

Abstract
Neuron identity transformations occur upon removal of specific regulatory factors in many different cellular contexts, thereby revealing the fundamental principle of alternative cell identity choices made during nervous system development. One common molecular interpretation of such homeotic cell identity transformations is that a regulatory factor has a dual function in activating genes defining one cellular identity, and repressing genes that define an alternative identity. We provide here evidence for an alternative, competition-based mechanism. We show that the MEC-3 LIM homeodomain protein can outcompete the execution of a neuropeptidergic differentiation program by direct interaction with the UNC-86/Brn3 POU homeodomain protein. MEC-3 thereby prevents UNC-86 from collaborating with the Zn finger transcription factor PAG-3/Gfi to induce peptidergic neuron identity and directs UNC-86 to induce an alternative differentiation program toward a glutamatergic neuronal identity. Homeotic control of neuronal identity programs has implications for the evolution of neuronal cell types.
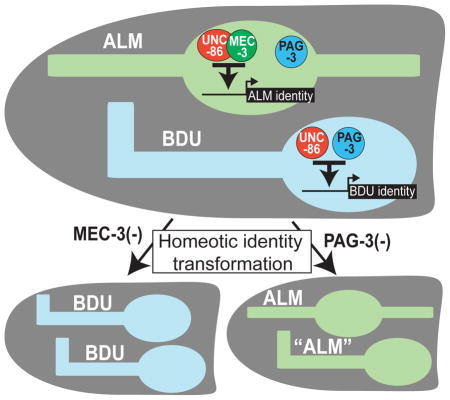
Graphical abstract
INTRODUCTION
In 1894, Bateson introduced the term homeosis to describe transformations of identities of homologous characters in a repeated series of animal characters (e.g. vertebrae). He observed these transformations as naturally occurring variants within many different species (Bateson, 1894). Homeotic transformations are not limited to segmented structures but can refer to different levels of organization, generally describing any transformation of one part of an organism into another (Sattler, 1988). In addition to whole tissues or organs, the homeosis concept has been applied to the level of single cells. For example, many classic lineage mutants in the nematode Caenorhabditis elegans, cause cellular identity transformations that have been described as homeotic (Sternberg and Horvitz, 1984). Photoreceptor identity transformations in the Drosophila retina, observed upon removal of the sevenless gene, have also been characterized as homeotic transformations (Tomlinson and Ready, 1986).
A variety of studies have shown that loss of expression or ectopic expression of a regulatory factor can bring about cell identity switches in the nervous system that are essentially homeotic in nature. For example, in mouse striatal interneurons, the LIM homeobox gene Lhx7 promotes cholinergic fate; loss of Lhx7 causes those neurons to instead adopt GABAergic fate (Lopes et al., 2012). In the dorsal horn of the spinal cord, Lbx1 selects GABAergic cell fate over glutamatergic cell fate (Cheng et al., 2005), while in the mesencephalon, Helt induces GABAergic fate while repressing glutamatergic fate (Nakatani et al., 2007). Distinct cortical neuron types in different cortical layers switch their identity upon removal of different types of TFs (Srinivasan et al., 2012).
The mechanistic basis of transformations in cell identity is often not clear. In principle, a transcription factor (TF) can simultaneously operate as an activator for some targets and a repressor of other target genes. In such cases, genetic removal of the TF results in failure to activate gene batteries that define one cellular state and a derepression of gene batteries that define an alternative state. Indeed, it has been shown that in the context of neocortical projection neurons, Fezf2 can activate genes that define the glutamatergic phenotype while directly repressing genes that define the GABAergic phenotype (Lodato et al., 2014). Cross-repressive interactions between TF inducers of specific identity programs have also been observed outside the nervous system, for example in the immune system (Graf and Enver, 2009).
In this paper we describe a principle that underlies a homeotic neuronal identity transformation in the nervous system of the nematode C. elegans, involving the ALM and BDU sister neuron pairs (Fig. 1A,B). The axonal projection patterns and synaptic connectivity patterns of the BDU and ALM are distinct (Fig. 1A)(White et al., 1986). Moerover, the ALM neurons contain specialized microtubules required for the light touch receptor function (Chalfie et al., 1985), while BDU neurons do not show any specific morphological features that would suggest a sensory neuron function; nevertheless, recent cell ablation studies have demonstrated that the BDU neurons are involved in a harsh touch response to the anterior half of the animals (Li et al., 2011). Whether the BDU neurons are themselves mechanoreceptors or act downstream of a mechanosensory neuron is presently not clear. Apart from morphology, there are also notable differences in the connectivity and neurotransmitter choice of the ALM and BDU neurons. The ALM neurons are glutamatergic (Lee et al., 1999). In contrast, unlike most C. elegans neurons, all of the synaptic outputs of the BDU neurons contain striking, darkly staining vesicles, suggesting that the BDU neurons make prominent use of neuropeptides (White et al., 1986). Indeed, five neuropeptide-encoding genes, producing at least 11 different neuropeptides are known to be expressed in BDU (Kim and Li, 2004; Li and Kim, 2010; Nathoo et al., 2001)(Fig. 1C). Moreover, a systematic mapping of neurotransmitter systems suggests that BDU may not use any classic, fast-acting neurotransmitter system, such as acetylcholine, glutamate, GABA or monoamines (Duerr et al., 2001; McIntire et al., 1993; Serrano-Saiz et al., 2013)(our unpubl. data). This indicates that the BDU neuron class may be akin to other neurons in vertebrate and invertebrate nervous systems which exclusively utilize neuropeptides for communication with downstream target neurons, such as vertebrate oxytocin/vasopressin-expressing magnocellular neurons (Salio et al., 2006). How peptidergic neurotransmitter identity of a neuron is controlled and coupled to other identity features of a neuron is not well understood.
Fig. 1. Control of BDU neuron identity.

A: Schematic drawing of ALM and BDU morphology.
B: Timing of TF expression. See Fig. S1B for expression data and cross-regulation of TFs.
C: Lineage & terminal markers of ALM and BDU. ‘+’ = expressed, ‘−’ = not expressed.
D: Responses to harsh touch (see Experimental Procedures). n is given at the bottom of each bar. Error bars expressed as s.e.m. **p<.001 ***p<.0001 (t-test with Bonferroni correction). See Fig. S1A for additional mutant analysis, rescue experiments and further controls.
E: The effect of unc-86, pag-3 and ceh-14 on BDU identity. n≥50 for each reporter. Significance is indicated in relation to wild type tested with Fischer’s Exact Test. ***:p≤.0001, *:p≤.05. See Supplementary Material for information on reporter transgenes.
Previous work has identified two TFs, the unc-86 POU homeobox and the mec-3 LIM homeobox genes, as critical inducers of ALM identity (Duggan et al., 1998; Way and Chalfie, 1988, 1989; Xue et al., 1992; Xue et al., 1993; Zhang et al., 2002), The C. elegans Senseles/Gfi ortholog pag-3, represses aberrant expression of two ALM marker genes in BDU (Jia et al., 1997), but how BDU identity is established is not known. We examine here how BDU neuropeptidergic identity is controlled and how the BDU differentiation program relates to the adoption of ALM glutamatergic identity. We show that BDU and ALM display homeotic transformations in mec-3 and pag-3 mutant backgrounds. We define a mechanism in which mec-3 is able to both induce ALM fate and repress BDU fate that involves competition for access to UNC-86 which is required for both ALM and BDU differentiation.
RESULTS
The ceh-14 LIM homeobox gene controls the peptidergic identity of the BDU neurons
While numerous studies have elucidated the mechanisms that genetically control classic neurotransmitter identity (Hobert, 2011), little is known about the genetic control and coordination of peptidergic neuron identity features. We first corroborated the importance of the neuropeptidergic features of the BDU neurons by finding that at least two of the BDU-expressed neuropeptide genes (flp-10 and nlp-1) are required for the previously described harsh touch sensory functions of the BDU neurons (Fig. 1D; Fig. S1A). Using fosmid-based reporter constructs, we then corroborated the previously noted expression of three TFs in BDU, the unc-86 POU homeobox gene (Finney and Ruvkun, 1990), the pag-3 gene, an ortholog of the Senseless/Gfi Zn finger TF (Jia et al., 1997) and the ceh-14 LIM homeobox gene, the ortholog of vertebrate Lhx3/4 (Cassata et al., 2000)(Fig. 1B; Fig. S1B). The unc-86 gene is turned on in the mother of the BDU neuron (Finney and Ruvkun, 1990), while ceh-14 and pag-3 start to be expressed after the birth of the BDU neurons (Fig. S1B, summarized in Fig. 1B). Each TF is expressed throughout the life of the BDU neurons (Fig. S1B). Consistent with its earlier onset of expression, unc-86 is required for expression of pag-3 and ceh-14 (Fig. S1B).
The availability of eight molecular markers of terminal BDU identity (listed in Fig. 1C) allowed us to probe the effect of BDU-expressed TFs on terminal BDU identity. We found that ceh-14 null mutant animals lose the expression of only a subset of the BDU identity markers, including of all three neuropeptide genes examined, flp-10, nlp-1 and nlp-15 (Fig. 1E). As expected from the loss of nlp-1 expression, we found that ceh-14 mutants display harsh touch response defects (Fig. 1D). These defects are not further enhanced by removal of either nlp-1 or a gene, egl-3, that is generally required for neuropeptide processing (Fig. 1D), indicating that the defects in ceh-14 mutants can be ascribed to their loss of neuropeptide signaling. ceh-14 also controls its own expression (Fig. 1E), suggesting that ceh-14 may be continuously required to maintain neuropeptidergic identity. The expression of unc-86 and pag-3 are unaffected in ceh-14 null mutants (Fig. S1B).
unc-86 and pag-3 affect all aspects of BDU identity
In contrast to the restricted defects of ceh-14 null mutants, loss of the unc-86 POU homeobox affects all neuron type-specific molecular features of BDU terminal identity (Fig. 1E). unc-86 mutants also display defects in the BDU-mediated harsh touch response (Fig. 1D). unc-86 is not required for the generation of BDU or adoption of its generic neuronal identity, as assessed by intact expression of a pan-neuronal marker in BDU (rab-3; data not shown). pag-3 also has a very broad effect on BDU identity. Expression of all of the terminal BDU identity genes described above is strongly affected in pag-3 mutants (except those that are also expressed in ALM, for reasons that will become evident later), including the expression of ceh-14 and its neuropeptide targets (Fig. 1E). However, pag-3 does not have an impact on unc-86 expression in BDU (Fig. S1B), demonstrating that in pag-3 mutants, the BDU neuron is formed and expresses unc-86, but is not able to induce terminal BDU differentiation.
To examine whether unc-86 and pag-3 directly control terminal BDU identity features, we analyzed the cis-regulatory architecture of two genes that define terminal BDU identity and that are both unc-86- and pag-3-dependent, the tyramine receptor-encoding ser-2 locus and ceh-14 LIM homeobox gene. Transgenic reporter animals that contain 5′ regions of the ser-2 and ceh-14 loci showed expression in BDU and other neurons. Through deletion analysis, we narrowed down the informational content of these reporters to ~ 400 bp of 5′ sequences (Fig. 2) and found predicted UNC-86/POU binding sites and PAG-3/Senseless binding motifs (Lee et al., 2010; Xiang et al., 1995) in these fragments. We deleted these motifs and found that they are required for expression of ser-2 and ceh-14 in BDU (Fig. 2). In the case of ser-2, deletion of one PAG-3 binding site has intermediate effects on ser-2 reporter gene expression, while deletion of both predicted PAG-3 binding sites abolishes expression (Fig. 2A). Deletion of either POU homeodomain binding site alone abolished ser-2 reporter expression (Fig. 2A). In the case of the cis-regulatory controls regions of the ceh-14 locus, we found three predicted binding sites for either TF and observed a synergistic requirement for these sites. Deletion of single UNC-86 binding sites had no effect on reporter expression, while deletion of all UNC-86 sites partially disrupted expression (Fig. 2B). Combining the mutation of all three UNC-86 sites with a mutation in a presumptive PAG-3 site, which alone has no effect on reporter expression, completely abolished reporter expression (Fig. 2B). We verified that the predicted UNC-86 binding sites neighboring the ceh-14 locus are indeed genuine UNC-86 binding sites in vitro using electrophoretic mobility shift assay (Fig. S2).
Fig. 2. Cis-regulatory elements controlling BDU-expressed genes.

A: Mutational analysis of cis-regulatory elements controlling expression of the ser-2 locus.
B: Mutational analysis of cis-regulatory elements controlling expression of the ceh-14. For mutation of PAG-3 sites, the core AATC was mutated to CCCC, for UNC-86 site, the central TAAT was mutated to CCAT. (+): at least 80% of animals showed bright expression in BDU. (+/−): between 10 and 30% of animals showed dim expression in BDU. (−): less than 10% of animals showed dim expression in BDU. n≥50 for each line.
C: Summary of genetic interaction data.
The finding that ser-2 and ceh-14 contain functionally required UNC-86 and PAG-3 binding sites suggests that unc-86 does not merely work through pag-3 to affect BDU gene expression. We rather conclude that unc-86 and pag-3 are terminal selector-type TFs (Hobert, 2011) that cooperate to induce terminal differentiation of the BDU neurons (summarized in Fig. 2C).
Transformation of BDU to ALM identity in pag-3 mutants
In spite of their similar effects on the induction of terminal features of BDU identity, there are striking differences in the unc-86 and pag-3 mutant phenotypes. In the initial identification of pag-3 mutants, the BDU neurons were noted to display ectopic expression of two terminal markers of the identity of the ALM sister cell, the mec-4 ion-channel-encoding gene and the mec-7-tubulin-expressing gene (Jia et al., 1996). We examined a potential transformation of BDU to ALM identity in more detail. First, we reexamined the ectopic expression of mec-4 and mec-7 in BDU results with different reporters and observed the same results as previously reported (Fig. 3A). Second, we examined additional molecular markers for ALM identity, namely the tubulin acetyltransferase-encoding mec-17 gene and the vesicular glutamate transporter-encoding gene eat-4, which defines the glutamatergic neurotransmitter identity of ALM. Both markers are also ectopically expressed in the BDU neurons of pag-3 mutants (Fig. 3A). Third, we examined whether the MEC-4 mechanosensory channel, which normally is targeted to discrete dots along the length of the ALM neuron (Chelur et al., 2002), will cluster along the axon of the transformed BDU neuron and found this to indeed be the case (Fig. 3B). Fourth, we examined axonal morphology of the BDU neurons in pag-3 mutants and found that they lose their long posteriorly directed processes and rather extend short posterior processes much alike the ALM neurons (Fig. 3F). The ventral turn of the BDU axons into the deirid commissure, normally undertaken by BDU but not ALM, is still executed normally in pag-3 mutants (Fig. 3H) and the axon remains associated with the excretory canal (Fig. 3I). We conclude that BDU identity is largely, but not completely transformed into ALM identity in pag-3 mutants.
Fig. 3. Reciprocal homeotic transformation of BDU to ALM in pag-3 mutants and ALM to BDU in mec-3 mutants.

A: ALM markers are ectopically expressed in BDU in pag-3(ls20).
B: Transformed BDU shows MEC-4 receptor cluster, as assessed with an mec-4::rfp translational fusion which appears in puncta along the axon of both ALM and BDU in pag-3(ls20).
C: BDU markers are ectopically expressed in ALM in mec-3(e1338).
D: Ectopic ALM marker expression is genetically in pag-3(−) animals is dependent on mec-3. The single mutant data is reiterated from panel A for comparison.
E: Ectopic BDU marker expression in mec-3(−) animals is dependent on pag-3. The single mutant data is reiterated from panel B for comparison.
F: The length of the posterior processes of ALM and BDU are altered in pag-3(ls20) and mec-3(e1338) mutants. Measurements were done on ser-2::gfp (otIs358) and mec-4::gfp (zdIs5)-expressing animals at the L4 stage.
G: The cell body position of ALM is more anterior in mec-3(e1338), while the position of BDU is unaffected by pag-3(ls20). Measurements refer to distance from the vulva during L4 stage, with higher measurements indicating a more anterior position.
H: ALM is located more laterally in mec-3(e1338), while BDU lateral position is unaffected. Position was examined in relation to the excretory canal (red, glt-3::rfp). The ALM cell body and axon are located more centrally in wild type but colocalize with the excretory canal in mec-3(e1338). BDU is associated with the excretory tract in both wild type and pag-3(ls20).
I: The ventral turn of the BDU process (visualized with ser-2::gfp) is unaffected in pag-3(ls20), while mec-3(e1338) causes the ALM process (visualized with mec-4::gfp) to undergo a ventral turn. The image shown for mec-3(e1338) is not typical: in most animals, the ALM axon closely follows the BDU axon in the ventral turn.
J: Schematic of changes to ALM and BDU in mec-3(e1338) and pag-3(ls20) mutants.
In all panels, significance refers to comparison to wild type. n≥50. Significance for A, C–E measured using Fischer’s Exact Test. ***p<.0001
Previous work had shown that the ectopic expression of mec-4 and mec-7 in the BDU neurons of pag-3 mutants genetically requires mec-3 (Jia et al., 1996), the ALM-expressed LIM homeobox gene that is required for ALM differentiation We independently confirmed this epistatic relationship with our reporter reagents (Fig. 3D). However, the previous pag-3 study did not provide evidence for ectopic expression of mec-3 in the BDU neurons of pag-3 mutants (Jia et al., 1996), as might be expected given the mec-3 dependence of the BDU to ALM transformation. Using a mec-3 reporter transgene not previously available, we observed ectopic expression of mec-3 in the “BDU” neurons of pag-3 mutants (Fig. 3A). Expression of mec-3 in ALM is unaffected in pag-3 mutants (Fig. 3A). We conclude that pag-3 not only drives BDU terminal identity but also represses ALM identity by repressing expression of the ALM identity driver mec-3 in BDU.
To assess whether the activating effect of pag-3 on BDU identity could be solely explained by a double-inhibitory mechanism in which pag-3 inhibits a repressor effect of mec-3 on BDU identity genes, we analyzed BDU identity in pag-3; mec-3 double mutants, and found that BDU identity is still lost (Fig. 3E). Therefore, as already suggested by our cis-regulatory analysis described above (Fig. 2), pag-3 rather appears to positively induce expression of BDU markers and independently, through repression of mec-3, inhibit the expression of ALM identity.
While the loss of BDU identity is shared by pag-3 and unc-86 mutants, the BDU to ALM transformation is only observed in pag-3 mutants and not in unc-86 mutants. In these animals, no ectopic expression of ALM identity markers can be observed in the BDU neurons (data not shown). This is expected since ALM differentiation requires unc-86 (Chalfie and Sulston, 1981; Duggan et al., 1998).
Reciprocal, homeotic ALM to BDU transformation in mec-3 mutants
Previous work has shown that unc-86 cooperates with mec-3 to induce terminal differentiation of the ALM neurons (Chalfie and Sulston, 1981; Duggan et al., 1998; Way and Chalfie, 1988, 1989; Xue et al., 1992; Xue et al., 1993). Even though the effect of unc-86 and mec-3 on the induction of ALM features are similar, there are striking differences in the unc-86 and mec-3 mutant phenotypes. The axons of the ALM neurons of mec-3 mutants were previously noted to extend more posteriorly than in wild-type animals and appear more ventrally positioned, thereby appearing more BDU-like; a more anterior position of the cell body of ALM was also noted (Way and Chalfie, 1988). Using previously unavailable gfp markers that label BDU morphology, we confirmed and quantified the presence of the posteriorly directed process of ALM in mec-3 mutants (Fig. 3F), the anterior position of the cell body (Fig. 3G) and the ventral shift of the ALM axons in mec-3 mutants (Fig. 3I). By colabeling the excretory canal, we found that the transformed ALM axon now occupies the same tract along the excretory canal that the normal BDU axon occupies (Fig. 3I). Moreover, we found that the ALM axons of mec-3 mutants now undergo the ventral turn into the deirid commissure, much alike what BDU axons do (Fig. 3H). The morphology transformations are summarized in Fig. 3J.
The availability of BDU identity markers allowed us to further examine the extent of transformation of ALM to BDU. We found that all BDU identity markers examined, including two neuropeptide-encoding genes, the tyramine receptor ser-2 and the IgSF zig-3, are ectopically expressed in the ALM neurons of mec-3 mutants (Fig. 3C). The ectopic expression of BDU markers in ALM in mec-3 mutants genetically depends on pag-3, since in mec-3; pag-3 double mutants, the flp-10 gene fails to be expressed in ALM (Fig. 3E).
Taken together, in mec-3 mutants, the ALM neurons display a homeotic transformation to the identity of the BDU neurons, based on morphology (summarized in Fig. 3J) and molecular markers. Unlike in pag-3 mutants, where only one morphological feature was transformed, the ALM to BDU transformation in mec-3 mutants extends to all morphological features that we could examine. In contrast, to mec-3 mutants, unc-86 mutants do not display an ALM to BDU transformation, simply because unc-86 is not only required for ALM, but also BDU differentiation, as shown above.
mec-3 is restricted to ALM by transcriptional repression in BDU via pag-3 and a non-canonical Wnt signaling system
The reciprocal homeotic transformations of neuronal identity in mec-3 and pag-3 mutants and the genetic epistasis experiments that we described above demonstrate that mec-3 and pag-3 antagonize each others activity. mec-3 promotes ALM identity and, by antagonizing pag-3 activity, inhibits BDU identity (i.e. the gain of BDU identity in ALM of mec-3 mutants genetically depends on pag-3), while pag-3 promotes BDU identity and, by antagonizing mec-3 activity, inhibits ALM identity (i.e. the gain of ALM identity in BDU of pag-3 mutants genetically depends on mec-3). In principle, such mutual antagonism could occur on the level of a mutual inhibition of each others expression. As described above, we indeed found that pag-3 represses mec-3 expression in BDU (Fig. 3A). However, both PAG-3 antibody staining (Cameron et al., 2002), a fosmid-based reporter for pag-3 expression (Fig. S1B) and single molecule mRNA fluorescence in situ hybridization (Fig. S1B) shows that pag-3 expression not only in BDU, but also in ALM, at similar levels. Why does pag-3 not inhibit mec-3 expression in ALM and how can mec-3 antagonize pag-3 activity in ALM?
Seeking to address the first question, we hypothesized that pag-3 may require other, factor(s) to repress mec-3 expression which may only be present in BDU, but not in ALM. mec-3 repression occurs on the transcriptional level as the derepression of mec-3 in pag-3 is inferred from a transcriptional reporter of the mec-3 locus (Fig. 3A). Since a non-canonical Wnt pathway is activated in the posterior daughter cell upon many asymmetric cell divisions along the anterior/posterior axis in the developing embryo (Mizumoto and Sawa, 2007) and since BDU is the posterior daughter of the embryonic neuroblast division that generates ALM and BDU (Fig. 1B), we examined two key indicators of the activity of this non-canonical Wnt pathway, the TCF-like protein POP-1 and the β-catenin-like protein SYS-1. In posterior cells in which the Wnt signal is active, the TCF-like protein POP-1 is exported from the nucleus (Mizumoto and Sawa, 2007), resulting in lower nuclear POP-1 in the posterior nucleus, compared to the anterior nucleus. Moreover, the Wnt signaling system stabilizes the β-catenin-like protein SYS-1 in posterior cells, compared to anterior cells (Mizumoto and Sawa, 2007). We indeed found that after division of the ALM/BDU mother in the embryo, high levels of POP-1 are present in the anterior ALM neuron and low levels in the posterior BDU neuron (Fig. 4A). Conversely, we observed low SYS-1 in ALM and high SYS-1 in BDU (Fig. 4A).
Fig. 4. A non-canonical Wnt signal represses mec-3 expression in BDU.

A: Expression of pop-1::gfp and sys-1::gfp after the ALM/BDU cell division in the embryo. Cells are marked by unc-86fosmid::rfp.
B: mec-3prom::gfp expression is derepressed in BDU in mom-4(ne1539); lit-1(t1512) temperature sensitive mutants. Embryos were shifted from the permissive temperature to the restrictive temperature after the birth of the ALM/BDU mother but before the ALM/BDU cell division and analyzed at 2-fold stage. Expression of mec-3prom::gfp is seen in both ALM and BDU after temperature shift but only in ALM in animals at the permissive temperature. ALM and BDU are marked with arrows.
C: Schematic summary. The Wnt receptor employed predominantly employed in the “anterior/posterior” coordinate system is MOM-5, the nature (and source) of the ligands is unknown (Mizumoto and Sawa, 2007).
To examine whether the Wnt signaling system in BDU indeed is involved in repressing mec-3 expression, we altered the activity of the kinases MOM-4 and LIT-1 which are required for the Wnt signaling-dependent export of POP-1 from the posterior sister nucleus (Takeshita and Sawa, 2005). We used the temperature-sensitive double mutant mom-4(ne1539); lit-1(t1512) to disrupt the Wnt/β-catenin asymmetry pathway (Takeshita and Sawa, 2005) and found that in temperature-shifted animals, mec-3 is indeed derepressed in the posterior BDU neuron (Fig. 4B). We conclude that the pag-3-dependent, BDU-specific repression of mec-3 expression involves a Wnt signaling system (Fig. 4C). The Wnt signal may result in the induction of expression of a BDU-specific cofactor with which PAG-3 works together to repress mec-3 expression; or, alternatively, PAG-3 may cooperate more directly with BDU-enriched SYS-1 to repress mec-3 expression specifically in BDU.
MEC-3 outcompetes PAG-3 for UNC-86 access
As mentioned above, the expression of pag-3 in both BDU and ALM does not only prompt the question how pag-3 can repress mec-3 expression in BDU, but also prompts the question of how mec-3 can antagonize pag-3 activity in ALM. We first tested whether mec-3 is not only required to antagonize pag-3 in ALM, but whether it is also sufficient to antagonize pag-3 upon ectopic expression in BDU. We found that unc-86 promoter-driven mec-3 expression is able to convert BDU into ALM, as assessed by examination of several terminal identity markers (Fig. 5A). The mec-3-induced BDU to ALM conversion can even be achieved long after the two neurons have differentiated in the embryo, as assessed by inducing mec-3 in larval stage using the heat-shock promoter (Fig. 5A). Overexpression of pag-3 under control of the heat-shock promoter is, in contrast, not able to convert ALM to BDU (data not shown) and overexpression of pag-3 under the unc-86 promoter is also only mildly able to convert ALM into BDU (Fig. 5B). We conclude that mec-3 is a true homeotic gene in the sense that it is not only required to prevent a homeotic transformation, but also sufficient to induce a homeotic transformation (Fig. 5C).
Fig. 5. mec-3 is sufficient to transform BDU to ALM.

A: Ectopic expression of mec-3 causes a BDU to ALM transformation, irrespective of the timing of mec-3 expression (lower panel), but dependent on an unc-86 interaction (u5 allele). See Experimental Procedure for the heat-shock experiments.
B: Ectopic pag-3 expression does not result in significant reciprocal transformations.
C: Schematic summary.
Significance indicates comparison to WT or no heat shock using Fischer’s Exact Test. ***:p≤.0001, **:p≤.001, *:p≤.05. n≥20.
The “recessive” nature of pag-3 compared to mec-3 does not appear to relate to limited levels of pag-3 expression since unc-86 promoter-driven pag-3 overexpression in ALM does not convert ALM to BDU. The inability of endogenously expressed pag-3 to drive BDU identity in ALM could be explained by the presence of an as yet unknown cofactor that is present in BDU, but not ALM. Since ectopic expression of MEC-3 in BDU is able to antagonize the activities of UNC-86 and PAG-3, we disfavor such a possibility. Instead we considered the possibility that UNC-86 and PAG-3 are sufficient in principle to induce BDU identity in ALM, but are actively prevented from doing so by MEC-3. Two observations lead us to formulate a hypothesis of how MEC-3 may antagonize UNC-86/PAG-3. MEC-3 and UNC-86 directly interact with one another as a heterodimer in vivo and in vitro and bind to directly adjacent sites on DNA (Rohrig et al., 2000; Xue et al., 1993). In contrast, as we have shown in Fig. 2, the UNC-86 and PAG-3 binding sites are spaced by many nucleotides, making heterodimer formation on DNA still possible, but less likely. We therefore hypothesized that MEC-3 may be able to recruit UNC-86 and thereby prevent UNC-86 from cooperating with PAG-3 to induce BDU genes. Two previously described missense alleles of unc-86, u5 and u168 (Chalfie and Au, 1989) allowed us to test this hypothesis. These alleles selectively affect the physical association of UNC-86 with MEC-3 and do not affect other, UNC-86-dependent, but MEC-3-independent neuronal differentiation events (Rohrig et al., 2000). We found that in unc-86(u5) mutants, the activation of unc-86/mec-3-dependent ALM-specific genes mec-17 and mec-4 is indeed disrupted (Fig. 6A). The same effect is observed in u168 mutants (data not shown). However, the BDU-specific genes ceh-14, flp-10 and zig-3 are now ectopically activated specifically in ALM and this activation depends on pag-3 (Fig. 6A). These results support the hypothesis that if endogenous, wild-type MEC-3 protein is not able to physically interact with UNC-86, the UNC-86 protein will cooperate with PAG-3 to drive BDU fate. These results also indicate that the components to induce BDU identity in ALM are present in ALM but that MEC-3, by specific binding to UNC-86, is able to antagonize PAG-3 and thereby inhibit the execution of BDU fate.
Fig. 6. Evidence in support of a competition model.

A: Mutations that impair UNC-86/MEC-3 binding cause ALM to BDU transformations which depend on pag-3.
B: A mec-3 hypomorph shows dual expression of ALM and BDU reporters in ALM.
C: Increased levels of unc-86 in ALM cause dual expression of ALM and BDU reporters in ALM.
Significance indicates comparison to WT (A) or no heat shock (C) using Fischer’s Exact Test. ***:p≤.0001, **:p≤.001, *:p≤.05, NS: not significant. n≥40 in panel A, B, n=15 for panel C.
D: Electrophoretic mobility shift assays reveal impact of MEC-3 on DNA binding by UNC-86. UNC-86 binds to the ceh-14 and tph-1 promoters; this binding is eliminated with the addition of MEC-3, but not by the addition of the homeodomain protein CEH-43. Probes are as described in Experimental Procedures; the probes each contain one UNC-86 binding site but no MEC-3 or PAG-3 binding sites. EMSA was performed with 100 nM UNC-86; 50, 100, and 200 nM (for ceh-14 probe) or 100 and 200 nM (for tph-1 probe) MEC-3; and 50, 100, and 200 nM CEH-43. In addition to the probe shown, each reaction contained an equal concentration of unlabeled mec-3 promoter. See additional data in Fig. S2.
If MEC-3 indeed outcompetes PAG-3 for access to UNC-86, then the BDU to ALM transformation observed upon ectopic expression of mec-3 in BDU should not occur in an unc-86(u5) mutant background in which UNC-86 is not able to interact with MEC-3. We indeed found that ectopic mec-3 can not induce mec-17::rfp expression in BDU in unc-86(u5) animals (Fig. 5A).
A model in which MEC-3 competes with PAG-3 to direct all UNC-86 to induce ALM identity (rather than allowing UNC-86 to interact with PAG-3 to induce BDU identity) makes another prediction: lowering the level of mec-3 expression may still provide enough MEC-3 protein to operate together with unc-86, but not enough to successfully outcompete PAG-3 cooperation with UNC-86. In this scenario, ALM markers may still be expressed in ALM, but there may now be ectopic expression of BDU identity markers in ALM as well. We tested this possibility using the mec-3(u298) allele, a previously described weak allele with incompletely penetrant mechanosensory defects. These allele contains a transposon insertion upstream of the mec-3 locus, thought to lower mec-3 expression (Way and Chalfie, 1989), a notion we independently confirmed using smFISH (data not shown). We found that in mec-3(u298) mutants, the ALM markers mec-4 and mec-17 are still expressed in ALM while the BDU markers zig-3, ceh-14 and flp-10 are derepressed in ALM (Fig. 6B). This “mixed fate” is different from the null mutant phenotype of mec-3, which shows complete loss of ALM markers in ALM, and genetically separates the adoption of ALM identity from the repression of BDU identity. Apparently, different levels of mec-3 are required for ALM induction and competition with PAG-3 for UNC-86 access.
The competition model predicts that a mixed ALM/BDU fate should be observed not only upon lowering the expression of mec-3, but also upon increasing the expression of unc-86. Higher levels of unc-86 expression would provide enough UNC-86 protein to physically interact with MEC-3 and to collaborate with PAG-3, thus allowing both sets of identity genes to be expressed simultaneously. We tested this prediction using a heat shock promoter to ubiquitously increase unc-86 expression beginning at the 3-fold stage, after the ALM/BDU division. We found that after induction of unc-86, the BDU marker zig-3 was ectopically expressed in ALM, while expression of the ALM marker mec-17 was unaffected (Fig. 6C).
We used electrophoretic mobility shift assays (EMSAs) to examine the competition model in molecular detail. We found that bacterially produced UNC-86 protein is capable of binding to a 90 bp, double-stranded DNA sequence from the promoter of the BDU-expressed gene ceh-14, which contains predicted UNC-86 binding sites. UNC-86 binding to this site can be competed by adding bacterially produced MEC-3 protein together with an DNA probe that contains an UNC-8/MEC-3 binding site from an ALM expressed gene (Fig. 6D; Fig. S4). Adding equal concentrations of the homeodomain protein CEH-43 (Fig. 6D) or equal concentrations of PAG-3 does not result compete for UNC-86 binding to the probe from the BDU gene (Fig. 6D; Fig. S4). The MEC-3-mediated competition was dependent upon UNC-86/MEC-3 interactions, as UNC-86(L195F), a point mutation that corresponds to the unc-86(u5) allele and abolishes the MEC-3 binding (Rohrig et al., 2000), did not show changes in binding upon the addition of MEC-3 (Fig. S4). In addition, the competition was not seen without the presence of unlabeled oligos from an ALM promoter, indicating that recruitment of UNC-86 away from BDU gene promoters is dependent on alternative DNA binding (Fig. S4). We also found that MEC-3 also reduces UNC-86 binding to a 90 bp DNA sequence from the locus of tph-1 (Fig. 6D), a gene controlled by UNC-86 and distinct cofactors in other neuron types (Sze et al., 2002; Zhang et al., 2014)(N. Flames and O.H., unpubl. data).
Taken together, our data suggests that unc-86 and pag-3 drive a “default” BDU state and that this state can, in principle, be induced in both the ALM and BDU neurons. In ALM, however, the presence of mec-3 diverts from the ground state since MEC-3 can, by direct interaction with UNC-86, prevent the execution of the UNC-86/PAG-3 program and rather induce the ALM differentiation program.
The competition mechanism operates in other cellular contexts
We tested the generality of the competition mechanism in two different manners. We first considered the PLM/ALN sister neurons in the tail of the animal (Fig. 7A). PLM is a light touch receptor neuron that is analogous to ALM in several ways, including its function, overall molecular composition and reliance on the UNC-86/MEC-3 heterodimer for its differentiation. Its sister cell is the cholinergic ALN neuron, a neuron that is distinct from the peptidergic BDU neuron, both in terms of overall morphology, synaptic connectivity, molecular profile and neurotransmitter identity. However, like the BDU neuron pair, the ALN neuron pair also expresses unc-86 throughout its lifetime (Finney and Ruvkun, 1990) and requires unc-86 for its generation (Chalfie et al., 1981). A factor that collaborates with UNC-86 to induce ALN differentiation – in analogy to PAG-3 in BDU - is not currently known.
Fig. 7. The competition mechanism operates in other cellular contexts.

A: Schematic drawing of ALM, BDU, ALN, and PLM neurons. Note that pag-3 is not expressed in ALNL (Jia et al., 1997) (data not shown).
B: lad-2prom::gfp (otIs439) expression is altered by mec-3 and unc-86. Each dot indicates one animal with the given number of lad-2prom::gfp(+) cells.
C: cho-1fosmid::mCherry (otIs544) expression is altered by mec-3(e1338).
D: Ectopic mec-3 causes the ALM terminal marker mec-17prom::rfp to be ectopically expressed in the tail. Horizontal bars in panels B, C, and D indicate average number of cells.
E: Ectopic mec-3 alters expression of tph-1prom::gfp (zdIs13). Expression was examined in HSN and NSM neurons in wild type and with ectopic mec-3 driven by the unc-86 5.2kb promoter.
To ask whether in analogy to the ALM/BDU sister neuron pair, mec-3 also operates in PLM to prevent the a homeotic transformation to ALN fate by competing for UNC-86 access, we tested two predictions: First, in mec-3 mutants, ectopic expression of ALN markers in PLM should be observed and we indeed found this to be the case (Fig. 7B,C). Second, ectopic expression of mec-3 in ALN should convert ALN to PLM identity. Using the unc-86 promoter to drive mec-3 in ALN, we indeed found that expression of an ALN marker is abrogated while a PLM marker is ectopically expressed (Fig. 7B,D). We conclude that even though the ALN neuron class is very distinct from the BDU neuron class, there are fundamental similarities in the way that their identity is controlled. unc-86 controls identity of ALN and PLN. In PLM, mec-3 does not only induce PLM identity but prevents a homeotic transformation to ALN identity, likely by competing with an as yet unknown unc-86 cofactor expressed in both ALN and PLM. This cofactor (in analogy to pag-3) normally drives ALN identity in conjunction with unc-86 but is prevented by mec-3 in doing so in PLM.
We assessed whether other unc-86-dependent cell fate decisions could also be disrupted by MEC-3 titrating UNC-86 away from its respective, cell type-specific target genes. In the serotonergic NSM neurons, unc-86 cooperates with the LIM homeobox gene ttx-3 to drive NSM terminal differentiation (Zhang et al., 2014), while in HSN unc-86 cooperates with the ETS domain TF ast-1 and the Zn finger TF sem-4 (N. Flames and O.H., unpubl. data). We indeed found that ectopic expression of mec-3 in NSM and HSN disrupts the unc-86-dependent expression of the serotonergic marker tph-1 (Fig. 7E). Ectopic expression of mec-3 under control of the unc-86 promoter not only disrupts the respective differentiation programs of other unc-86-expressing neurons, but induces touch marker expression in many of the 57 unc-86 expressing neurons. For example, ectopic mec-17 expression can be observed in up to nine additional cells in the tail ganglia of the worm (Fig. 7D) which precisely matches the number of unc-86 expressing neurons in the tail (Finney and Ruvkun, 1990). Taken together, these findings indicate that mec-3 can operate in very distinct cellular context to “divert” unc-86 from its normal function, converting neurons into alternative states.
DISCUSSION
In the first part of this paper, we have described a gene regulatory program that defines the differentiated state of the BDU neurons. The neuropeptidergic identity of BDU, which we found to be critical for its function, constitutes a “subroutine” under control of the ceh-14 LIM homeobox gene, the C. elegans ortholog of vertebrate Lhx3/4. This subroutine is in turn under control of two TFs, the POU homeobox gene unc-86 (C. elegans ortholog of vertebrate Brn3) and the Zn finger TF pag-3 (C. elegans ortholog of vertebrate Gfi), which jointly regulate not only neuropeptidergic identity, but also all other tested molecular identity features of the BDU neurons. The coregulation of distinct identity features by this combination of two TFs, likely occurring by direct binding and activation of terminal identity genes, provides further support for the broad applicability of the concept of neuronal identity control by terminal selectors (Hobert, 2011). As previously observed in several other C. elegans neuron classes (Hobert, 2010), terminal selector TFs coregulate many distinct identity features of a specific neuron type. Such coregulation contrasts the alternative, theoretical model of neuronal identity features being controlled in a piece-meal manner by distinct TFs.
Previous work had already established that the ALM neurons, the sister neurons of the BDU neurons, are also controlled by two closely cooperating terminal selector-type TFs, unc-86 and mec-3 (Chalfie and Sulston, 1981; Duggan et al., 1998; Way and Chalfie, 1988, 1989; Xue et al., 1992; Xue et al., 1993). Notably, the terminal selector combinations for ALM and BDU share a common factor, the unc-86 POU homeobox gene. Yet the target gene spectrum of UNC-86 is distinct in ALM and BDU and apparently dictated by UNC-86’s collaboration with distinct cofactors, the MEC-3 LIM homeodomain protein in ALM and PAG-3 in BDU. Previous work has shown that UNC-86 operates as terminal selector in combination with yet other TFs in completely distinct neuron classes as well, for example, the cholinergic IL2 sensory neurons (Zhang et al., 2014), the serotonergic NSM and HSN neurons (Zhang et al., 2014)(N. Flames and O.H., unpubl. Data) or the glutamatergic PVR neurons (Serrano-Saiz et al., 2013).
In the second part of this paper, we have explored the effects of removal of terminal selectors on neuronal identity. Genetic removal of TFs that drive specific neuronal identity programs in either C. elegans or other animal species can have remarkably distinct consequences, depending on cellular context; in some cases, neurons will merely remain in an ill-defined, undifferentiated state (e.g. (Altun-Gultekin et al., 2001; Kratsios et al., 2011; Liu et al., 2010)), in other cases neurons may die (e.g. (Beby et al., 2010)) while in a number of cases, the identity of a neuron switches to the identity of another neuron type (e.g. (Lopes et al., 2012; Sagasti et al., 1999)). Often such identity transformations are just inferred by changes in very select identity features, such as neurotransmitter identity, and it is therefore not entirely clear how extensive such transformations are (e.g. (Lopes et al., 2012)). The availability of a host of molecular markers as well as the ability to visualize anatomy in detail allowed us to show that removal of either the BDU terminal selector pag-3 or the ALM terminal selector mec-3 results in either complete (mec-3 mutant) or almost complete (pag-3 mutant) identity transformations. Corroborating the notion of mec-3 being a homeotic regulator is our observation that mec-3 is not only required to prevent a homeotic transformation but also sufficient to promote a homeotic transformation upon ectopic misexpression. The homeotic phenotypes observed in mec-3 mutants are a testament to the broad impact that a terminal selector has on defining the identity of a neuron type.
The conventional interpretation of homeotic identity transformations is that a given TF promotes expression of genes that define one identity, while inhibiting the expression of genes defining an alternative identity (Fig. 8; left panels). Indeed in some cases, it has been shown that a TF can have a dual function as an activator and repressor. For example, in the neocortex Fezf2 directly promotes expression of Vglut1, the key identity determinant of glutamatergic neurons and directly represses expression of Gad1, the key identity determinant of GABAergic neurons (Lodato et al., 2014). In the dorsal spinal cord, Ptf1 directly activates structural identity determinant of GABAergic neurons (e.g. Gad1), and directly inhibits TFs that regulate the glutamatergic phenotype (Borromeo et al., 2014). How broadly applicable such dual functionality of a single TF is remains unclear. We have described here evidence that supports a distinct mechanism enabling simultaneous activation of one cellular identity and repression of another. As schematically illustrated in a generalized way in Fig. 8 (right panels), MEC-3 induces a differentiation program (ALM identity) by binding to a cooperating TF, UNC-86. Due to limiting amounts of UNC-86 in a cell, this binding makes UNC-86 unable to engage in cooperation with the TF PAG-3. In BDU, UNC-86 is left unperturbed by MEC-3, and can collaborate with PAG-3 and induces BDU identity.
Fig. 8. Mechanisms to control alternative cell fate choices.

In the conventional interpretation of alternative cell fate choices, TF Z works to promote one fate while simultaneously inhibiting another fate. The model described here operates by a distinct principle.
The interaction of UNC-86 with MEC-3 and with PAG-3 are likely to be fundamentally distinct. UNC-86 and MEC-3 directly interact with one another and the heterodimer binds to a specific DNA sequence motif with adjacent UNC-86 and MEC-3 binding sites (Duggan et al., 1998; Xue et al., 1993). In contrast, the presumptive UNC-86 and PAG-3 binding sites are physically spaced apart by many dozen nucleotides and their spacing differs in distinct target promoters. Therefore, UNC-86 and PAG-3 may not directly interact in BDU but rather co-conspire to recruit additional factors required for transcriptional activation. Such a scenario applies to many other regulatory elements in which TFs are displayed in a so-called “billboard manner” to recruit transcriptional machinery (Arnosti and Kulkarni, 2005). In this billboard architecture, cooperativity is not a necessity and indeed we found that the removal of multiple UNC-86 and/or PAG-3 sites is required before an effect is observed. By binding directly to UNC-86, MEC-3 is apparently able to not only disrupt this billboard architecture, but also recruit UNC-86 to a distinct set of targets, resulting in a homeotic transformation.
As a note of caution, we have not formally ruled out the possibility that mec-3 acts by either inducing the expression of a factor in ALM that prevents unc-86/pag-3 from inducing BDU fate or represses the expression of a BDU-expressed cofactor that unc-86/pag-3 require to induce BDU fate. We disfavor the existence of unknown factors in light of (a) the gene dosage experiments (particularly the unc-86 dosage), (b) the gel shift experiments and (c) the disruption of unc-86-mediated differentiation events in completely distinct cellular contexts by ectopic expression of mec-3. It appears less parsimonious to argue that in all these distinct cellular contexts, mec-3 is capable of controlling the activity of cell-type specific regulatory cofactors.
Ectopic expression of specific TFs are known to result in disrupting the activity of endogenous TFs with which the misexpressed TF physically interacts. For example, the phenomenon of “phenotypic suppression” in which ectopic Hox gene expression dominates over the function of another Hox gene, is based on a cofactor competition mechanism (Noro et al., 2011). However, previous studies only considered competition in artificial misexpression contexts while in the example that we describe here the competition mechanism occurs among endogenous proteins (UNC-86, MEC-3 and PAG-3) that normally coexist in a specific neuron type (ALM). It is attractive to speculate that the phenomenon of “posterior dominance” in which there is a phenotypic dominance between endogenously coexpressed Hox proteins (Duboule and Morata, 1994) is also explicable by a competition for a cofactor.
The competition mechanism operating in the ALM neurons to prevent a homeotic transformation is contrasted by a distinct mechanism that acts in the BDU neurons to prevent a (partial) homeotic transformation in ALM. This mechanism involves transcriptional repression of a homeotic regulator (mec-3), which is mediated by two factors, a Wnt signal and the PAG-3 TF. Based on dual activator/repressor functions of its vertebrate and Drosophila orthologs (Jafar-Nejad and Bellen, 2004), we hypothesize that PAG-3 may have a dual role as an activator of the BDU terminal gene battery and a Wnt-dependent repressor of mec-3 expression.
We view the application of the homeosis concept to the ALM versus BDU cell fate decision useful in light of the deep roots that homeosis has in evolutionary thought (Akam, 1998; Bateson, 1894; Sternberg and Horvitz, 1984). Since Bateson defined the homeotic transformation in the late 1800’s in the context of studying natural variations (Bateson, 1894), it has been speculated that homeotic transformations can be a driver for evolutionary change. This notion has already been discussed in the context of homeotic transformation in C. elegans (Sternberg and Horvitz, 1984). A key feature of such a hypothesis is the saltatory, discontinuous nature of evolutionary changes evoked by homeotic transformations (Sternberg and Horvitz, 1984). However, it has been argued that conventional homeotic mutations in meristic series (e.g. leg/antenna transformations) are too drastic to be evolutionarily beneficial (Akam, 1998). On the other hand, transformations in the identity of “only” individual cell types can easily be envisioned to have adaptive value. In the context of the ALM/BDU (and PLM/ALN) identities described here, one could envision that in an ancestral state unc-86 and pag-3 generated multiple BDU neurons after symmetric division of a neuroblast. Recruitment of mec-3 expression into the anterior daughter of the neuroblast then transformed BDU identity to ALM identity. The same scenario may apply to the ALN neurons, of which there may have been two originally, but recruitment of mec-3 again diverted one of them to a touch neuron (PLM) identity. A gain of mec-3 expression in distinct, unc-86-dependent neuronal cell types allows the generation of multiple, relatively homogenous unc-86/mec-3-dependent touch neurons in different parts of the body, thereby providing touch sensitivity to much of the length of the animal. In essence, the principle of homeosis allows the generation of novel neuronal cell types in discontinuous, saltatory manner in which an ancestral differentiation program is diverted to a distinct differentiation program through gain of expression of a terminal selector-type TF.
EXPERIMENTAL PROCEDURES
Transgenes
DNA constructs used to generate transgenic strains and a list of transgenes used in this study can be found in the Supplementary Information.
Harsh touch assay
Harsh touch assays were performed on gravid adults which were transferred to a Nematode Growth Medium (NGM) plate with OP50 bacteria one hour before testing. Animals were scored blind to genotype. The assay was performed on animals moving in a forward direction while off the bacteria lawn. Each animal was touched once in the anterior half of the body, just posterior to the pharynx, using a flattened platinum wire pick attached to a glass Pasteur pipette. Animals were scored by measuring the number of head swings each animal moved in a backwards direction before stopping, reversing direction, or performing an omega turn.
Heat shock experiments
For hsp::mec-3 experiments, animals were heat shocked at bean stage (~360 minutes), 3-fold stage (~550 minutes), or at L2 larval stage. For hsp::unc-86 experiments, animals were heat shocked at 3-fold stage. Each heat shock consisted of three rounds of 30 minutes at 37°C, followed by one hour of rest at 20 °C. Animals were then maintained overnight at 25°C and scored the following day.
Electrophoretic mobility shift assays
Full-length mec-3 cDNA was cloned into the pET21b His tag bacterial expression vector (EMD Millipore) and transformed into BL21(DE3) cells (NEB). Protein expression was induced using 1 mM IPTG for four hours and purified with Ni-NTA resin (Qiagen) under denaturing conditions as previously described (Wenick and Hobert, 2004). unc-86 cDNA in pET21b was similarly purified, as previously described (Zhang et al., 2014). ceh-43 cDNA in pET21b was induced in 1 mM IPTG for four hours. To purify, bacteria was pelleted, frozen, and resuspended in 50 mM Tris (pH 7.5), 500 mM NaCl, 20 mM Imidazole with protease inhibitors. The solution was sonicated and purified with Ni-NTA resin (Qiagen). The same buffer plus 300 mM imidazole was used for elution, and protein was dialyzed into 20 mM HEPES (pH 7.9), 200 mM NaCl, 10% glycerol, 2 mM MgCl2.
To perform EMSAs, a short oligonucleotide was end-labeled with [Γ-32P]ATP using T4 polynucleotide kinase (NEB) according to the manufacturer’s specifications. A complementary sequence was added to the 3′ end of each oligonucleotide used in the EMSA. The radiolabeled short sequence was annealed to each long oligo and the remaining DNA was filled in using Klenow (NEB). Protein and DNA were incubated for 20 minutes at room temperature in 1x binding buffer (5x Binding Buffer (BB): 50 μl 1M Tris pH 7.5, 50 μl 5M NaCl, 5 μl 1M MgCl2, 250 ul 80% glycerol, 2.5 μl 1 M DTT, 5 μl 0.5M EDTA, 250 μl Poly dI-dC, 2.5mg/ml bovine serum albumin, 290 μl H2O), before loading on 4% (79:1 acrylamide:bis-acrylamide) gel and run at 165V at 4°C for 2–3 hour s. Purified UNC-86 was run at 100 nM concentration. MEC-3 concentrations for the ceh-14 probe were 50, 100, and 200 nM; for the tph-1 probe, MEC-3 concentrations were 100 and 200 nM. CEH-43 concentrations were 50, 100, and 200 nM.
Supplementary Material
Acknowledgments
We thank Q. Chen for expert assistance in generating transgenic strains, members of the C. elegans community and the knockout consortia for providing strains, N. Abe for help and advice on gel shifts, N. Flames for communicating unpublished results, I. Greenwald, R. Mann, M. Chalfie, N. Flames and members of the Hobert lab for comments on the manuscript. This work was funded by the NIH (R01NS039996-05; R01NS050266-03) and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Akam M. Hox genes, homeosis and the evolution of segment identity: no need for hopeless monsters. Int J Dev Biol. 1998;42:445–451. [PubMed] [Google Scholar]
- Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O. A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development. 2001;128:1951–1969. doi: 10.1242/dev.128.11.1951. [DOI] [PubMed] [Google Scholar]
- Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94:890–898. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- Bateson W. Materials for the study of variation, treated with especial regard to discontinuity in the origin of species. London: Macmillan; 1894. [Google Scholar]
- Beby F, Housset M, Fossat N, Le Greneur C, Flamant F, Godement P, Lamonerie T. Otx2 gene deletion in adult mouse retina induces rapid RPE dystrophy and slow photoreceptor degeneration. PLoS One. 2010;5:e11673. doi: 10.1371/journal.pone.0011673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borromeo MD, Meredith DM, Castro DS, Chang JC, Tung KC, Guillemot F, Johnson JE. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development. 2014;141:2803–2812. doi: 10.1242/dev.105866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S, Clark SG, McDermott JB, Aamodt E, Horvitz HR. PAG-3, a Zn-finger transcription factor, determines neuroblast fate in C. elegans. Development. 2002;129:1763–1774. doi: 10.1242/dev.129.7.1763. [DOI] [PubMed] [Google Scholar]
- Cassata G, Kagoshima H, Andachi Y, Kohara Y, Durrenberger MB, Hall DH, Burglin TR. The LIM homeobox gene ceh-14 confers thermosensory function to the AFD neurons in Caenorhabditis elegans. Neuron. 2000;25:587–597. doi: 10.1016/s0896-6273(00)81062-4. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Au M. Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science. 1989;243:1027–1033. doi: 10.1126/science.2646709. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Horvitz HR, Sulston JE. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell. 1981;24:59–69. doi: 10.1016/0092-8674(81)90501-8. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelur DS, Ernstrom GG, Goodman MB, Yao CA, Chen L, ROH, Chalfie M. The mechanosensory protein MEC-6 is a subunit of the C. elegans touch-cell degenerin channel. Nature. 2002;420:669–673. doi: 10.1038/nature01205. [DOI] [PubMed] [Google Scholar]
- Cheng L, Samad OA, Xu Y, Mizuguchi R, Luo P, Shirasawa S, Goulding M, Ma Q. Lbx1 and Tlx3 are opposing switches in determining GABAergic versus glutamatergic transmitter phenotypes. Nat Neurosci. 2005;8:1510–1515. doi: 10.1038/nn1569. [DOI] [PubMed] [Google Scholar]
- Duboule D, Morata G. Colinearity and functional hierarchy among genes of the homeotic complexes. Trends Genet. 1994;10:358–364. doi: 10.1016/0168-9525(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Duerr JS, Gaskin J, Rand JB. Identified neurons in C. elegans coexpress vesicular transporters for acetylcholine and monoamines. Am J Physiol Cell Physiol. 2001;280:C1616–1622. doi: 10.1152/ajpcell.2001.280.6.C1616. [DOI] [PubMed] [Google Scholar]
- Duggan A, Ma C, Chalfie M. Regulation of touch receptor differentiation by the Caenorhabditis elegans mec-3 and unc-86 genes. Development. 1998;125:4107–4119. doi: 10.1242/dev.125.20.4107. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Graf T, Enver T. Forcing cells to change lineages. Nature. 2009;462:587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- Hobert O. Neurogenesis in the nematode Caenorhabditis elegans. WormBook. 2010:1–24. doi: 10.1895/wormbook.1.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol. 2011;27:681–696. doi: 10.1146/annurev-cellbio-092910-154226. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Bellen HJ. Gfi/Pag-3/senseless zinc finger proteins: a unifying theme? Mol Cell Biol. 2004;24:8803–8812. doi: 10.1128/MCB.24.20.8803-8812.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Xie G, Aamodt E. pag-3, a Caenorhabditis elegans gene involved in touch neuron gene expression and coordinated movement. Genetics. 1996;142:141–147. doi: 10.1093/genetics/142.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Xie G, McDermott JB, Aamodt E. The C. elegans gene pag-3 is homologous to the zinc finger proto-oncogene gfi-1. Development. 1997;124:2063–2073. doi: 10.1242/dev.124.10.2063. [DOI] [PubMed] [Google Scholar]
- Kim K, Li C. Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. The Journal of comparative neurology. 2004;475:540–550. doi: 10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- Kratsios P, Stolfi A, Levine M, Hobert O. Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene. Nat Neurosci. 2011;15:205–214. doi: 10.1038/nn.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in caenorhabditis elegans. J Neurosci. 1999;19:159–167. doi: 10.1523/JNEUROSCI.19-01-00159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Doddapaneni K, Hogue A, McGhee L, Meyers S, Wu Z. Solution structure of Gfi-1 zinc domain bound to consensus DNA. J Mol Biol. 2010;397:1055–1066. doi: 10.1016/j.jmb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Li C, Kim K. Neuropeptide Gene Families in Caenorhabditis elegans. In: Geary TG, editor. Neuropeptide Systems as Targets for Parasite and Pest Control. 2010. [Google Scholar]
- Li W, Kang L, Piggott BJ, Feng Z, Xu XZ. The neural circuits and sensory channels mediating harsh touch sensation in Caenorhabditis elegans. Nat Commun. 2011;2:315. doi: 10.1038/ncomms1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Maejima T, Wyler SC, Casadesus G, Herlitze S, Deneris ES. Pet-1 is required across different stages of life to regulate serotonergic function. Nat Neurosci. 2010;13:1190–1198. doi: 10.1038/nn.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, Molyneaux BJ, Zuccaro E, Goff LA, Chen HH, Yuan W, Meleski A, Takahashi E, Mahony S, Rinn JL, et al. Gene co-regulation by Fezf2 selects neurotransmitter identity and connectivity of corticospinal neurons. Nat Neurosci. 2014 doi: 10.1038/nn.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes R, Verhey van Wijk N, Neves G, Pachnis V. Transcription factor LIM homeobox 7 (Lhx7) maintains subtype identity of cholinergic interneurons in the mammalian striatum. Proc Natl Acad Sci U S A. 2012;109:3119–3124. doi: 10.1073/pnas.1109251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- Mizumoto K, Sawa H. Two betas or not two betas: regulation of asymmetric division by beta-catenin. Trends Cell Biol. 2007;17:465–473. doi: 10.1016/j.tcb.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Nakatani T, Minaki Y, Kumai M, Ono Y. Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development. 2007;134:2783–2793. doi: 10.1242/dev.02870. [DOI] [PubMed] [Google Scholar]
- Nathoo AN, Moeller RA, Westlund BA, Hart AC. Identification of neuropeptide-like protein gene families in Caenorhabditis elegans and other species. Proc Natl Acad Sci U S A. 2001;98:14000–14005. doi: 10.1073/pnas.241231298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro B, Lelli K, Sun L, Mann RS. Competition for cofactor-dependent DNA binding underlies Hox phenotypic suppression. Genes Dev. 2011;25:2327–2332. doi: 10.1101/gad.175539.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrig S, Rockelein I, Donhauser R, Baumeister R. Protein interaction surface of the POU transcription factor UNC-86 selectively used in touch neurons. EMBO J. 2000;19:3694–3703. doi: 10.1093/emboj/19.14.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagasti A, Hobert O, Troemel ER, Ruvkun G, Bargmann CI. Alternative olfactory neuron fates are specified by the LIM homeobox gene lim-4. Genes Dev. 1999;13:1794–1806. doi: 10.1101/gad.13.14.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salio C, Lossi L, Ferrini F, Merighi A. Neuropeptides as synaptic transmitters. Cell Tissue Res. 2006;326:583–598. doi: 10.1007/s00441-006-0268-3. [DOI] [PubMed] [Google Scholar]
- Sattler R. Homeosis in Plants. Am J Bot. 1988;75:1606–1617. [Google Scholar]
- Serrano-Saiz E, Poole RJ, Felton T, Zhang F, de la Cruz ED, Hobert O. Modular Control of Glutamatergic Neuronal Identity in C. elegans by Distinct Homeodomain Proteins. Cell. 2013;155:659–673. doi: 10.1016/j.cell.2013.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Leone DP, Bateson RK, Dobreva G, Kohwi Y, Kohwi-Shigematsu T, Grosschedl R, McConnell SK. A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc Natl Acad Sci U S A. 2012;109:19071–19078. doi: 10.1073/pnas.1216793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW, Horvitz HR. The genetic control of cell lineage during nematode development. Annu Rev Genet. 1984;18:489–524. doi: 10.1146/annurev.ge.18.120184.002421. [DOI] [PubMed] [Google Scholar]
- Sze JY, Zhang S, Li J, Ruvkun G. The C. elegans POU-domain transcription factor UNC-86 regulates the tph-1 tryptophan hydroxylase gene and neurite outgrowth in specific serotonergic neurons. Development. 2002;129:3901–3911. doi: 10.1242/dev.129.16.3901. [DOI] [PubMed] [Google Scholar]
- Takeshita H, Sawa H. Asymmetric cortical and nuclear localizations of WRM-1/beta-catenin during asymmetric cell division in C. elegans. Genes Dev. 2005;19:1743–1748. doi: 10.1101/gad.1322805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson A, Ready DF. Sevenless: a cell-specific homeotic mutation of the Drosophila eye. Science. 1986;231:400–402. doi: 10.1126/science.231.4736.400. [DOI] [PubMed] [Google Scholar]
- Way JC, Chalfie M. mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell. 1988;54:5–16. doi: 10.1016/0092-8674(88)90174-2. [DOI] [PubMed] [Google Scholar]
- Way JC, Chalfie M. The mec-3 gene of Caenorhabditis elegans requires its own product for maintained expression and is expressed in three neuronal cell types. Genes Dev. 1989;3:1823–1833. doi: 10.1101/gad.3.12a.1823. [DOI] [PubMed] [Google Scholar]
- Wenick AS, Hobert O. Genomic cis-Regulatory Architecture and trans-Acting Regulators of a Single Interneuron-Specific Gene Battery in C. elegans. Dev Cell. 2004;6:757–770. doi: 10.1016/j.devcel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society of London B Biological Sciences. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15:4762–4785. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Finney M, Ruvkun G, Chalfie M. Regulation of the mec-3 gene by the C. elegans homeoproteins UNC-86 and MEC-3. Embo J. 1992;11:4969–4979. doi: 10.1002/j.1460-2075.1992.tb05604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Tu Y, Chalfie M. Cooperative interactions between the Caenorhabditis elegans homeoproteins UNC-86 and MEC-3. Science. 1993;261:1324–1328. doi: 10.1126/science.8103239. [DOI] [PubMed] [Google Scholar]
- Zhang F, Bhattacharya A, Nelson JC, Abe N, Gordon P, Lloret-Fernandez C, Maicas M, Flames N, Mann RS, Colon-Ramos DA, et al. The LIM and POU homeobox genes ttx-3 and unc-86 act as terminal selectors in distinct cholinergic and serotonergic neuron types. Development. 2014;141:422–435. doi: 10.1242/dev.099721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M. Identification of genes expressed in C. elegans touch receptor neurons. Nature. 2002;418:331–335. doi: 10.1038/nature00891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.