence the name) of the basement membrane, a protein network foundation for most cells, tissues, and organs, and they influence cell differentiation, migration, adhesion and survival, as well as overall organ phenotype. They bind to other cell membrane and ECM molecules and help anchor organized tissue cells to the basement membrane. Fibronectins are large, cell surface and ECM glycoproteins, which exhibit structural and adhesive properties in cell-associated fibrillar matrices [54]. Like other ECM proteins, they are produced mainly by fibroblasts and are one of the primary cell adhesion molecules, promoting attachment of cells to the ECM by binding ECM collagen and other components to integrin trans-membrane receptors. Within the ECM, super-molecular fibronectin assemblies can be dynamically unfolded and stretched to multiple times their resting length by myocyte-generated or external forces [55].
3. The key role of integrin cell surface receptors in ECM–cardiomyocyte interactions
The aforementioned ECM components form a flexible scaffold for cell membrane adhesion that transforms mechanical loading into intracellular signals [14]. During ventricular filling, endocardium and relaxing/relaxed myocytes are normally deformed by chamber volume expansion and by variable diastolic shear, torque, and “squeeze” forces associated with intraventricular vortex flow, as discussed in the companion paper [3]. Deformation forces are transmitted and balanced within the cells by cytoskeletal and nucleoskeletal tensegrity action (see Sections 2.2 and 6–6.2); they may also be transmitted to and from other interlacing RV/LV mural cells through both cell–cell junctions and cell–ECM adhesions [1,2]. In fact, tensegrity accounts for the capacity of cells, tissues, organs, and the body to absorb impacts without necessarily being damaged. Mechanical energy flows away from a site of impact through the tensegrity achieving disposition of the extracellular, cytoskeletal, and nucleoskeletal lattices/matrices. The better their tensional integrity, the more readily these structures absorb shocks and deformations throughout the whole interconnected global matrix (cf. Figure 4) instead of at one localized region.
Growth factors stored within the ECM are released following mechanical stimulation [56]; they encourage cell proliferation and production of ECM molecules by fibroblasts [57]. Many cell types, including cardiomyocytes, normally require integrin-mediated adhesion to the ECM to maintain their survival, and signals from integrin engagement and clustering within focal adhesions/costameres contribute to prevention of apoptosis or “anoikis” (from Gk. for “homelessness”), viz., apoptosis induced by lack of correct cell/ECM attachment [58,59].
The ECM provides a supporting physical framework for cells during cardiac tissue and organ morphogenesis, homeostasis and remodeling. By means of integrin cell surface receptors, it also acts as an informational apparatus inasmuch as it detects, transduces and coordinates outside-in as well as inside-out mechanical signals originating from adjacent cells and the tissue microenvironment [60,61]. Integrins (see below) effectively transduce mechanical information, amplified by fibrous components of the ECM, to the embedded cells while also revealing the status of the cells to the outside, allowing rapid and flexible interactive responses to changes in the Bernardian environment. Complex, dynamic, regulated interactions that link extracellular and intracellular protein networks serve to adjust cellular responses to extracellular forces and related mechanical cues, such as vortical shear-induced stretching and vortical squeeze-induced compressions and distortions [1–3]. Consequently, networks of ECM proteins and associated cell surface receptors ought to exert subtle dynamic influences on gene expression and cell and organ morphomechanics, such as pertain to cardiac remodeling. It is indeed generally recognized now that the cardiac ECM is an essential determinant of tissue-specific gene expression [22,57,62].
As noted above, signals likely provided by the ECM in response to normal diastolic vortical shear and “squeeze” are transduced by integrin cell-surface matrix receptors, aptly named [63] for their role in integrating the intracellular CSK with the ECM, and vice-versa. Integrins can also be viewed as integrators of structural-spatial cues communicated by the ECM with those supplied by soluble ligands. A sizable and growing family of rod-like membrane-spanning heterodimeric (αβ) glycoprotein cell surface receptors [64], the integrins pin cells onto the ECM (Figure 1, lower left inset). The intracellular portion of integrins binds to actin filaments of the CSK; the extracellular, to various ECM proteins, including collagens, laminins and fibronectin [65–67].
ECM proteins bind to integrins as well as other cell surface receptors, activating signaling pathways that regulate cellular adhesion, growth and apoptosis, and tissue morphology [64,68]. In the myocardium, they couple the ECM to structural and functional elements within the endocardial and myocardial cells [1,2] (Figure 2, top). The forces transmitted through the ECM both activate cellular paracrine signaling pathways and initiate cytoskeletal rearrangements [1,60,69]. Integrins detect deformation forces, such as hydrodynamic tractive shear and torques. They then bind specific signal transducing ECM ligands, leading to rapid activation of intracellular signaling cascades and of the large assembly of “solid-state,” F-actin-based, cytoskeletal structures that directly propagate externally applied cell-deforming forces. Focal adhesion kinase (FAK) is a cytoplasmic enzyme that plays a major role in integrin-mediated signaling cascades [70,71]. The size of the signal cascade can increase rapidly, allowing for a large response, to relatively small initiating mechanical cues – including diastolic vortical shear and “squeeze” forces [1–3] (see Section 4). Signal cascades improve efficiency of signal transfer, facilitate interactions among different signal pathways, and control localization of signal actions within the cardiomyocytes [72].
4. Mechanotransduction and signaling affect myocardial function and adaptations
To respond promptly to changing circulatory demands, cardiomyocytes contain several mechanotransductory regulatory receptors connected to an advanced intracellular signaling system. Hence, they can transduce imposed mechanical forces/deformations into differentiated biochemical signals; e.g., an external shear-force on an endocardial cell can change the conformation of a transmembrane protein that acts as a stress-sensitive ion channel, causing it to open, thus allowing a transient K+ influx [73]. This is one archetypical example of the many ways a cell can physically “feel” its surroundings and respond to environmental cues. Mechanosensitive feedbacks can modify cellular and extracellular tissue and organ function and structure [74]. They can modulate myocardial architecture and diverse cellular properties and processes (e.g., cardiomyocyte adhesiveness, hypertrophy, fibrosis and apoptosis/anoikis) involved in cardiac homeostasis and RV/LV adaptations [44,50,51,57,70,71,75]. Any disturbance to typical myocardial mechanosensing and signaling activities, such as those likely subserved through normal diastolic vortical shear, torques, and “squeeze”, could lead to cardiac abnormalities [1–3].
Electrical excitation and excitation–contraction coupling entail compound and fast ionic fluxes. Consequently, cardiomyocyte membranes contain a large number of different ion-transporting proteins and channels. These stretch-sensitive structures and stretch-activated channels (SAC) open upon increased stretch of the membrane (see Figure 3), allowing the passage of cations, K+, Na+ and Ca2+ [76,77], and can thus modulate calcium handling and cardiomyocyte rhythmicity. Then again, the membrane is in some measure spared from inordinate stretch by caveolae. These are cytoskeletally controlled flask-shaped invaginations or dimples, which can append additional “reserve” material to the sarcolemma under tension, through rapid unfolding [78]. Caveolar flattening helps avoid rupture, when a cardiomyocyte is stretched. For geometric reasons [1], this protective mechanism is most valuable for the subendocardial RV/LV myocytes, which undergo the most marked cyclic strains during normal pumping and, especially, with augmentations of the stroke volume. The caveolae contain several signaling components, and their mechanical deformation has been linked to the activation of stretch-induced signaling cascades [78]. Thus, diastolic vortical shear stress may dynamically regulate both caveolar structure and signaling [79]. Caveolar dysfunction has been linked to a wide range of human diseases, including cardiac disease and muscular dystrophies [80].
Figure 3.
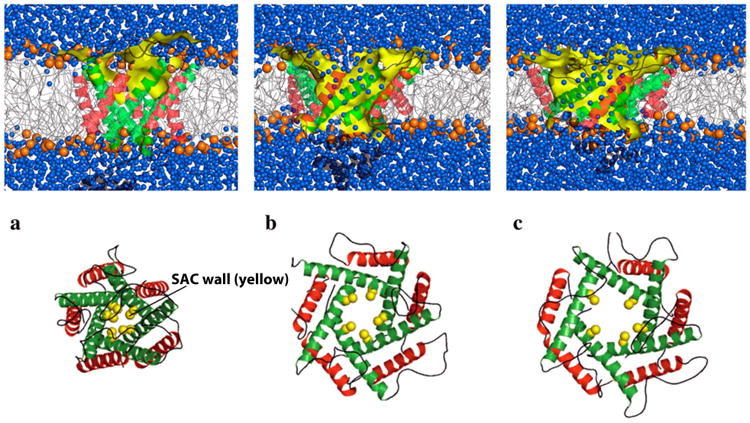
Schematic of numerical simulation of the gating process of a sarcolemmal mechanosensitive ion channel. Putting the membrane under tension, the stretch-activated channels (SAC) undergo significant conformational changes in accordance with an iris-like dilation mechanism, reaching a conducting state on a microsecond timescale. Diagrammed is a channel with (a) small, (b) intermediate, and (c) large opening under the action of rising membrane tension. Top: side view showing the lipid bilayer (blue denotes an aqueous environment, yellow represents the SAC wall). Bottom: End view showing the simulated membrane pore. Reproduced, slightly modified, with permission from Yefimov S, et al [77].
As summarized in the preceding Sections, the machinery of mechanotransduction in the myocardium encompasses an interdependent network of cell–cell and cell–ECM interactions and signaling responses. Although commonly they are spatially discrete, transmembrane cadherin and integrin complexes activate many of the same signaling pathways and provoke similar cellular activities, acting as interdependent functional nodes in a larger cytomechanics network; thus, they appear as functionally equivalent. Intriguing ongoing research examines how extracellular forces are transmitted into individual cells [81–83]. Cellular capacity to sense and respond to external forces involves diverse cytoskeletal and biochemical mechanisms [84–86]. Sensing vortical shear and squeeze forces and the ensuing responses must encompass similar intricate interactions.
Established endothelial mechanotransduction research addresses how external shear gets converted into biochemical signals [1,14,73,74,81]. However, endocardial cytoskeletal filament bundles can also transmit deforming vortical forces physically, through transmembrane integrin proteins in FAs, to the basal lamina of the basement membrane and the ECM, and also to lateral intercellular cadherin attachments, where further mechanotransduction events go on [1,2,81–86]. Myocardial longitudinal intercellular junctions include the intermediate filament-anchoring desmosomes and the actin filament anchoring adherens junctions, which – being tied up through linker proteins to the CSK proteins of neighboring cardiomyocytes – support structural stability through cell–cell adhesion (see Figure 2) and, in addition, regulate transcription of genes. Both junction types are formed by various cadherins, most of which are single-pass transmembrane adhesion glycoproteins. The variable, Ca2+-dependent, cadherin adhesiveness allows for an adjustable strength in the mechanical linkage across cell–cell junctions [87,88].
The generic focal adhesion complex is a macromolecular heterocomplex [89] including membrane-spanning integrins [65] and syndecans [90], and intracellular vinculin, paxillin, and talin [91] anchoring ECM-linked integrin to cytoskeletal F-actin. The dynamic linkage at FAs has been modeled as a macromolecular clutch–declutch mechanism, by which a modular interface of adaptor proteins mediates a dynamic mechanical connection between the F-actin–integrin– ECM complexes [92]. The clutch releases or connects the actin to the integrin–ECM complexes in order to deliver stress or relieve it, as follows: When the clutch is disengaged, the actin CSK is uncoupled from the ECM; when it is partially engaged, forces are transmitted only to some extent between the ECM and CSK; but when it is fully engaged, forces are transmitted totally between the ECM and the actin CSK.
Most importantly, FAs self-assemble and stretch upon application of pulling forces and disband when these forces decline [93]. Mechanical distortion of FAs elicits mechanical “solid-state” signaling, and synergistic signaling by integrins and syndecans subserves cellular and paracrine responses to external forces [94,95]. These, in turn, activate biochemical reactions and affect gene expression, protein synthesis, and cellular phenotype. Defects in such “solid-state” pathways, through which mechanics cues regulate cytomechanical cellular responses, could link impaired diastolic filling vortex dynamics and strength levels to RV/LV morphomechanical abnormalities, as I describe in detail in Section 5.
5. Advantages of cytoskeletal “solid-state” mechanotransduction and force transmission across the interlocking myocardial cells
Advances in nanotechnology for cytomechanical investigations nowadays allow direct measurement of forces at cellular, subcellular, and macromolecular scales [48]. The mechanical microenvironment of the myocardial cells that are being subjected to the cyclic (quasi-periodic) diastolic vortical forces is specified by way of forces transmitted across cell-to-cell junctions and through myocardial cell attachments to the ECM. Cell–ECM and cell-to-cell interactions take place mainly via integrins and cadherins, respectively [96]. Cardiomyocytes are coupled longitudinally by the electromechanically active intercalated discs and transversely, at the Z-disc, by special FA structures identified as costameres [31] (Figure 2, top).
Integrins, and other specialized proteins radially arrayed at costameres, anchor the Z-discs of the myofibrils to the sarcolemma (see Figure 2, top) and couple mechanically a cardiomyocyte's internal contractile machinery to the ECM [64,97], and, ultimately, to that of contiguous muscle cells. N-cadherins are transmembrane adhesion proteins that mediate cell-to-cell junctions through their extracellular domains and form the desmosomes of the intercalated discs, which link the interlaced cardiomyocytes together end-to-end [98,99]. It looks as if integrins, N-cadherins and related cell–ECM and cell-to-cell adhesion proteins act like key logic gates in the mighty biological computer of the RV/LV myocardium, functioning as key intersections for the overall flow of energy and information in the pumping heart. They are focal points where many approaches converge in terms of intracardiac fluid dynamics, energetics, physiology, biochemistry, systems biology and, in due course, therapeutic approaches.
Mechanotransduction in the myocardium is not a single-cell phenomenon. Instead, mechanical forces and cues can be transmitted through cellular and ECM tissue components and can be operational over hundreds of microns to millimeters of myocardial tissue thickness. Intercalated discs connecting cardiac muscle cells contain desmosomes (see Figure 2, middle & bottom), which hold the cardiomyocytes together during contraction [100,101], induced by the sliding of the sarcomeric myofibrils that is regulated by the intracellular concentration of calcium ions released by the sarcoplasmic reticulum. With cadherin proteins acting as the strong adhering link between adjacent sarcolemma membranes, desmosomes mechanically attach the CSK of a cardiomyocyte to those of adjoining cells, as well as to the ECM [76]. The desmosomal cadherin desmoglein 2 participates in the intercellular attachment of the force-transmitting intermediate filaments of the adjacent cells. Mutations of desmoglein 2 result in decreased numbers of cardiomyocyte desmosomes and a widening of intercalated disc gaps; impaired cell-to-cell adhesion then leads to cardiomyocyte detachment and cell death, proceeding to desmosomal dilated cardiomyopathy [102].
5.1 Main advantages of cytoskeletal force transduction and signaling
The advantages of the cytoskeletal force transduction and “solid-state” signaling across the interlacing ventricular myocardial cells are manifold [1,2,103]:
Without cytoskeletal intermediation involving amplification by molecular motor mechanisms generating forces and torques [104], the intensities of vortex-associated shear and squeeze forces on myocardial cells might not suffice for conformational rearrangements (e.g., macromolecular unfolding in 3D) and biochemical activity modifications. Unfolding decreases drastically the binding affinity between two folded macromolecules [105]. A noteworthy example of this regulatory operation in cardiomyocytes involves the sarcoendoplasmic reticulum (SR) calcium transport ATPase (SERCA) and the SR membrane protein phospholamban (PLN), which regulate cardiac contractility. SERCA regulation depends on PLN structural transitions (i.e., folding/unfolding) [106]. Activation of SR and plasma-membrane Ca2+ channels causes increased cytoplasmic Ca2+ concentration and leads to cardiomyocyte contraction. SERCA, a SR calcium pump, mediates transport of cytoplasmic Ca2+ into the SR lumen, causing the ensuing relaxation [107]. PLN binding to SERCA inhibits SERCA-mediated Ca2+ flux from the cytoplasm into the SR.
-
CSK-based “solid-state” mechanotransmission of stress signals is much speedier as well as more precise and efficient in conveying information across multiple cells via intercellular junctions, or over long cardiomyocyte intracellular distances, than diffusion-based chemical signaling. A tensed cytoskeleton is necessary for long-distance force propagation in the cytoplasm (see next Section). Long-distance stress propagation in the cytoplasm and into the nucleus of living cells has been examined and the results show that stress-induced signal mechanotransduction is at least 40 times faster than growth factor-induced chemical signal transduction [108,109]. Thus, mechanical action at a distance is accomplished extremely rapidly by the propagation of physical cues (input deformation energy) through transmembrane integrins and cadherins, the associated FAs and junctional complexes, and the network of load-bearing cytoskeletal filaments that link up to the nucleus, its internal scaffolding assemblies, anchoring, regulatory, and signaling proteins, and associated chromatin fibers [110].
Cytoskeletal filaments can be tensed to different degrees as they are traversing long intracellular distances. Thus, the speed and fidelity of the intracellular signaling response can be regulated through CSK stress fiber and intermediate filament associated molecular motors, by modulating cytoskeletal prestress and stiffness, which are proportional to the signal wave propagation velocity [1,111]. Cytoskeletal mechanotransduction and mechanotransmission provide for very nearly synchronously altering activities of assorted molecules at widespread intracellular and nuclear sites; this could be crucial for multifaceted control of cell function and adaptations by the diastolic laminar vortical shear, torque, and squeeze forces.
Channeling forces along prestresd cytoskeletal filaments can focus stresses on specific mechanosensory molecules and structures, while protecting most other cellular components from excessive strains. Forces exerted on RV/LV myocardium (such as those due to the diastolic filling vortices) will be channeled over the ECM fibers and linked integrins, and will in that way be focused on focal adhesions/costameres and the CSK, where mechanochemical transduction processes may then proceed. Various substrates and enzymes facilitating signal transduction, DNA synthesis, transcription, RNA processing, translation for protein production, protein folding, and other processes are not floating free in the nucleoplasm, or in the sarcoplasm; instead, they function when immobilized on the insoluble “solid-state” cytoskeletal/nucleoskeletal scaffolding within the cytoplasm and the nucleus [112]. Thus, forces focused on the scaffolding filaments may result in physical deformations of these immobilized molecules; such molecular deformations may directly influence biochemical activities and kinetics—cf. the effects of tension and compression on polymerization dynamics of cytoskeletal microtubules [113].
6. External force transmission to the nucleus and epigenetic influences of the vortical shear and “squeeze” on gene expression
Molecules in the working myofibril sarcomeres of cardiomyocytes can act as mechanosensors for outside-in mechanotransduction, which are capable of nuclear signaling. This allows adaptive transcriptional responses (mechano-transcription coupling) to long- or short-term changes in loading, as in pressure and volume overload or in experimentally stretched cardiomyocytes in vitro, bringing about reactions such as increased sarcomerogenesis [114,115], or stepped up mRNA expression for Connective Tissue Growth Factor (CTGF) [116]. Cardiomyocytes encompass numerous components implicated in mechanotransduction of external forces, including load-bearing subcellular structures, such as the dynamic cytoskeletal filament networks, transmembrane cell–cell and cell–ECM junctions, and stretch-sensitive membrane structures, including SACs and caveolae. Each of these is involved in imparting structural integrity as well as in the “sensation,” mechanotransduction, transmission, and/or modulation of myocardial “environmental” forces and mechanical cues. For instance, FAs are external force mechanosensors that can activate both biochemical and cytoskeletal signaling pathways for cell function/structure control and organ remodeling [28,84,89–95].
The nucleus may effectively be construed as a mechanosensor, whose envelope, internal nuclear scaffolds (nucleoskeleton, NSK, Figure 4), and chromatin are firmly coupled to the ECM by the CSK and FA receptors, mainly integrins. Highly ordered chromatin structure represents a physical obstacle for transcriptional machinery and transcription factors to bind DNA [1–3,117]. It is, therefore, important that externally applied forces can modify nuclear microarchitecture, including chromatin organization and consequently gene expression, much more rapidly than biochemical signaling transduction cascades [103,108,109]. Forces exerted through the ECM on FA integrin receptors catalyze phosphorylation of signaling proteins but are also propagated via the hard-wired cytoskeletal filaments directly to the nucleus, nucleoskeleton [118], and linked chromatin [2,103,119].
6.1 Global mechanical interconnectedness
According to mechanics premises (St. Venant's principle) for homogeneous continuum materials, stress decays rapidly as the reciprocal of the distance squared; thus, one would predict that a local mechanical load of physiologic magnitude should cause simply a local deformation, and only local direct mechanotransduction. However, since prestressed tensegrity assemblies resist externally applied forces by geometric readjustments of their structural members, any local deformation should beget a global rearrangement of the CSK “solid-state” lattice and should be distributed to points remote from the applied force. In a prestressed inhomogeneous material (tensegrity cell model), the stresses are actually channeled preferentially over CSK filaments that are stiffened owing to prestresses and, therefore, they decay at a slower rate [108,109]. Macromolecular cytoskeletal motors can pull, move, reshape and organize the CSK filament system and can adjust its regional prestress levels. Anchoring the motors (e.g., to the subsarcolemmal cell cortex) results in them tugging on the CSK filaments and increasing their tension. This allows for long-distance force propagation and rapid “reach” into the nucleus. Cadherins, integrins, and related cell-to-cell and cell–ECM adhesion proteins bond every myocardial cell to neighboring cells and to the surrounding ECM (Figure 2).
The interlinked extracellular, cytoskeletal, and nucleoskeletal matrices represent an all-encompassing web of high-speed interconnections and communications within the myocardium, which stretches from nuclei to cells and tissues. It extends into the “environment” and senses mechanical stimuli/signals, involving vortical shear, torque, and “squeeze” forces, originating therein (see Figure 4). This global matrix connects all myocardial cells and tissues to their environment and allows the dynamic fields of energy and information linking them to form swift, bidirectional feedback paths between the environment outside, and the cells within—cf. “outside-in” and “inside-out” signal transmission discussion, in Section 2.1. Individual component tensegrities are integral – all parts are connected in a non-hierarchical association.
Forces and deformations are transferred bidirectionally globally across the entire network:
DNA ⇔ Nucleoskeletal Matrix ⇔ Cytoskeletal Matrix ⇔ ECM ⇔ Environment
In this fashion, all myocardial tissues and structures interconnect and criss-cross. This can be viewed as a rapid solid-state communication system. The fibrous protein structures of the ECM and cyto-/nucleoskeleton in the myocardial cells represent much more than scaffolding holding the synergistic complex together. They form a continuous and interconnected dynamic 3D network that is similar to a great information superhighway. They are constituents of a tensegrity based, interdigitated, interconnected and cooperative continuum that enables all parts of the system to communicate, rapidly and bidirectionally, with all others.
A tensegrity tissue matrix system allows for detailed transfer of information throughout the myocardium by direct transmission of mechanical energy through mechanical wave propagation, extending from the endocardial surfaces exposed to quasi-periodical vortical flow forces all the way down to the myocardial cells, nuclei and DNA. Since the tensegrity network is a mechanical continuum, vortical shear, torque, and “squeeze” forces applied in one part can have morphomechanical consequences for the entire myocardium and cardiac pumping performance. Movements, tensions, and mechanical energy fluxes propagated through this system interact epigenetically with the genetic material to shape the dynamic myocardial/cardiac phenotype (G×E interactions = Phenotype), as is shown in Figure 1.
6.2 Nucleus, where mechanical forces regulate gene expression
The LINC (Linker of Nucleoskeleton and Cytoskeleton) complex comprises two protein families, KASH (Klarsicht, ANC-1, and Syne homology) and SUN (Sad1/UNC-84), which span the nuclear envelope and link physically the CSK (KASH family) to the network of filaments within the nucleus (SUN family) that compose the nucleoskeleton [120–123]. SUN-domain proteins of the inner interact with various KASH-domain cohorts of the outer nuclear membrane to make ‘bridges’ across the inner and outer nuclear membranes [120] of the nuclear envelope (see Figure 4). These bridges, in turn, physically connect the nucleus to every major constituent of the CSK, and have been likened to the ‘Velcro’ in the nuclear envelope that mechanically links the CSK to the NSK [124].
By rearranging the configurations of cytoplasmic and nuclear macromolecular assemblies (cf. Figure 4), KASH and SUN can quickly induce mechanochemical conversions, modify (relax) the repressive structure of chromatin, promote nucleosome disruption and chromatin relaxation, and alter gene activities [1–3,86,117,125]. The nuclear matrix, or nucleoskeleton/chromatin scaffold [126], comprises the non-chromatin nuclear proteins and consists of: (a) the nucleoskeleton, an ordered and highly compartmentalized network of anastomosing protein fibers, similar in size to cytoskeletal intermediate filaments, which is linked to (b) the proteins of the nuclear lamina, which are anchored to the inner membrane of the nuclear envelope (Figure 4) and are known as lamins, not to be confused with the earlier discussed laminins (see Section 2.3). Lamins in the nuclear interior may act as conveyors of mechanical signals to regulate gene expression [121].
Both cytoskeletal “solid-state” and biochemical signaling are potent inputs into myocardial control mechanisms whose adaptive (remodeling) responses must counter considerable deviations from ordinary operating circumstances [84–86,127,128]. In biochemical signal transduction, most cell membrane receptors stimulate intracellular enzymes linked to them by guanine nucleotide-binding G proteins [129]; these G protein-coupled receptors (GPCRs) are responsible for regulating various important cellular responses including cell proliferation and survival. They amplify and transmit signals elicited by extracellular mechanical stimuli (stresses and strains) and ligand binding to downstream intracellular targets; signal targets include nuclear transcription factors [130], which modulate gene expression.
Cytoskeletal lattices also link transmembrane integrins to subnuclear elements [131], physically transmitting external forces to produce intranuclear deformations (Figure 4) and conformational modifications of DNA chromatin, modulating transcriptional activities [1– 3,125,132–134]. This suggests again that “environmental” mechanical stresses and strains can regulate the function of nuclear effectors. CSK solid-state signaling may yield rapid metabolic changes or trigger longer term responses, by altered transcription and translation of proteins – via nuclear/intracellular receptors. Filling vortex-associated shear and squeeze forces transmitted to the nucleus (Figure 4) could, therefore, modify nuclear protein self-assembly, DNA replication, gene transcription and RNA processing, affecting translation and the proteome. Notably, disruption of the F-actin CSK inhibits shear-mediated signaling and changes in gene expression [135], and there is evidence for direct functional involvement of actin, and of nuclear myosin 1 (NM1), in transcription [136–138].
7. Translating vortical mechanical forces into phenotypic adaptive (remodeling) responses
Mechanical forces are central in the maintenance and remodeling of normal tissues, including the myocardium, under physiological circumstances, as well as in the initiation and development of morphomechanical abnormalities and disease [139]. The ECM and its coupled cell adhesion protein receptors, including integrins, transduce biomechanical signals to the cell interior and allow interaction of cells with their mechanical (micro) environment. Furthermore, the expression of ECM protein genes is also modulated by mechanical forces. Complementarily, the cell shape-sustaining CSK is vital in myocardial responses to vortical shear, torques, and squeeze, and in related RV/LV adaptations [1–3,140].
Shear-induced endothelial gene transcription modulation, e.g., through changes in Ca2+ signaling and NO [141,142] can produce wide-ranging auto- and paracrine effects in the proteome [1,2]. They include rapid and reversible upregulation of mitogen-activated protein kinase (MAPK) cascades regulating complex cellular programs like cell adhesion, growth and apoptosis, and thus modulating genomic expression profiles, cell phenotype and histoarchitectonics [143–146]. Research on this mechanochemical link has focused on the CSK and its linkages to the ECM [85,147]. Additionally, internal cytoskeletal tension forces transmitted to adjacent cells by cell–cell adhesion cadherin receptors [99,148], can give rise to long-range mechanical communication and interactions among cells. Such interactions could dictate the mutual alignment of cells and affect histoarchitectonics in large cellular assemblies [149,150], such as the RV/LV myocardium.
As suggested in Figure 5, myocardial cells in general, and cardiomyocytes in particular, can take not necessarily heritable transgenerationally (i.e., here I am using Waddington's [151] designation of epigenetic) epigenetic external force inputs and integrate them with other signals to adapt their phenotype; e.g., choosing to upregulate the expression of proteins, potentially undergoing hypertrophy [152,153]. Cardiac mural cells can also respond by remodeling their ECM, modifying the abundance and composition of ECM proteins like collagen, potentially to shore up cardiac chambers against overdistension/dilatation [154]. Such responses lead directly to epigenetic phenotypic alterations and resultant functional changes in cardiac mechanics; e.g., RV/LV myocardium translates mechanical requirements of pressure or volume overloads into concentric and eccentric hypertrophic growth and remodeling [1,111,155–160].
Figure 5.
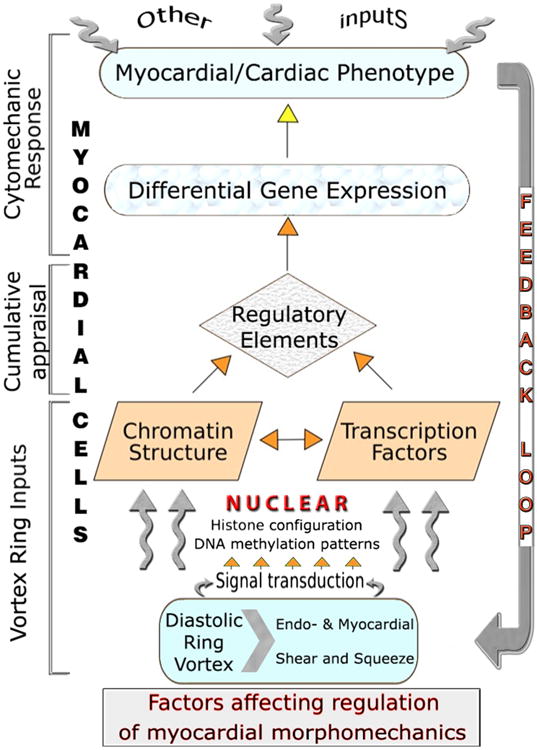
Summary of epigenetic dynamic actions of RV/LV diastolic toroidal vortex: Cytoskeletal “tensegrity” dynamics create equilibrium between strut compression and string tension, allowing internal structural balance and shape maintenance. Cells sense their physical 3D “environment,” including variable diastolic vortical shear, torque and “squeeze” forces, by transducing mechanical deformations and forces into differentiated transcription and translation signals, which can adjust cellular and extracellular tissue and organ phenotype; the latter, in turn, affects (feedback loop) diastolic filling vortex strength and vortical shear and “squeeze” forces. Mechanosensitive regulatory controls modulate myocyte shape and intracellular architecture, and processes as diverse as proliferation, hypertrophy, and apoptosis, involved in cardiac homeostasis and adaptations.
Application of tractive and tension forces, akin to those corresponding to diastolic vortical shear, torque, and squeeze (see Figure 5), to cells through their integrin-intermediated costameric adhesions modulates a broad array of signaling pathways, genes and differentiation processes, with effects that are of considerable (patho) physiological significance. A stretch-induced actin reinforcement response and remodeling has been shown experimentally [16,17,161]. Similarly, FAs self-assemble and expand upon experimental application of pulling forces and disband when these forces decline [93–97]. Thus, normal or hyperdynamic cyclic (quasi-periodic) diastolic vortical strength and associated forces acting across the endocardial and myocardial FAs and costameres (FA/CM) could give rise, through mechanical (“solid-state”) cyto-/nucleoskeletal and paracrine pathways, to thickened F-actin stress fibers and could cause integrin clustering into large FA/CM; conversely, under hypodynamic diastolic intraventricular rotatory flow conditions in which vortex strength and forces are low, F-actin would be reorganized into thinner bundles and FA/CM would be smaller and fewer. Accordingly, the high force/oversized FA/CM state would then be shifted to a weak force/undersized FA/CM state. Moreover, myocardial wall cells under higher mechanical forces typically put together stronger ECMs [162,163], ensuing partially from the effect of force magnitude on fibroblast gene expression and also because of direct force-induced effects on ECM fibrous components' assembly and 3D configuration. Recent work has shown that the mechanical strength of actin-to-ECM linkages rises in reaction to forces loaded there, and that several ECM component proteins change their 3D configuration in response to the forces applied at FAs, and this modifies their interactions with binding partners [164].
Contemporary research has ascertained one mechanism that regulates adhesion site strengthening versus disassembly under different levels of applied force [93,165]. A fluorescence-based tension sensor for vinculin, whose recruitment to FAs is regulated by externally or internally generated mechanical forces [166,167], revealed that FA strengthening ensues from higher force levels across this molecule and is followed by production and apposition of more vinculin and expansion in size and numbers of the FAs, culminating in reduced force per vinculin molecule. It appears, therefore, that FAs themselves are a major determinant of the signaling output [168]; the extent of integrin clustering could be an important variable governing signal output. FA stability could also affect signaling [169]. Ventricular enlargement/remodeling represents an example of FA/integrin-dependent (mal) adaptation [170], which is of particular significance in the context of vortex-engendered forces, as we shall see next.
7.1 Myocardial creep in dilated ventricles
In our integrative diastolic function investigations in canine animal models of heart disease and failure at the Duke Center for Emerging Cardiovascular Technologies [1,40,46,47,111,155,156,171–175], we demonstrated, using a sigmoidal diastolic passive filling pressure–volume relationship, that myocardial creep develops with RV dilatation in the course of surgically induced RV volume overload [1,40,47,155,156]. In human clinical investigations and canine experimental models of LV dilatation resulting from LV volume overload, or after ischemic myocardial injury or pacing-induced heart failure, myocardial creep was also found, in the form of a rightward shift of diastolic and end-systolic pressure-volume curves [176–180]. Ross and his coworkers also demonstrated that creep induced by volume overload correlated with both increased sarcomere length and “disarray” of the myofilaments [177]. It is also known, since the pioneering work of Bellhouse at Oxford, that the filling vortex strength is reduced with ventricular chamber dilatation [1,2,40,171–174].
Therefore, it seems that diminished vortical shear and squeeze forces, because of reduced vortex strength secondary to chamber enlargement with or without hypodynamic circulatory conditions (postischemic or pacing-induced dysfunction), may work as follows (cf. discussion in Sections 4 and 7): In contrast to normal or hyperdynamic cyclic (quasi-periodic) diastolic vortical strength and associated forces acting across the endocardial and myocardial FA/CM, which give rise, through mechanical cyto-/nucleoskeletal and paracrine pathways, to thickened actin stress fibers and cause integrin clustering into large FAs, hypodynamic diastolic intraventricular rotatory flow with low vortex strength and forces cause actin to be reorganized into thinner bundles and FA/CM to become smaller and fewer. This could then be associated with weakening of ECM proteins, side-to-side slippage, and elongation of cardiomyocytes, reorganization of myocardial sheets across the RV/LV wall thickness, and progressive myocardial creep and chamber dilatation, until strong recruitment of tensed collagen fibers ensues. This proposed sequence of events needs further investigation in forthcoming studies.
7.2 Potential effects of vortical stresses and strains on the Hippo pathway in the myocardium
In recent years, the Hippo kinase signaling pathway, which regulates cell proliferation and growth, differentiation, survival, and organ size, has attracted major attention in studies of regenerative medicine. Its two nuclear effectors, YAP (Yes-associated protein) and TAZ (WW domain containing transcription regulator 1, or WWTR1), are proteins that in experiments on 2D cell cultures have been shown to respond to complex mechanical cues represented by the rigidity of the extracellular matrix, cell geometry, cell density, and the status of the F-actin cytoskeleton [181,182,183]. In the developing heart, Hippo is a negative regulator of the pro-growth Wnt signaling pathway; it acts through a cascade of protein kinases, namely MST and LATS, which phosphorylate TAZ and YAP causing them to be sequestered in the cytoplasm, inactivated and degraded through proteasome activity [184]. When dephosphorylated, YAP and TAZ translocate to the nucleus and can interact with various transcription factors to induce gene expression, functioning as transcriptional coactivators. As transcriptional coactivators, they need to bind transcription factors to stimulate gene expression. The main transcriptional target of YAP and TAZ is the TEAD/TEF family of transcription factors, which have emerged as the prime mediators of YAP/TAZ function in Hippo signaling [185,186]. TEAD/TEF drive the expression of proliferative genes, as well as genes encoding inhibitors of apoptosis [182].
The regulation of cardiomyocyte proliferation is essential for normal heart development. Spatiotemporal patterns of cardiomyocyte proliferation are directly linked to cardiac morphogenesis, size, and function, since the heart is a pump, and a suitable size is required at every embryonic stage. The Hippo pathway helps control the heart size in mammalian embryos by repressing cardiomyocyte proliferation, by inhibiting Wnt signaling [187], and it may regulate tissue size in many animals by adjusting cell proliferation and apoptosis. It may help couple myocardial tissue growth to mechanical stresses or cell–cell contact, which might be central to cardiac size regulation. However, although cardiomyocyte proliferation levels are pronounced during early embryogenesis, soon after birth, mammalian cardiomyocytes exit the cell-cycle irreversibly. Although during the first 2 postnatal weeks many cardiomyocytes undergo mitosis, it only brings about binucleated myocytes because it occurs without cytokinesis [188]. The cell cycle exit impedes cardiac regeneration in mammals.
At present, there is no replacement therapy for dead or injured myocardium other than cardiac transplantation. However, therapy employing stem/progenitor cells holds out the prospect of myocardial repair, and may possess enormous potential in managing myocardial loss or heart injury and failure [189]. The discovery of heart resident stem/progenitor cells, a minor fraction of the total cardiac cell complement, has unlocked the potential for autologous myocardial repair, although their actual capability to improve heart function is still uncertain [190]. These cells have been demonstrated to gradually replenish cardiomyocytes when necessary [191], and their maintenance to be tightly regulated by biological and mechanical cues arising from the adjoining cells and the ECM [192]. Accordingly, methods for their segregation and proliferation to quantities adequate for therapeutic objectives are being investigated, along with methods for activation of endogenous stem cells or for transdifferentiation (metaplasia) of other cell types into cardiac cells [193,194]. YAP/TAZ can play an important role in controlling human cardiac progenitor cell differentiation toward cardiomyocyte lineage, and the YAP/TAZ axis has been described as a central regulator of human embryonic stem cell self-renewal, with experimental TAZ knockdown resulting in the loss of cell pluripotency [195]. YAP partakes in regulation of cardiomyocyte proliferation and embryonic heart development, its lack during cardiogenesis being associated with embryonic lethality. As noted above, YAP/TAZ shuttle to the nucleus when the 2D cell culture substrate becomes stiffer and cells can spread their cytoskeleton [181], effectively operating as general relays in cell mechanosensing. In addition, YAP and TAZ can play beneficial roles in stimulating tissue repair and cell regeneration when the F-actin CSK is undergoing cyclic tensional/compressional deformations/strains (cf. Figure 4). In the development of myocardial regeneration therapies, therefore, attention in future studies ought to be given to the application, on myocardial tissues encompassing stem/progenitor cells, of hydrodynamic shear, torque and “squeeze” forces analogous to those applied by the RV/LV diastolic filling vortex [1–3] both ante- and postpartum.
8. Future: correlation between RV/LV filling vortex-induced myocardial deformations and gene expression patterns regulating cardiac structure and function
As I suggested in Part 1 [3] of the present two-part survey and elsewhere [1,2,174], mechanisms analogous to Murray's law—endothelial cell modulation of vascular geometry by flow shear—are likely to exist between endocardial diastolic vortical shear, torque, and squeeze and RV/LV adaptations and remodeling. Mechanistic insights linking filling vortex patterns and attendant forces to variable expressions of gene regulation in RV/LV myocardium should reveal epigenetic mechanisms supporting ventricular function and adaptability through phenotypes not encoded in a rigid sense in the DNA.
RV/LV filling vortices can be visualized and analyzed as summarized in the companion paper [3], and are closely influenced [1] by heart rate, a primary determinant of both diastolic ventricular inflow and vortical velocities and of the diastolic time interval during which these vortices exert their regulatory actions on the Genotype × Environment = Phenotype equation (Figures 1 and 5). However, experimental evidence on cellular and molecular myocardial processes regulated by intraventricular vortical forces is currently nonexistent. Pertinent studies are needed, and should also include responses to interventions, so that functional models might be generated in due course. In a vascular context, analogous inquiries have identified scores of genes regulated by laminar shear whose transcriptional activity and expression increase or decline in response to flow [196–198]. Such studies have also provided a mechanistic explanation for endothelial gene regulation by flow, and led to the discovery of promoter sequences named shear stress responsive elements (SSREs), which mediate transcriptional responses to shear stress [199]. Analogous promoter sequences are likely to mediate RV/LV adaptive transcriptional feedbacks linked to diastolic vortical shear, torques, and squeeze.
8.1 Myocardial phenotypic plasticity
Myocardial phenotype accrues from selective expression of the genome, and embodies cell “experiences” and responses to extracellular/environmental forces [1–3]. To characterize how the variable strengths and durations of cyclic vortex shear and squeeze yield different myocardial “phenomes,” we need specialized research methodologies. High-density DNA microarrays (e.g., Affymetrix GeneChip), Northern blotting and serial analysis of gene expression (SAGE) for mRNA profiling, Western blotting for detection of specific proteins in tissue homogenates or cell extracts, and other gene expression and protein identification methods, afford powerful tools to study the expression/regulation of myocardial genes [116, 200–214]. Moreover, since 2005, Next Generation Sequencing (NGS) technologies for genome, epigenome and RNA sequencing have become available commercially [215]. NGS encompasses high-throughput DNA and RNA (coding and non-coding) sequencing methods which parallelize massively the sequencing process, producing thousands or millions of sequences at once. They can sequence human genomes and transcriptomes, and explore gene expression changes, epigenomic patterns, etc, on a genome-wide scale and with single base precision [216]. NGS technology begins by breaking up long pieces of DNA (or RNA converted to cDNA, by reverse transcription) into a great number of short sections of DNA; the resultant set of DNA is designated a “library” and the short pieces are “fragments” or “reads;” the fragments in the library are then sequenced individually and in parallel [215]. The parallel nature of NGS means that longer reads can be constructed by aligning and merging many contiguous short reads, a process defined as sequence assembly [217,218].
The term NGS applies to a number of different modern sequencing platforms, including Roche's pioneering 454 GS FLX System (currently being phased out), Illumina's Genome Analyzer II (GAII, also called Illumina Solexa) and HiSeq Systems, Life Technologies' Ion Torrent Personal Genome Machine (PGM), Ion Proton I and II, and 5500 SOLiD Systems, Pacific Biosciences' PacBio RS II, and others [219,220]. Remarkable progress has been achieved in terms of speed and throughput, along with a sharp reduction in per-base cost. Collectively, these advances have paved the way for the growth of a burgeoning number of applications in basic science and in translational research, including high quality rapid and inexpensive whole human genome sequencing (WGS), whole exome [protein-coding exon] sequencing (WES), and transcriptome sequencing [221,222].
As noted above, transcriptome sequencing entails sequencing cDNA fragments generated by reverse transcription of RNA; “RNA-Seq” for transcript quantification and characterization can determine RNA expression and splicing profiles under different operating (patho) physiological conditions [218,223]. Accordingly, it could be an emerging NGS application to characterize the epigenetics of diastolic RV/LV vortical stresses and strains, yielding potential diagnostic, therapeutic and prognostic insights. The above investigational tools are invaluable in upcoming pursuits of quantitative understanding of what RV/LV myocardial genes are differentially expressed, and how, and in tracking multiple ventricular myocardial constituent proteins under diverse circumstances of changing vortical strength and corresponding shear and “squeeze” patterns [1–3]. In interpreting such gene expression (mRNA) data, we should recall (Table 2) that a gene's mRNA level does not necessarily predict its active protein level [224–227]; differences in translation efficiency and posttranslational protein modifications or interactions may also be co-determinants in regulating active protein levels, myocardial structure and function.
Table 2. A gene's mRNA level does not necessarily reflect its protein level.
A. The processes determining steady-state protein
concentrations encompass [1–3]:
|
| The translation and protein degradation processes are no less important determinants of steady-state protein levels than mRNA transcription and stability [226]. |
|
|
| B. All mRNAs are not equal with regard to translation into proteins, and translational rate constants are the dominant factor in controlling protein levels [227]. Moreover, regulatory microRNAs (miRNAs) can exert posttranscriptional simultaneous repression of hundreds of genes, by inhibiting mRNA translation into protein [228]. |
| The commonplace housekeeping proteins would most likely exhibit relatively high correlation between mRNA and protein, whereas most important and remarkable regulators of cellular division, differentiation and development would be anticipated to show poor correlation between mRNA and protein. |
|
|
C. A multitude of post-translational mechanisms exists for
controlling protein turnover, half-lives, and abundance, through the action of:
|
|
|
| D. Mechanisms for maintenance of protein levels are better than those for mRNA levels. |
| Post-transcriptional, translational and protein-degradational regulation helps to compensate for drift of mRNA levels and bring protein levels back to preferred ranges [226]. |
|
|
| E. The mRNA levels cannot be used as surrogates for the corresponding protein levels without verification. |
| To understand the relationship between mRNA and protein levels, the dynamic processes involved in protein synthesis and degradation need to be understood better. An array of genomic and proteomic assessments would be anticipated to be more effective than either approach alone, in investigations to elucidate (patho) physiological adaptations and morphomechanical changes involving G×E interactions. |
The role of intranuclear deformations (Figure 4), nuclear microarchitecture and chromatin organization modifications in gene function is still imprecise [2,3,123]. Gene expression is an intricate multistep process, regulated strongly within the nucleus; the positioning of genes within the nuclear landscape is modified by dynamic intranuclear deformations (Figure 4), and plays an important role in gene regulation [228]. Correlating nuclear mechanics and structural organization with large-scale studies profiling gene expression, using NGS technologies for transcriptome sequencing, will ultimately lead to a more cohesive understanding of how the nuclear deformations induced by variable diastolic RV/LV vortical shear, torques, and “squeeze” contribute to myocardial phenotypic plasticity and morphomechanical changes. In addition, new nanotechnological approaches [229] to elucidating spatial genome organization and its link to function need to be developed.
9. Conclusions
Epigenetic mechanisms are fundamental in cardiac adaptations and disease. In the current post-genomic era, elusive epigenetic control mechanisms can begin to get unraveled. They promise to drive great growth and a veritable transformation in our appreciation of cardiac form-function and its adaptations to complex hydrodynamic forces, like the shear, torques, and “squeeze” associated with large-scale intraventricular filling vortex motions. Because of the Human Genome Project, translational cardiologists can already search a database to see what a gene specifies. In the future, they may also be able to look up when a gene is run— specifically, to learn how intraventricular vortex epigenetics control chemical modifications to DNA and to DNA-spooling proteins that regulate how cells deploy gene expression patterns. Notable in this context is the Encyclopedia of DNA Elements (ENCODE) project, a public research consortium launched by the National Human Genome Research Institute in Bethesda, MD, in 2003. ENCODE researchers intend to map which portions of human chromosomes are transcribed, how their transcription is regulated, and how it is affected by the way the DNA is packaged and positioned in the nucleus. Recently, the consortium made known [230,231] that besides the approximately 1% of our DNA that encompasses some 20,000 protein-coding genes, at least 80% of the human genome is transcribed into RNA that may well have a role in the regulation of gene expression, challenging the longstanding notion that much of our genome is junk.
This two-part survey has proposed that RV/LV diastolic filling vortex flow patterns are an overlooked epigenetic factor regulating/influencing functional and morphological changes in the heart. It has set down integrative concepts pertinent to its comprehensive investigation and relevant information from discrete but convergent disciplines. It is at the intersections among these synergetic disciplines where many of the novel concepts emerge.
Acknowledgments
Sources of Funding: Research support, for work from my Laboratory surveyed here, was provided by: National Heart, Lung, and Blood Institute, Grant R01 HL 050446; National Science Foundation, Grant CDR 8622201; and North Carolina Supercomputing Center and Cray Research.
Glossary
- compression (compressional stress)
The stress that squeezes something; the opposite of tension. The “pushing” stress component that acts perpendicular to a surface; compression in one (axial) direction induces tension in the other two (lateral) directions
- creep
The deformation (stretch) of a material under constant load over time; myocardial creep entails extracellular matrix (ECM) and cardiomyocyte elongation, myofiber slippage, and other histoarchitectonic changes
- cytoskeleton (CSK)
The fibrous proteins within a cell which provide tensegrity type structural support to the cell and are involved in mechanotransduction. It provides physical, high-speed, linkage between the cell membrane and putative intracellular stress-sensing components, including the nucleus, which respond to mechanical cues through multiple pathways
- epigenetics
(Gk. for “above/on top of” genetics) Refers to external modifications to DNA and its associated histone proteins that turn genes “on” or “off,” without altering the DNA base sequence
- epigenome
The complete set of epigenetic modifications (e.g., DNA methylation, histone acetylation, and chromatin remodeling) on the genome and allied histone proteins of a cell, tissue or organ
- extracellular matrix (ECM)
The dynamic macromolecular complex synthesized primarily by fibroblasts, which assembles into a network that surrounds cells. It not only provides essential physical scaffolding but also initiates crucial biomechanical and biochemical cues required for tissue function and adaptations
- focal adhesion kinase (FAK)
Cytoplasmic enzyme that plays a major role in integrin-mediated signaling cascades. The size of the signal cascade can increase rapidly, allowing for a large response, to small initiating mechanical cues, such as diastolic vortical shear and “squeeze” forces
- gene expression
The phenomenon whereby a gene is transcribed into an RNA molecule
- genome (human)
All the DNA—in humans, encompassing about 3 billion DNA base pairs—coiled and recoiled around aggregated proteins (histones) in 23 distinct (single or haploid set) chromosomes within the nucleus, allowing control of the accessibility of DNA information. Mitochondrial DNA, distinct from nuclear DNA, is a small genome contained in all mitochondria
- genotype
The inherited genetic makeup of a cell
- Hippo pathway
Signaling pathway that plays a restraining role in the control of organ growth and size via a cascade of protein kinases, which phosphorylates TAZ and YAP causing them to be retained in the cytoplasm; in the nucleus, TAZ and YAP act as transcriptional regulators. In the developing heart, Hippo is a negative regulator of the pro-growth Wnt signaling pathway
- histoarchitectonics
(Gk. ιστóς = “tissue” + αρχιτεκτονική = “architecture”) The adjustable cellular and non-cellular composition, changing intricate form patterns, and dynamic geometric arrangements of organs and tissues; investigated using various modalities, recognizing that form, structure, and function are inseparable
- KASH
One of two protein families (KASH/SUN domains) that are jointly responsible for holding together the two nuclear envelope membranes and for attaching them to the cytoskeleton and the nucleoskeleton. A branched F-actin cytoskeleton permeating the cytoplasm is linked to the nuclear envelope via the KASH/SUN integral membrane protein complexes and to the cell membrane via membrane-embedded integrin proteins
- LINC (Linker of Nucleoskeleton and Cytoskeleton)
Complexes built from members of two protein families, SUN and KASH. SUN domain proteins are integral to the inner nuclear membrane, whereas KASH domain proteins reside in the outer nuclear membrane. SUN and KASH proteins directly bind each other, thereby forming a bridge across the nuclear envelope, which is essential for enhanced CSK/NSK force-coupling and for physical and functional linkage between the cytoplasm and nucleoplasm
- mechanotransduction
How mechanical forces/cues are sensed and transduced into biochemical signals affecting cellular, tissue, and organ function and morphomechanical adaptations; involved are stretch-activated channels (SAC), cell adhesion receptors, extracellular matrix, cytoskeletal, and nucleoskeletal molecules, etc
- Next Generation Sequencing (NGS)
High-throughput DNA and RNA (coding and non-coding) sequencing methods which parallelize massively the sequencing process, producing thousands or millions of sequences at once; they can explore gene expression changes, epigenomic pattern, etc, on a genome-wide scale and with single base precision
- nuclear envelope
A double lipid bilayer membrane with multiple pores that surrounds the nucleus. The pores regulate the passage of macromolecules like proteins and RNA; since information is carried by the macromolecules, the envelope exerts some control over its flow
- nucleoskeleton (NSK)
The fibrillar substructure of the nuclear matrix, primarily composed of intermediate filaments that consist of lamin proteins; it supports signaling, chromatin remodeling, DNA replication, and mRNA synthesis, processing and transport
- paralogs
(Gk. para: “in parallel”) Genes [proteins] related by duplication within a genome and passed on side-by-side
- phenotype
The observable morphomechanical characteristics of a cell, tissue, organ, or organism, which are based on the genotype and environmental factors
- phenotypic plasticity
The capacity of a cell, tissue, organ, or organism, to change its morphomechanical state in response to “environmental” stimuli
- remodeling
The (mal)adaptive change of tissue and organ morphology due to external or internal stimuli
- sarcolemma
(sarco-, Gkpertaining to flesh; + lemma, Gk.: sheath) myocyte cell membrane
- shear stress
A stress state where the stress is acting parallel to a given surface, as opposed to a normal (compression or tension) stress where the stress is vertical to the surface. When viscous fluids (e.g., blood) flow along surfaces, they generate shear stresses “pulling” tangentially on them causing angular distortions
- strain
A description of deformation (extension, shortening, volume change, or angular distortion) in terms of relative displacement of particles in a body under stress, which excludes rigid-body motions
- stress
The force per unit area acting upon an object or material tending to cause it to change shape or volume
- SUN
One of two protein families (KASH/SUN domains) that are jointly responsible for holding together the two nuclear envelope membranes and for attaching them to the cytoskeleton and the nucleoskeleton. A branched F-actin cytoskeleton permeating the cytoplasm is linked to the nuclear envelope via the KASH/SUN integral membrane protein complexes and to the cell membrane via membrane-embedded integrin proteins
- TAZ (WW domain containing transcription regulator 1 or WWTR1)
Transcriptional coactivator and downstream effector of the Hippo pathway. Similarly to its paralog YAP, phosphorylated TAZ is sequestered in the cytoplasm and degraded; when dephosphorylated, TAZ/YAP translocates to the nucleus and interacts with other transcription factors to drive transcription, inducing gene expression
- tensegrity structures
Arrangements consisting of components in tension and compression in stable equilibrium. The application of forces to a tensegrity structure will deform it into a slightly different shape in a way that supports the applied forces. Buckminster Fuller coined the word “tensegrity” from the words “tension” and “integrity”
- tension (tensile stress)
The stress state leading to elongation in the tensile direction, while the volume stays constant; accordingly, the other 2 directions must decrease in size; i.e., a tension in one (axial) direction induces a compression in the other two (lateral) directions, and conversely. Tension is the opposite of compression
- torque
The tendency of a force to rotate an object about a pivot. A torque can be thought of as a twist to an object, which tends to produce rotation; e.g., pushing or pulling the handle of a wrench coupled to a nut or bolt produces a torque (turning force)
- traction (tractive force)
The force generating tangential motion of a surface, through viscous shear or dry friction exerted on the surface
- transdifferentiation
Conversion of one differentiated cell type into another. It is a kind of transformation called metaplasia (Gk. for “change in form”)
- vortex
This is the rotating motion of a multitude of fluid particles around a common center; the paths of the individual particles do not have to be circular, but may also be asymmetric. The RV/LV diastolic filling vortex is a toroidal vortex, a torus- or ring-shaped vortex, where blood spins around an imaginary axis line that forms a closed loop
- YAP (Yes-associated protein)
Transcriptional co-activator and downstream effector of the Hippo pathway. Similarly to its paralog TAZ, when phosphorylated YAP is isolated in the cytoplasm and degraded; when dephosphorylated, YAP/TAZ translocates to the nucleus and interacts with various transcription factors to induce gene expression
References
- 1.Pasipoularides A. Heart's Vortex: Intracardiac Blood Flow Phenomena. Shelton, CT: People's Medical Publishing House; 2010. p. 960. [Google Scholar]
- 2.Pasipoularides A. Diastolic filling vortex forces and cardiac adaptations: probing the epigenetic nexus. Hellenic J Cardiol. 2012;53:458–69. [PMC free article] [PubMed] [Google Scholar]
- 3.Pasipoularides A. Mechanotransduction mechanisms for intraventricular diastolic vortex forces and myocardial deformations: Part 1. J Cardiovasc Transl Res. 2015;8:76–87. doi: 10.1007/s12265-015-9611-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hines LM, Stampfer MJ, Ma J, et al. Genetic variation in alcohol dehydrogenase and the beneficial effect of moderate alcohol consumption on myocardial infarction. N Engl J Med. 2001;344:549–555. doi: 10.1056/NEJM200102223440802. [DOI] [PubMed] [Google Scholar]
- 5.Talmud PJ, Stephens JW, Hawe E, et al. The significant increase in cardiovascular disease risk in APOEepsilon4 carriers is evident only in men who smoke: potential relationship between reduced antioxidant status and ApoE4. Ann Hum Genet. 2005;69:613–622. doi: 10.1111/j.1529-8817.2005.00205.x. [DOI] [PubMed] [Google Scholar]
- 6.Brull DJ, Dhamrait S, Moulding R, et al. The effect of fibrinogen genotype on fibrinogen levels after strenuous physical exercise. Thromb Haemost. 2002;87:37–41. [PubMed] [Google Scholar]
- 7.Abu-Amero KK, Al-Boudari OM, Mohamed GH, Dzimiri N. T null and M null genotypes of the glutathione S-transferase gene are risk factor for CAD independent of smoking. BMC Med Genet. 2006;7:38. doi: 10.1186/1471-2350-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pimstone SN, Sun XM, du Souich C, Frohlich JJ, Hayden MR, Soutar AK. Phenotypic variation in heterozygous familial hypercholesterolemia: a comparison of Chinese patients with the same or similar mutations in the LDL receptor gene in China or Canada. Arterioscler Thromb Vasc Biol. 1998;18:309–315. doi: 10.1161/01.atv.18.2.309. [DOI] [PubMed] [Google Scholar]
- 9.Garrod A. The incidence of alkaptonuria: a study in chemical individuality. Lancet. 1902;2:1616–20. [PMC free article] [PubMed] [Google Scholar]
- 10.Qin Z, Buehler MJ, Kreplak L. A multi-scale approach to understand the mechanobiology of intermediate filaments. J Biomech. 2010;43:15–22. doi: 10.1016/j.jbiomech.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279(5350):514–9. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- 12.Calaghan SC, Le Guennec JY, White E. Cytoskeletal modulation of electrical and mechanical activity in cardiac myocytes. Prog Biophys Mol Biol. 2004;84:29–59. doi: 10.1016/s0079-6107(03)00057-9. [DOI] [PubMed] [Google Scholar]
- 13.Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313:2063–76. doi: 10.1016/j.yexcr.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 14.Deroanne CF, Lapiere CM, Nusgens BV. In vitro tubulogenesis of endothelial cells by relaxation of the coupling extracellular matrix-cytoskeleton. Cardiovasc Res. 2001;49:647–58. doi: 10.1016/s0008-6363(00)00233-9. [DOI] [PubMed] [Google Scholar]
- 15.Buss F, Kendrick-Jones J. Multifunctional myosin VI has a multitude of cargoes. Proc Natl Acad Sci U S A. 2011;108:5927–8. doi: 10.1073/pnas.1103086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol Biol Cell. 2012;23:1846–59. doi: 10.1091/mbc.E11-12-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mengistu M, Brotzman H, Ghadiali S, Lowe-Krentz L. Fluid shear stress-induced JNK activity leads to actin remodeling for cell alignment. J Cell Physiol. 2011;226:110–21. doi: 10.1002/jcp.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–44. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregorio CC, Antin PB. To the heart of myofibril assembly. Trends Cell Biol. 2000;10:355–62. doi: 10.1016/s0962-8924(00)01793-1. [DOI] [PubMed] [Google Scholar]
- 20.Zile M, Koide M, Sato H, et al. Role of microtubules in the contractile dysfunction of hypertrophied myocardium. J Am Coll Cardiol. 1999;33:250–60. doi: 10.1016/s0735-1097(98)00550-6. [DOI] [PubMed] [Google Scholar]
- 21.Tagawa H, Wang N, Narishige T, Ingber DE, Zile MR, Cooper Gt. Cytoskeletal mechanics in pressure overload cardiac hypertrophy. Circ Res. 1997;80:281–89. doi: 10.1161/01.res.80.2.281. [DOI] [PubMed] [Google Scholar]
- 22.Haggart CR, Ames EG, Lee JK, Holmes JW. Effects of stretch and shortening on gene expression in intact myocardium. Physiol Genomics. 2014;46:57–65. doi: 10.1152/physiolgenomics.00103.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003;116:1157–73. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Maxwell IZ, Heisterkamp A, et al. Viscoelastic retraction of single living stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanics. Biophys J. 2006;90:3762–73. doi: 10.1529/biophysj.105.071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trepat X, Deng L, An SS, et al. Universal physical responses to stretch in the living cell. Nature. 2007;447:592–595. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soares e Silva M, Depken M, Stuhrmann B, Korsten M, MacKintosh FC, Koenderink GH. Active multistage coarsening of actin networks driven by myosin motors. Proc Natl Acad Sci. 2011;108:9408–13. doi: 10.1073/pnas.1016616108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–80. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 28.Geiger B, Bershadsky A. Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell. 2002;110:139–42. doi: 10.1016/s0092-8674(02)00831-0. [DOI] [PubMed] [Google Scholar]
- 29.Sarantitis I, Papanastasopoulos P, Manousi M, Baikoussis NG, Apostolakis E. The cytoskeleton of the cardiac muscle cell. Hellenic J Cardiol. 2012;53:367–79. [PubMed] [Google Scholar]
- 30.Kostin S, Hein S, Arnon E, Scholz D, Schaper J. The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev. 2000;5:271–80. doi: 10.1023/A:1009813621103. [DOI] [PubMed] [Google Scholar]
- 31.Ervasti JM. Costameres: the Achilles' heel of Herculean muscle. J Biol Chem. 2003;278:13591–4. doi: 10.1074/jbc.R200021200. [DOI] [PubMed] [Google Scholar]
- 32.Samarel AM. Focal adhesion signaling in heart failure. Pflügers Arch - Eur J Physiol. 2014;466:1101–11. doi: 10.1007/s00424-014-1456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–35. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 34.Kim SA, Tai CY, Mok LP, Mosser EA, Schuman EM. Calcium dependent dynamics of cadherin interactions at cell-cell junctions. Proc Natl Acad Sci U S A. 2011;108:9857–62. doi: 10.1073/pnas.1019003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yap AS, Crampton MS, Hardin J. Making and breaking contacts: the cellular biology of cadherin regulation. Curr Opin Cell Biol. 2007;19:508–14. doi: 10.1016/j.ceb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Resink TJ, Philippova M, Joshi MB, Kyriakakis E, Erne P. Cadherins and cardiovascular disease. Swiss Med Wkly. 2009;139:122–34. doi: 10.4414/smw.2009.12429. [DOI] [PubMed] [Google Scholar]
- 37.Koenderink GH, Dogic Z, Nakamura F, et al. An active biopolymer network controlled by molecular motors. Proc Natl Acad Sci. 2009;106:15192–7. doi: 10.1073/pnas.0903974106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen Q, Janmey PA. Polymer physics of the cytoskeleton. Curr Opin Solid State Mater Sci. 2011;15:177–82. doi: 10.1016/j.cossms.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasipoularides A, Vlachos PP, Little WC. Vortex formation time is not an index of ventricular function. J Cardiovasc Transl Res. 2015;8:54–8. doi: 10.1007/s12265-015-9607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasipoularides A. Fluid dynamics of ventricular filling in heart failure: overlooked problems of RV/LV chamber dilatation. Hellenic J Cardiol. 2015;56:85–95. [PMC free article] [PubMed] [Google Scholar]
- 41.Yurchenco P. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3:a004911. doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sivakumar P, Gupta S, Sarkar S, Sen S. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol Cell Biochem. 2008;307:159–67. doi: 10.1007/s11010-007-9595-2. [DOI] [PubMed] [Google Scholar]
- 43.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–33. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tyagi SC, Joshua IG. Exercise and nutrition in myocardial matrix metabolism, remodeling, regeneration, epigenetics, microcirculation, and muscle. Can J Physiol Pharmacol. 2014;92:521–3. doi: 10.1139/cjpp-2014-0197. [DOI] [PubMed] [Google Scholar]
- 45.Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int J Cardiol. 2001;79:277–86. doi: 10.1016/s0167-5273(01)00449-1. [DOI] [PubMed] [Google Scholar]
- 46.Pasipoularides AD, Shu M, Womack MS, Shah A, Von Ramm O, Glower DD. RV functional imaging: 3-D echo-derived dynamic geometry and flow field simulations. Am J Physiol Heart Circ Physiol. 2003;284:H56–H65. doi: 10.1152/ajpheart.00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasipoularides A. Right and left ventricular diastolic pressure–volume relations: a comprehensive review. J Cardiovasc Transl Res. 2013;6:239–52. doi: 10.1007/s12265-012-9424-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasipoularides A. LV twisting-and-untwisting in HCM: ejection begets filling. Diastolic functional aspects of HCM. Am Heart J. 2011;162:798–810. doi: 10.1016/j.ahj.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muiznieks LD, Weiss AS, Keeley FW. Structural disorder and dynamics of elastin. Biochem Cell Biol. 2010;88:239–50. doi: 10.1139/o09-161. [DOI] [PubMed] [Google Scholar]
- 50.Castaldo C, Di Meglio F, Miraglia R, et al. Cardiac fibroblast-derived extracellular matrix (biomatrix) as a model for the studies of cardiac primitive cell biological properties in normal and pathological adult human heart. Biomed Res Int. 2013;2013:352370. doi: 10.1155/2013/352370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–76. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson SA, Blazeski A, Copeland CR, et al. Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through N-cadherin. J Mol Cell Cardiol. 2014;68:29–37. doi: 10.1016/j.yjmcc.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schéele S, Nyström A, Durbeej M, Talts JF, Ekblom M, Ekblom P. Laminin isoforms in development and disease. J Mol Med (Berl) 2007;85:825–36. doi: 10.1007/s00109-007-0182-5. [DOI] [PubMed] [Google Scholar]
- 54.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 55.Smith ML, Gourdon D, Little WC, et al. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 2007;5(10):e268. doi: 10.1371/journal.pbio.0050268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–9. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15–28. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang P, Valentijn AJ, Gilmore AP, Streuli CH. Early events in the anoikis program occur in the absence of caspase activation. J Biol Chem. 2003;278:19917–25. doi: 10.1074/jbc.M210337200. [DOI] [PubMed] [Google Scholar]
- 59.Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002;90:1282–9. doi: 10.1161/01.res.0000023201.41774.ea. [DOI] [PubMed] [Google Scholar]
- 60.Provenzano PP, Keely PJ. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci. 2011;124:1195–205. doi: 10.1242/jcs.067009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009;122(Pt 2):159–63. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhat R, Bissell MJ. Of plasticity and specificity: dialectics of the microenvironment and macroenvironment and the organ phenotype. WIREsDev Biol. 2013 doi: 10.1002/wdev.130. [DOI] [PubMed] [Google Scholar]
- 63.Tamkun JW, DeSimone DW, Fonda D, et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell. 1986;46:271–82. doi: 10.1016/0092-8674(86)90744-0. [DOI] [PubMed] [Google Scholar]
- 64.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 67.Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeletal linkages. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 68.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215.1–215.9. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–23. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng X, Kraus MS, Wei H, et al. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–27. doi: 10.1172/JCI24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng X, Wu X, Druso JE, et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci U S A. 2008;105:6638–43. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397–418. doi: 10.1101/gad.1758709. [DOI] [PubMed] [Google Scholar]
- 73.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–70. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 74.Brakemeier S, Eichler I, Hopp H, Kohler R, Hoyer J. Up-regulation of endothelial stretch-activated cation channels by fluid shear stress. Cardiovasc Res. 2002;53:209–18. doi: 10.1016/s0008-6363(01)00476-x. [DOI] [PubMed] [Google Scholar]
- 75.Bray MA, Sheehy SP, Parker KK. Sarcomere alignment is regulated by myocyte shape. Cell Motil Cytoskeleton. 2008;65:641–51. doi: 10.1002/cm.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salameh A, Dhein S. Effects of mechanical forces and stretch on intercellular gap junction coupling. Biochim Biophys Acta. 2013;1828:147–56. doi: 10.1016/j.bbamem.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 77.Yefimov S, van der Giessen E, Onck PR, Marrink SJ. Mechanosensitive membrane channels in action. Biophys J. 2008;94:2994–3002. doi: 10.1529/biophysj.107.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gervasio OL, Phillips WD, Cole L, Allen DG. Caveolae respond to cell stretch and contribute to stretch-induced signaling. J Cell Sci. 2011;124:3581–90. doi: 10.1242/jcs.084376. [DOI] [PubMed] [Google Scholar]
- 79.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am J Physiol Heart Circ Physiol. 2003;285:H1720–9. doi: 10.1152/ajpheart.00344.2002. [DOI] [PubMed] [Google Scholar]
- 80.Parton RG, del Pozo M. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 81.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tamada M, Sheetz MP, Sawada Y. Activation of a signaling cascade by cytoskeleton stretch. Dev Cell. 2004;7:709–18. doi: 10.1016/j.devcel.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 83.Roca-Cusachs p, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc Natl Acad Sci USA. 2013;110:E1361–E1370. doi: 10.1073/pnas.1220723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vogel V. Mechanotransduction involving multimodular proteins: converting force into biochemical signals. Annu Rev Biophys Biomol Struct. 2006;35:459–88. doi: 10.1146/annurev.biophys.35.040405.102013. [DOI] [PubMed] [Google Scholar]
- 85.Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265–75. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 86.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–7. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 87.Shapiro L, Fannon AM, Kwong PD, et al. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–37. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 88.Kim SA, Tai CY, Mok LP, Mosser EA, Schuman EM. Calcium-dependent dynamics of cadherin interactions at cell-cell junctions. Proc Natl Acad Sci. 2011;108:9857–62. doi: 10.1073/pnas.1019003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Calderwood DA. Integrin activation. J Cell Sci. 2004;117:657–66. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- 90.Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. 2006;259:339–50. doi: 10.1111/j.1365-2796.2006.01620.x. [DOI] [PubMed] [Google Scholar]
- 91.Critchley DR. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem Soc Trans. 2004;32:831–6. doi: 10.1042/BST0320831. [DOI] [PubMed] [Google Scholar]
- 92.Giannone G, Mège RM, Thoumine O. Multi-level molecular clutches in motile cell processes. Trends Cell Biol. 2009;19:475–86. doi: 10.1016/j.tcb.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 93.Liu Z, Tan JL, Cohen DM, et al. Mechanical tugging force regulates the size of cell—cell junctions. Proc Natl Acad Sci USA. 2010;107:9944–9. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. 2007;8:957–69. doi: 10.1038/nrm2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exper Cell Res. 2000;261:25–36. doi: 10.1006/excr.2000.5043. [DOI] [PubMed] [Google Scholar]
- 96.Martinez-Rico C, Pincet F, Thiery JP, Dufour S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J Cell Sci. 2010;123:712–22. doi: 10.1242/jcs.047878. [DOI] [PubMed] [Google Scholar]
- 97.Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 289:H2291–301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- 98.McCain ML, Parker KK. Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pfluegers Arch - Eur J Physiol. 2011;462:89–104. doi: 10.1007/s00424-011-0951-4. [DOI] [PubMed] [Google Scholar]
- 99.Chopra A, Tabdanov E, Patel H, Janmey PA, Kresh JY. Cardiac myocyte remodeling mediated by N-cadherin- dependent mechanosensing. Am J Physiol Heart Circ Physiol. 2011;300:H1252–66. doi: 10.1152/ajpheart.00515.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson AJ, Schoenauer R, Ehler E, Agarkova I, Bennett PM. Cardiomyocyte growth and sarcomerogenesis at the intercalated disc. Cell Mol Life Sci. 2014;71:165–81. doi: 10.1007/s00018-013-1374-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perriard JC, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc Med. 2003;13:30–8. doi: 10.1016/s1050-1738(02)00209-8. [DOI] [PubMed] [Google Scholar]
- 102.Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–79. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 103.Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 104.Kull FJ, Endow SA. Force generation by kinesin and myosin cytoskeletal motor proteins. J Cell Sci. 2013;126:9–19. doi: 10.1242/jcs.103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kastritis PL, Bonvin AM. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface. 2012;10:20120835. doi: 10.1098/rsif.2012.0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gustavsson M, Traaseth NJ, CB Karim CB, et al. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca2+-ATPase. J Mol Biol. 2011;408:755–65. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–77. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 108.Hu S, Chen J, Butler JP, Wang N. Prestress mediates force propagation into the nucleus. Biochem Biophys Res Commun. 2005;329:423–8. doi: 10.1016/j.bbrc.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 109.Na S, Collin O, Chowdhury F, et al. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc Natl Acad Sci USA. 2008;105:6626–31. doi: 10.1073/pnas.0711704105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 111.Pasipoularides A. Complementarity and competitiveness of the intrinsic and extrinsic components of the total ventricular load: Demonstration after valve replacement in aortic stenosis Editorial. Am Heart J. 2007;153:4–6. doi: 10.1016/j.ahj.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ingber DE. Tensegrity-based mechanosensing from macro to micro. Prog Biophys Mol Biol. 2008;97:163–79. doi: 10.1016/j.pbiomolbio.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 114.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–95. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 115.Dorn GW, II, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ Res. 2003;92:1171–5. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 116.Blaauw E, Lorenzen-Schmidt I, Babiker FA, et al. Stretch-induced upregulation of connective tissue growth factor in rabbit cardiomyocytes. J Cardiovasc Transl Res. 2013;6:861–9. doi: 10.1007/s12265-013-9489-5. [DOI] [PubMed] [Google Scholar]
- 117.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–7. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 118.Dahl KN, Kalinowski A. Nucleoskeleton mechanics at a glance. J Cell Sci. 2011;124:675–8. doi: 10.1242/jcs.069096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Webster M, Witkin KL, Cohen-Fix O. Sizing up the nucleus: nuclear shape, size and nuclear-envelope assembly. J Cell Sci. 2009;122:1477–86. doi: 10.1242/jcs.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Starr DA, Fridolfsson HN. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu Rev Cell Dev Biol. 2010;26:421–44. doi: 10.1146/annurev-cellbio-100109-104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Janmey PA, Wells RG, Assoian RK, McCulloch CA. From tissue mechanics to transcription factors. Differentiation. 2013;86:112–20. doi: 10.1016/j.diff.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stroud MJ, Banerjee I, Veevers J, Chen J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res. 2014;114:538–48. doi: 10.1161/CIRCRESAHA.114.301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simon DN, Wilson KL. The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat Rev Mol Cell Biol. 2011;12:695–708. doi: 10.1038/nrm3207. [DOI] [PubMed] [Google Scholar]
- 124.Tzur YB, Wilson KL, Gruenbaum Y. SUN-domain proteins: ‘Velcro’ that links the nucleoskeleton to the cytoskeleton. Nat Rev Mol Cell Biol. 2006;7:782–8. doi: 10.1038/nrm2003. [DOI] [PubMed] [Google Scholar]
- 125.Dahl KN, Ribeiro AJ, Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. 2008;102:1307–18. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nickerson J. Experimental observations of a nuclear matrix. J Cell Sci. 2001;114:463–74. doi: 10.1242/jcs.114.3.463. [DOI] [PubMed] [Google Scholar]
- 127.Orr AW, Helmke BP, Blackman BR, Schwartz MA. Mechanisms of mechanotransduction. Dev Cell. 2006;10:11–20. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 128.De R, Zemel A, Safran SA. Do cells sense stress or strain? Measurement of cellular orientation can provide a clue. Biophys J. 2008;94:L29–31. doi: 10.1529/biophysj.107.126060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevates endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation. 1993;88:193–7. doi: 10.1161/01.cir.88.1.193. [DOI] [PubMed] [Google Scholar]
- 130.Dzau VJ. Transcription factor decoy. Circ Res. 2002;90:1234–6. doi: 10.1161/01.res.0000025209.24283.73. [DOI] [PubMed] [Google Scholar]
- 131.Georgatos SD. Towards an understanding of nuclear morphogenesis. J Cell Biochem. 1994;55:69–76. doi: 10.1002/jcb.240550108. [DOI] [PubMed] [Google Scholar]
- 132.Zlatanova J, Leuba SH. Stretching and imaging single DNA molecules and chromatin. J Muscle Res Cell Motil. 2002;23:377–95. doi: 10.1023/a:1023498120458. [DOI] [PubMed] [Google Scholar]
- 133.Marko JF, Poirier MG. Micromechanics of chromatin and chromosomes. Biochem Cell Biol. 2003;81:209–20. doi: 10.1139/o03-047. [DOI] [PubMed] [Google Scholar]
- 134.Trinkle-Mulcahy L, Lamond AI. Toward a high-resolution view of nuclear dynamics. Science. 2007;318:1402–7. doi: 10.1126/science.1142033. [DOI] [PubMed] [Google Scholar]
- 135.Knudsen HL, Frangos JA. Role of cytoskeleton in shear stress-induced endothelial nitric oxide production. Am J Physiol Heart Circ Physiol. 1997;273:H347–55. doi: 10.1152/ajpheart.1997.273.1.H347. [DOI] [PubMed] [Google Scholar]
- 136.Visa N. Actin in transcription. EMBO reports. 2005;6:218–9. doi: 10.1038/sj.embor.7400362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bettinger BT, Gilbert DM, Amberg DC. Actin up in the nucleus. Nat Rev Mol Cell Biol. 2004;5:410–5. doi: 10.1038/nrm1370. [DOI] [PubMed] [Google Scholar]
- 138.Olave IA, Reck-Peterson SL, Crabtree GR. Nuclear actin and actin-related proteins in chromatin remodeling. Annu Rev Biochem. 2002;71:755–81. doi: 10.1146/annurev.biochem.71.110601.135507. [DOI] [PubMed] [Google Scholar]
- 139.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Helmke BP. Molecular control of cytoskeletal mechanics by hemodynamic forces. Physiology. 2005;20:43–53. doi: 10.1152/physiol.00040.2004. [DOI] [PubMed] [Google Scholar]
- 141.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–71. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 143.Bellin RM, Kubicek JD, Frigault MJ, et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc Natl Acad Sci. 2009;106:22102–7. doi: 10.1073/pnas.0902639106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li C, Hu Y, Mayr M, Xu Q. Cyclic strain stress-induced mitogen-activated protein kinase (MAPK) phosphatase 1 expression in vascular smooth muscle cells is regulated by Ras/Rac-MAPK pathways. J Biol Chem. 1999;274:25273–80. doi: 10.1074/jbc.274.36.25273. [DOI] [PubMed] [Google Scholar]
- 145.Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of cJun kinases. Nature. 1994;369:156–60. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 146.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–6. [PubMed] [Google Scholar]
- 147.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–24. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 148.Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–31. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 149.Faust U, Hampe N, Rubner W, et al. Cyclic stress at mHz frequencies aligns fibroblasts in direction of zero strain. PLoS ONE. 2011;6(12):e28963. doi: 10.1371/journal.pone.0028963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De R, Zemel A, Safran SA. Dynamics of cell orientation. Nat Phys. 2007;3:655–9. [Google Scholar]
- 151.Waddington CH. An introduction to modern genetics. New York: Macmillan; 1939. [Google Scholar]
- 152.Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006;27:86–94. doi: 10.1152/physiolgenomics.00028.2006. [DOI] [PubMed] [Google Scholar]
- 153.Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–41. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 154.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–72. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- 155.Pasipoularides AD, Shu M, Shah A, Glower DD. Right ventricular diastolic relaxation in conscious dog models of pressure overload, volume overload and ischemia. J Thorac Cardiovasc Surg. 2002;124:964–72. doi: 10.1067/mtc.2002.126677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pasipoularides AD, Shu M, Shah A, Silvestry S, Glower DD. Right ventricular diastolic function in canine models of pressure overload, volume overload and ischemia. Am J Physiol Heart Circ Physiol. 2002;283:H2140–50. doi: 10.1152/ajpheart.00462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mirsky I, Pasipoularides A. Clinical assessment of diastolic function. Prog Cardiovas Dis. 1990;32:291–318. doi: 10.1016/0033-0620(90)90018-w. [DOI] [PubMed] [Google Scholar]
- 158.Pasipoularides A, Mirsky I, Hess OM, Grimm J, Krayenbuehl JP. Muscle relaxation and passive diastolic properties in man. Circulation. 1986;74:991–1001. doi: 10.1161/01.cir.74.5.991. [DOI] [PubMed] [Google Scholar]
- 159.Gelpi RJ, Pasipoularides A, Lader AS, et al. Changes in diastolic cardiac function in developing and stable perinephritic hypertension in conscious dogs. Circ Res. 1991;68:555–67. doi: 10.1161/01.res.68.2.555. [DOI] [PubMed] [Google Scholar]
- 160.Mirsky I, Pasipoularides A. Elastic properties of normal and hypertrophied cardiac muscle. Fed Proc. 1980;39:156–61. [PubMed] [Google Scholar]
- 161.Colombelli J, Besser A, Kress H, et al. Mechanosensing in actin stress fibers revealed by a close correlation between force and protein localization. J Cell Sci. 2009;122:1665–79. doi: 10.1242/jcs.042986. [DOI] [PubMed] [Google Scholar]
- 162.Wille JJ, Elson EL, Okamoto RJ. Cellular and matrix mechanics of bioartificial tissues during continuous cyclic stretch. Ann Biomed Eng. 2006;34:1678–90. doi: 10.1007/s10439-006-9153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Balestrini JL, Billiar KL. Equibiaxial cyclic stretch stimulates fibroblasts to rapidly remodel fibrin. J Biomech. 2006;39:2983–90. doi: 10.1016/j.jbiomech.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 164.Hirata H, Sokabe M, Lim CT. Molecular mechanisms underlying the force-dependent regulation of actin-to-ECM linkage at the focal adhesions. Prog Mol Biol Transl Sci. 2014;126:135–54. doi: 10.1016/B978-0-12-394624-9.00006-3. [DOI] [PubMed] [Google Scholar]
- 165.Grashoff C, Hoffman BD, Brenner MD, et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–6. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Riveline D, Zamir E, Balaban NQ, et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–86. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kuo JC. Focal adhesions function as a mechanosensor. Prog Mol Biol Transl Sci. 2014;126:55–73. doi: 10.1016/B978-0-12-394624-9.00003-8. [DOI] [PubMed] [Google Scholar]
- 169.Haase K, Al-Rekabi Z, Pelling AE. Mechanical cues direct focal adhesion dynamics. Prog Mol Biol Transl Sci. 2014;126:103–34. doi: 10.1016/B978-0-12-394624-9.00005-1. [DOI] [PubMed] [Google Scholar]
- 170.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: The tugof-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–33. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 171.Pasipoularides A, Shu M, Shah A, Womack MS, Glower DD. Diastolic right ventricular filling vortex in normal and volume overload states. Am J Physiol Heart Circ Physiol. 2003;284:H1064–72. doi: 10.1152/ajpheart.00804.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pasipoularides A, Shu M, Shah A, Tucconi A, Glower DD. RV instantaneous intraventricular diastolic pressure and velocity distributions in normal and volume overload awake dog disease models. Am J Physiol Heart Circ Physiol. 2003;285:H1956–65. doi: 10.1152/ajpheart.00372.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pasipoularides A. Analysis of vortex flow imaging in normal and dysfunctional RV's. American Society of Echocardiography 22nd Annual Scientific Sessions; Montreal. 2011; EE02d – Flow Vortex Imaging; PROLibraries.com. [Google Scholar]
- 174.Pasipoularides A. Evaluation of right and left ventricular diastolic filling. J Cardiovasc Transl Res. 2013;6:623–39. doi: 10.1007/s12265-013-9461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pasipoularides A. Invited commentary: Functional Imaging (FI) combines imaging datasets and computational fluid dynamics to simulate cardiac flows. J Appl Physiol. 2008;105:1015. doi: 10.1152/japplphysiol.zdg-8134-vpcomm.2008. [DOI] [PubMed] [Google Scholar]
- 176.Dodge HT, Hay RE, Sandler H. Pressure-volume characteristics of the diastolic left ventricle of man with heart disease. Am Heart J. 1962;64:503–11. doi: 10.1016/0002-8703(62)90036-4. [DOI] [PubMed] [Google Scholar]
- 177.Ross J, Jr, Sonnenblick EH, Taylor RR, Covell JW. Diastolic geometry and sarcomere length in the chronically dilated canine left ventricle. Circ Res. 1971;28:49–61. doi: 10.1161/01.res.28.1.49. [DOI] [PubMed] [Google Scholar]
- 178.Paulus WJ, Grossman W, Serizawa T, Bourdillon PD, Pasipoularides A, Mirsky I. Different effects of two types of ischemia on myocardial systolic and diastolic function. Am J Physiol Heart Circ Physiol. 1985;248:H719–28. doi: 10.1152/ajpheart.1985.248.5.H719. [DOI] [PubMed] [Google Scholar]
- 179.Glower DD, Schaper J, Kabas JS, et al. Relation between reversal of diastolic creep and recovery of systolic function after ischemic myocardial injury in conscious dogs. Circ Res. 1987;60:850–60. doi: 10.1161/01.res.60.6.850. [DOI] [PubMed] [Google Scholar]
- 180.Komamura K, Shannon RP, Pasipoularides A, et al. Alterations in left ventricular diastolic function in conscious dogs with pacing-induced heart failure. J Clin Invest. 1992;89:1825–38. doi: 10.1172/JCI115787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 182.Low BC, Pan CQ, Shivashankar CV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Let. 2014;588:2663–70. doi: 10.1016/j.febslet.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 183.Reddy P, Deguchi M, Cheng Y, Hsueh AJW. Actin Cytoskeleton Regulates Hippo Signaling. PLoS ONE. 2013;8(9):e73763. doi: 10.1371/journal.pone.0073763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–93. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pan D. The hippo signaling pathway in development and cancer. Developmental Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li Z, Zhao B, Wang P, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24:235–40. doi: 10.1101/gad.1865810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Heallen T, Zhang M, Wang J, et al. Hippo Pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 189.Hall JL, Terzic A. Heart failure transcriptome: when discoveries change practice? Circ Cardiovasc Genet. 2011;4:469–71. doi: 10.1161/CIRCGENETICS.111.961367. [DOI] [PubMed] [Google Scholar]
- 190.Stastna M, Abraham MR, Van Eyk JE. Cardiac stem/progenitor cells, secreted proteins, and proteomics. FEBS Lett. 2009;583:1800–7. doi: 10.1016/j.febslet.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pagliari S, Vilela-Silva AC, Forte G, et al. Cooperation of biological and mechanical signals in cardiac progenitor cell differentiation. Adv Mater. 2011;23:514–8. doi: 10.1002/adma.201003479. [DOI] [PubMed] [Google Scholar]
- 193.van der Bogt KE, Sheikh AY, Schrepfer S, et al. Comparison of different adult stem cell types for treatment of myocardial ischemia. Circulation. 2008;118:S121–9. doi: 10.1161/CIRCULATIONAHA.107.759480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 195.Varelas X, Miller BW, Sopko R, et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell. 2010;18:579–91. doi: 10.1016/j.devcel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 196.Zhang J, Friedman MH. Adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am J Physiol Heart Circ Physiol. 2012;302:H983–91. doi: 10.1152/ajpheart.00168.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38:1949–71. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 198.Garcia-Cardena G, Comander J, Anderson KR, Blackman BR, Gimbrone MA., Jr Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci. 2001;98:4478–85. doi: 10.1073/pnas.071052598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Silberman M, Barac YD, Yahav H, et al. Shear stress-induced transcriptional regulation via hybrid promoters as a potential tool for promoting angiogenesis. Angiogenesis. 2009;12:231–42. doi: 10.1007/s10456-009-9143-7. [DOI] [PubMed] [Google Scholar]
- 200.Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–36. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 201.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–46. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 202.Barnes J, Pat B, Chen YW, et al. Whole-genome profiling highlights the molecular complexity underlying eccentric cardiac hypertrophy. Ther Adv Cardiovasc Dis. 2014;8:97–118. doi: 10.1177/1753944714527490. [DOI] [PubMed] [Google Scholar]
- 203.Nygård S, Reitan T, Clancy T, et al. Identifying pathogenic processes by integrating microarray data with prior knowledge. BMC Bioinformatics. 2014 Apr 24;15:115. doi: 10.1186/1471-2105-15-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rowell J, Koitabashi N, Kass DA, Barth AS. Dynamic gene expression patterns in animal models of early and late heart failure reveal biphasic-bidirectional transcriptional activation of signaling pathways. Physiol Genomics. 2014;46:779–87. doi: 10.1152/physiolgenomics.00054.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nattel S, Frelin Y, Gaborit N, Louault C, Demolombe S. Ion-channel mRNA-expression profiling: Insights into cardiac remodeling and arrhythmic substrates. J Mol Cell Cardiol. 2010;48:96–105. doi: 10.1016/j.yjmcc.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 206.Horn S, Lueking A, Murphy D, et al. Profiling humoral autoimmune repertoire of dilated cardiomyopathy (DCM) patients and development of a disease-associated protein chip. Proteomics. 2006;6:605–13. doi: 10.1002/pmic.200401293. [DOI] [PubMed] [Google Scholar]
- 207.Anisimov SV. Serial Analysis of Gene Expression (SAGE): 13 years of application in research. Curr Pharm Biotechnol. 2008;9:338–50. doi: 10.2174/138920108785915148. [DOI] [PubMed] [Google Scholar]
- 208.Tarasov KV, Brugh SA, Tarasova YS, Boheler KR. Serial analysis of gene expression (SAGE): a useful tool to analyze the cardiac transcriptome. Methods Mol Biol. 2007;366:41–59. doi: 10.1007/978-1-59745-030-0_3. [DOI] [PubMed] [Google Scholar]
- 209.Patino WD, Mian OY, Hwang PM. Serial analysis of gene expression: technical considerations and applications to cardiovascular biology. Circ Res. 2002;91:565–9. doi: 10.1161/01.res.0000036018.76903.18. [DOI] [PubMed] [Google Scholar]
- 210.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–7. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 211.Liew CC, Dzau VJ. Molecular genetics and genomics of heart failure. Nat Rev Genet. 2004;5:811–25. doi: 10.1038/nrg1470. [DOI] [PubMed] [Google Scholar]
- 212.Haqqani AS, Kelly JF, Stanimirovic DB. Quantitative protein profiling by mass spectrometry using label-free proteomics. Methods Mol Biol. 2008;439:241–56. doi: 10.1007/978-1-59745-188-8_17. [DOI] [PubMed] [Google Scholar]
- 213.Hall JL, Grindle S, Han X, et al. Genomic profiling of the human heart before and after mechanical support with a ventricular assist device reveals alterations in vascular signaling networks. Physiol Genomics. 2004;17:283–91. doi: 10.1152/physiolgenomics.00004.2004. [DOI] [PubMed] [Google Scholar]
- 214.Hall JL, Birks EJ, Grindle S, et al. Molecular signature of recovery following combination left ventricular assist device (LVAD) support and pharmacologic therapy. Eur Heart J. 2007;28:613–27. doi: 10.1093/eurheartj/ehl365. [DOI] [PubMed] [Google Scholar]
- 215.Metzker ML. Sequencing technologies–the next generation. Nature Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 216.Anandhakumar C, Kizaki S, Bando T, Pandian GN, Sugiyama H. Advancing small-molecule-based chemical biology with next-generation sequencing technologies. Chembiochem. 2015;16:20–38. doi: 10.1002/cbic.201402556. [DOI] [PubMed] [Google Scholar]
- 217.Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Research. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li B, Fillmore N, Bai Y, et al. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biology. 2014;15:553. doi: 10.1186/s13059-014-0553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gut IG. New sequencing technologies. Clin Transl Oncol. 2013;15:879–81. doi: 10.1007/s12094-013-1073-6. [DOI] [PubMed] [Google Scholar]
- 220.Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila) 2012;5:887–900. doi: 10.1158/1940-6207.CAPR-11-0432. [DOI] [PubMed] [Google Scholar]
- 221.Marian AJ. Challenges in medical applications of whole exome/genome sequencing discoveries. Trends Cardiovasc Med. 2012;22:219–23. doi: 10.1016/j.tcm.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Barbato E, Lara-Pezzi E, Stolen C, et al. Advances in induced pluripotent stem cells, genomics, biomarkers, and antiplatelet therapy highlights of the year in JCTR 2013. J Cardiovasc Transl Res. 2014;7:518–25. doi: 10.1007/s12265-014-9555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mitchell A, Guan W, Staggs R, et al. Identification of differentially expressed transcripts and pathways in blood one week and six months following implant of left ventricular assist devices. PLoS One. 2013;8(10):e77951. doi: 10.1371/journal.pone.0077951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–73. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 225.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–32. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Schwanhausser B, Busse D, Li N, Dittmar G, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 227.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ferrai C, de Castro IJ, Lavitas L, Chotalia M, Pombo A. Gene positioning. Cold Spring Harbor perspectives in biology. 2010;2:a000588. doi: 10.1101/cshperspect.a000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wong IY, Bhatia SN, Toner M. Nanotechnology: emerging tools for biology and medicine. Genes Dev. 2013;27:2397–408. doi: 10.1101/gad.226837.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Maher B. ENCODE: The human encyclopaedia. Nature. 2012;489:46–8. doi: 10.1038/489046a. [DOI] [PubMed] [Google Scholar]
- 231.The ENCODE Project Consortium. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]