

Abstract
Background
Endothelial cell activation and injury by the terminal pathway of complement is important in various pathobiological processes, including xenograft rejection. Protection against injury by human complement can be induced in porcine endothelial cells (ECs) with IL-4 and IL-13 through metabolic activation. However, despite this resistance, the complement-treated ECs were found to lose membrane permeability control assessed with the small molecule calcein. Therefore to define the apparent discrepancy of permeability changes vis-à-vis the protection from killing we now investigated whether IL-4 and IL-13 influence the release of the large cytoplasmic protein lactate dehydrogenase (LDH) in ECs incubated with complement or the pore-forming protein melittin.
Methods
Primary cultures of ECs were pretreated with IL-4 or IL-13 and then incubated with human serum as source of antibody and complement or melittin. Cell death was assessed using neutral red. Membrane permeability was quantitated measuring LDH release.
Results
We found that IL-4/IL-13 induced protection of ECs from killing by complement or melittin despite loss of LDH in amounts similar to control ECs. However, the cytokine-treated ECs that were protected from killing rapidly regained effective control of membrane permeability. Moreover, the viability of the protected ECs was maintained for at least 2 days.
Conclusion
We conclude that the protection induced by IL-4/IL-13 in ECs against lethal attack by complement or melittin is effective and durable despite severe initial impairment of membrane permeability. The metabolic changes responsible for protection allow the cells to repair the membrane injury caused by complement or melittin.
Keywords: complement, melittin, endothelial cells, cytokines, cytotoxicity, xenotransplantation
Introduction
Vascular ECs are known to perform multiple critical functions and their response to stimuli that affect their functional integrity play a central role in inflammation, ischemia/reperfusion injury, and allograft and xenograft injury. In these conditions products of complement activation are known to often cause EC injury and hence it may be desirable to interfere with complement activation or to enhance the protection of the cells against complement attack.
In models of xenotransplantation where complement is a mediator of injury we have previously shown that IL-4 and IL-13 are efficient inducers of protection of porcine ECs from injury caused by the membrane attack complex (MAC) of complement, and also from the membrane pore-former protein melittin [1,2]. In an ex vivo model of xenotransplantation we have also reported protection of the endothelium from complement-mediated destruction in pig arteries that were transduced with adenoIL4 and then perfused with human blood [1]. In our previous studies we have also found that the protection induced with IL-4 and IL-13 was intrinsic and required activation of fatty acid and phospholipid synthesis, including cardiolipin, and preservation of mitochondrial integrity [3,4]. In addition, we found that IL-4 induced up-regulation of the junction protein claudin-5, which contributed to EC protection from complement injury [5].
In our previous studies, we defined ECs as “alive” (or “viable”) if they retained the ability to take up the vital dye neutral red [6] after complement or melittin treatment. However, despite the IL-4-induced resistance, the complement-treated ECs were found to lose control of membrane permeability assessed with the small molecule calcein to the same extent as cells not pre-treated with IL-4 [3]. Therefore it became important to ascertain whether large cytoplasmic proteins were also lost through the initial membrane disruption caused by complement and, if so, whether this injury was repaired promptly in the IL-4-pretreated ECs that survived the complement attack. Moreover, we also sought to determine whether the protection caused by IL-4 was durable. For these studies we investigated membrane permeability using the large cytoplasmic protein lactate dehydrogenase (LDH) [7] in IL-4-protected and control ECs incubated with complement and, for comparison, melittin. LDH release has often been used to quantify cell death caused by various agents [8]. In the current study we found that despite initial loss of cytoplasmic LDH, the IL-4-pretreated ECs are protected from killing long-term, with no initial impairment of the formation of large complement and melittin trans-membrane channels.
Materials and Methods
Reagents and endothelial cells
Reagents were as previously described [1,3]. We explanted ECs from pig aortae, cultured and identified them as previously described [2,9]. We performed experiments with cells in passages 3 to 7, 2 to 3 days post-confluence, in gelatin-coated 48-well tissue culture plates for EC killing and LDH assays. All incubations were carried out at 37°C in a 5% CO2 atmosphere.
Treatment of ECs with cytokines and killing assay
We incubated ECs with IL-4 or IL-13 for 48 h in 1% FBS-DMEM [2]. After washing with RPMI-1640 containing 0.5% BSA, ECs were incubated for 2 h with 150 μl of a human serum pool in RPMI-1640 at 7.5% as source of anti-pig antibodies and complement [2,3], or 0.1% Triton-X100 for 100% cell lysis. In other experiments, instead of human serum, we used melittin diluted in PBS, or NP-40 or Triton X100 diluted in RPMI-1640. In some experiments, we removed the complement solution after 2 h of incubation and washed the cells twice with 1% FBS-DMEM. Then, we added 250 μl of 1% FBS-DMEM medium and continued the incubation to complete 48 h. After washing, we tested the ECs for viability and permeability. We measured EC viability using the vital dye neutral red and calculated % specific killing as described previously [2,10]. Values are given as mean ± SE of triplicate samples. We previously established the validity of this assay to measure necrotic cell death caused by human complement at the serum concentrations and incubation times used in our current study [2].
Membrane permeability assay
We assessed EC membrane permeability using LDH released into the supernatants as OD units with a LDH assay kit, according to manufacturer’s instructions (Roche). We calculated LDH release as follows: % LDH release = (OD sample – OD reagent blank)/(OD Triton X100 maximum release – OD Triton X100 blank). Values are given as means ± SE of triplicate samples. We validated the measurements of LDH released in supernatants from cells that were incubated up to 48 h by showing that LDH, either released from ECs with Triton X100 or added extrinsically as a purified enzyme to EC monolayers or incubated alone in medium for various time periods up to 48 h, retained its full enzymatic activity.
Statistical analysis
Results are shown as means ± S.E.M. of 3-4 independent experiments that were performed in triplicate. P-values were obtained using a two-sample T-test assuming equal variance.
Results
IL-4 and IL-13 induce protection of ECs from complement-mediated killing despite loss of membrane permeability control
In our previous studies on EC protection from complement we assessed cell death using a viability assay based on the ability of live cells but not dead cells to take-up the dye neutral red. The protected ECs however demonstrated loss of membrane permeability when analyzed with the small molecule calcein (M = 622 D) to a degree similar to control ECs [3]. Therefore, given that complement causes cell death through channel formation with impairment of membrane permeability resulting in loss of cytoplasmic molecules and ultimately cell necrosis [11,12], we now assessed membrane permeability by measuring the release of the large tetrameric cytoplasmic protein LDH (M = 142,000 D [7]). We found that ECs pretreated with IL-4 or IL-13, while strongly protected from killing by complement assessed with neutral red, had similar loss of LDH as control cells over a wide range of cytokine concentrations (Fig. 1).
Fig. 1.
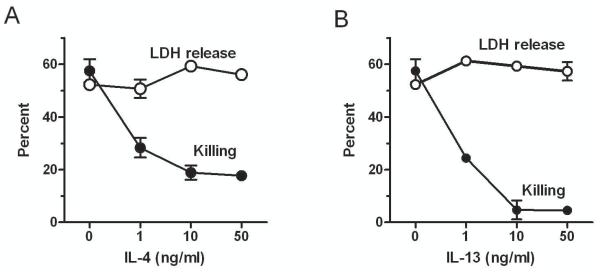
IL-4- and IL-13-protected ECs and control ECs exhibit similar loss of permeability control upon incubation with complement. ECs were incubated with various doses of IL-4 (A) or IL-13 (B) for 48 h, washed and incubated with 7.5% human serum for 2 h. Then the % LDH released in the supernatants and the % killing (cells that failed to take up neutral red) were measured. Results are means ± S.E.M. of 4 experiments (A) and 3 experiments (B) that were performed in triplicate. LDH release vs. killing, p < 0.01 for all doses of IL-4 and IL-13 tested.
The IL-4-treated ECs that are resistant to killing by complement rapidly regain efficient control of membrane permeability
We then investigated the kinetics of LDH loss during EC incubation with complement and found that the LDH loss progressed in a similar manner in IL-4-treated ECs and in untreated controls (Fig. 2A). The LDH loss was markedly reduced after 2 h of incubation in both experimental and control cells. This kinetic study shows that the additional release at 2 h compared to 90 min is very small for both experimental and control cells. For direct comparison with cell killing, experiments run simultaneously in which the degree of killing was measured with neutral red showed that the IL-4-treated ECs were strongly protected from complement in comparison to control ECs (Fig. 2B). These results suggest that in cells pretreated with IL-4 membrane permeability to LDH is fully regained after 2 h of incubation with complement, indicating that the membrane lesions have resealed.
Fig. 2.
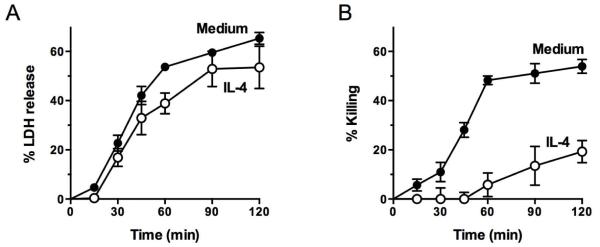
ECs treated with IL-4 followed by complement that are resistant to killing rapidly regain efficient control of membrane permeability. ECs were incubated with 10 ng/ml IL-4 or medium alone for 48 h, washed, and incubated with 7.5% human serum for the indicated times. Then % LDH release (A) and % killing using neutral red (B) were measured. Results are means ± S.E.M. of 3 independent experiments that were performed in triplicate. In (A) LDH release, IL-4 vs. medium p > 0.05 for all time points except at 60 min p < 0.05. In (B) % killing, IL4 vs. medium p < 0.01 for all time points except at 15 min and 30 min p < 0.05.
The viability of the ECs that are protected from complement by IL-4 is durable
We then asked whether complement attack on IL-4-protected cells had any effect on long-term membrane permeability. We treated ECs with human complement for 2 h and, after washing, either tested them immediately or after 48 h of incubation in medium without complement. In both instances we measured LDH in the supernatants and EC neutral red uptake. We found that LDH loss stopped after 2 h of incubation with complement in IL-4-treated ECs, as well as in the control ECs that had survived complement attack, with no additional LDH loss during 48 h of incubation in medium (Fig. 3). As indicated in Materials and Methods, after 48 h of incubation LDH activity is fully measurable by our assay method. Similar to LDH release, EC killing also stopped after 2 h of incubation with complement. As in experiments described above, after 2 h of incubation with complement there was markedly reduced cell killing with IL-4 pretreatment and only a slight reduction of LDH release. Thus, this experiment indicates that the IL-4-protected ECs are able to remain alive long-term after recovery from complement attack.
Fig. 3.
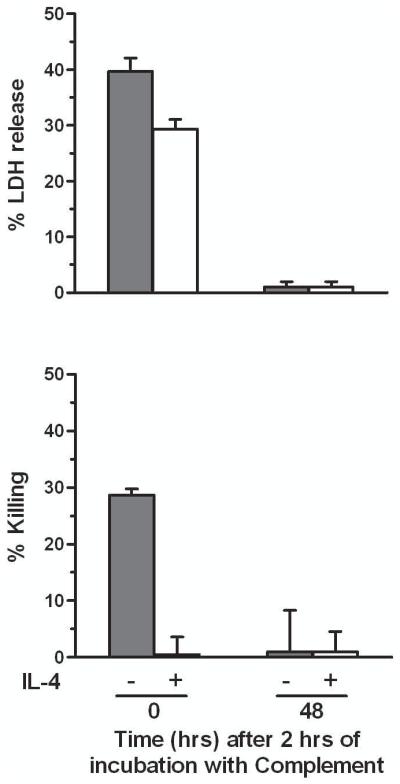
The viability of ECs protected from complement by pretreatment with IL-4 is durable and after 2 h of incubation with complement the ECs stop releasing LDH. ECs were incubated with 10 ng/ml IL-4 or medium for 48 h, washed, and then incubated with 7.5% human serum for 2 h. At this point the cells were either tested for LDH release and neutral red uptake (Killing) or washed and placed in 1% FBS-DMEM for an additional 48 h and then tested for LDH release and neutral red uptake. Results are means ± S.E.M. of 2 independent experiments that were performed in triplicate. After 2 h in human serum, IL-4-treated ECs vs. medium control ECs, for % LDH release p < 0.05 and for % killing p < 0.001; medium control ECs % LDH release vs. % killing p < 0.05. After 2 h in human serum plus 48 h in medium alone, IL-4-treated ECs vs. medium control, p > 0.05 for both % LDH release and % killing.
IL-4 and IL-13 also induce protection of ECs from killing by melittin despite loss of membrane permeability control
Like the complement TP, the protein melittin is a channel-former that causes necrotic cell death [13] and therefore we have previously used melittin in studies of IL-4/IL-13 EC protection. We now asked whether this protection would also allow the release of LDH in the face of protection from killing. We found that ECs pretreated with IL-4 or IL-13, while protected from killing by melittin assessed with neutral red, had similar LDH loss as control cells (Fig. 4). These results suggest that cytokine-induced protection from killing by the pore-former melittin, like the protection from complement, also develops despite initial disruption of membrane permeability.
Fig. 4.

IL-4 and IL-13 induced protection against killing of ECs by melittin but did not prevent melittin-induced loss of permeability control. ECs were incubated with medium alone or with IL-4 or IL-13 at 10 ng/mL for 48 h, and then tested for sensitivity to killing by 2.5 μM melittin for 2 h. EC killing was determined by neutral red uptake and permeability was measured by LDH release. Results are means ± S.E.M. of 3 experiments that were performed in triplicate. LDH release vs. killing for IL-4- and IL-13-treated, p < 0.001.
Discussion
In this study, we investigated control of EC permeability in cells that are protected from killing by complement or melittin as a consequence of pre-treatment with IL-4 or IL-13. We report here that the protected ECs, defined by their ability to take-up the vital dye neutral red, exhibit a severe but transient loss of permeability control, defined by the release of the large cytoplasmic protein LDH. After 2 h of incubation with complement there was no additional LDH leakage, both in cytokine-treated cells and in a small percentage of control cells that were not killed by the amount of complement used in these experiments. After 48 h of incubation in medium without complement the unlysed cells exhibited normal uptake of neutral red indicating that the protected cells exhibited full and durable viability. These observations demonstrate that the cytokine-treated cells repaired their complement-induced lesions, a process known as resealing [14]. In contrast, most ECs that were not pre-treated with IL-4 and had lost LDH during the 2 h of incubation with complement failed to repair their functional lesions and died.
Central to the interpretation of our findings of cytokine-induced protection from killing by complement is the ability of the terminal pathway of complement (TP) to form membrane permeability channels [11,12]. These channels are caused by the TP proteins C8 and C9 through domains that penetrate cell membranes [15,16]. In our current study we asked whether IL-4 and IL-13 induced protection from complement by impairing the ability of the TP to form large channels that cause major disruptions of membrane permeability. However, we found similar amounts of LDH released from ECs incubated with complement, whether or not the cells were pretreated with the cytokines, suggesting that these cytokines do not induce protection from killing by impairing the channel-forming ability of the TP. Our results using LDH shows that the extent of membrane disruption caused by the TP in cytokine-treated ECs is similar to that in untreated controls. Moreover, we have previously reported that pretreatment of ECs with IL-4 does not increase the membrane expression of complement regulators CD46, CD55 or CD59 and that incubation of those cells with complement results in similar binding of C3 and C5b-9 as controls not treated with IL-4 [1,2]. After 2 h of incubation with complement there was slightly less bound MAC on IL-4-treated cells than in controls [3], supporting our interpretation of membrane resealing discussed below.
LDH release has often been used in studies of necrotic cell death [8]. Our study, however, demonstrates that LDH release should not be used in isolation to assess cell death. Given that the ECs pre-treated with IL-4/IL-13 and then incubated with complement or melittin have shown similar levels of LDH losses as cells not pre-treated with the cytokines, it would not have been possible for us to discover the protective effect had we only used LDH release to assess cell death. We interpret the apparently discordant results obtained with LDH and neutral red as caused by membrane resealing, a process that must be addressed in studies of cell cytotoxicity mediated by pore-forming mechanisms.
In many studies to induce cytoprotection using in vitro models of xenotransplantation in which protection was assessed using only membrane permeability assays such as the release of LDH the results conclusively show that protection had been achieved [17-20]. On the other hand results from other studies employing only those assays appear to demonstrate failure of induction of protection [21]. Our current study, however, suggests that to ensure that those negative results were not due to resealing of the MAC lesions, measurement of true cell survival would be required. Therefore for these situations we suggest using the neutral red assay in conjunction with the LDH assay. Neutral red is a supravital dye that is well qualified to measure live cells since its uptake requires intact membranes and then it concentrates in the lysosomes, where it is retained [6]. The LDH assay is based solely on the loss of membrane permeability control. Together, these assays are well suited for studies of cytotoxicity on ECs arranged in monolayers. Monolayers, in contrast to ECs in suspension, mimic the anatomic organization of the ECs in the vascular system and have an intercellular junction that plays a role in IL-4-induced protection [5].
We have previously reported that protection induced by IL-4/IL-13 against complement and melittin is due to activation of intracellular mechanisms, as summarized in Table 1. We have shown that the IL-4-induced changes in mitochondrial membrane lipid composition preserve mitochondrial integrity during complement attack, allowing the cell to maintain its metabolism while the channels produced by the MAC in the plasma membrane are being repaired [3]. The resealing mechanism consists mainly of loss of membrane-bound MAC. IL-4 induced up-regulation of the junction protein claudin-5 may also contribute to protection.
Table 1.
Summary of changes induced by IL-4 in ECs that allow resealing of complement-induced membrane lesions and resistance against killing (based on refs. 2, 4 and 5)
| Stimulation of phospholipid synthesis resulting in: |
| Preserved mitochondrial ultrastructure |
| Preserved mitochondrial electrical conductance |
| Reduced loss of cellular ATP |
| Up-regulation of intercellular junction protein claudin-5 resulting in: |
| Decreased paracellular permeability |
| Reduced decrease in electrical membrane resistance caused by complement |
Of additional interest, we have previously reported that human vascular ECs, like porcine ECs, can also be made resistant to killing by complement when pretreated with IL-4 or IL-13 [3], implying that this induced protection is not limited to porcine ECs. Moreover, we have previously established that the protection of porcine ECs is not restricted to maintaining cell viability in the face of complement attack but can be extended to preserve several aspects of monolayer endothelial structure and function. Thus IL-4-treated ECs were protected from cell retraction and gap formation following incubation with sublytic doses of human complement [1], a protection that is due to IL-4-induced up-regulation of the intercellular adhesion molecule claudin-5, which also contributed to protection from complement-mediated cytotoxicity [5]. IL-4-treated ECs were also protected from apoptosis induced by TNF-α [22]. Our unpublished studies have also demonstrated that these cytokines induced protection of ECs against responses to activation and pro-inflammatory stimuli, including E-selectin expression induced by LPS or TNF-α, production of IL-1α by activation with sublytic complement, and expression of tissue factor induced with TNF-α or LPS. Since these multiple protective effects are of clinical interest, further delineation of the mechanisms mediating IL-4-induced protection may provide new strategies for preventing complement-mediated vascular injury as it occurs in vascular disease, such as atherosclerosis, ischemia reperfusion injury, and vasculopathy as observed in vascularized allograft and xenograft rejection.
Acknowledgments
This work was supported by Grant RO1 HL062195 from the National Institutes of Health (APD). We gratefully acknowledge Bjorn Batdorf and Megan Schott for their expert assistance in these studies.
Abbreviations
- EC/s
endothelial cell/s
- LDH
lactate dehydrogenase
- TP
terminal pathway of complement
- MAC
membrane attack complex of complement
References
- 1.Black SM, Grehan JF, Rivard AL, et al. Porcine endothelial cells and iliac arteries transduced with AdenoIL-4 are intrinsically protected, through Akt activation, against immediate injury caused by human complement. J Immunol. 2006;177:7355–63. doi: 10.4049/jimmunol.177.10.7355. [DOI] [PubMed] [Google Scholar]
- 2.Grehan JF, Levay-Young BK, Fogelson JL, et al. IL-4 and IL-13 induce protection of porcine endothelial cells from killing by human complement and from apoptosis through activation of a phosphatidylinositide 3-kinase/Akt pathway. J Immunol. 2005;175:1903–10. doi: 10.4049/jimmunol.175.3.1903. [DOI] [PubMed] [Google Scholar]
- 3.Black SM, Schott ME, Batdorf BH, et al. IL-4 induces protection of vascular endothelial cells against killing by complement and melittin through lipid biosynthesis. Eur J Immunol. 2010;40:803–12. doi: 10.1002/eji.200939488. [DOI] [PubMed] [Google Scholar]
- 4.Black SM, Schott ME, Benson BA, et al. Interleukin-4 induces lipogenesis in porcine endothelial cells, which in turn is critical for induction of protection against complement-mediated injury. Transplant Proc. 2008;40:638–40. doi: 10.1016/j.transproceed.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalmasso AP, Goldish D, Benson BA, et al. Interleukin-4 induces up-regulation of endothelial cell claudin-5 through activation of FoxO1: role in protection from complement-mediated injury. J Biol Chem. 2014;289:838–47. doi: 10.1074/jbc.M113.455766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–31. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 7.Jaenicke R, Knof S. Molecular weight and quaternary structure of lactic dehydrogenase. 3. Comparative determination by sedimentation analysis, light scattering and osmosis. Eur J Biochem. 1968;4:157–63. doi: 10.1111/j.1432-1033.1968.tb00187.x. [DOI] [PubMed] [Google Scholar]
- 8.Fotakis G, Timbrell JA. In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett. 2006;160:171–7. doi: 10.1016/j.toxlet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Dalmasso AP, Benson BA, Johnson JS, et al. Resistance against the membrane attack complex of complement induced in porcine endothelial cells with a Gal alpha(1-3)Gal binding lectin: up-regulation of CD59 expression. J Immunol. 2000;164:3764–73. doi: 10.4049/jimmunol.164.7.3764. [DOI] [PubMed] [Google Scholar]
- 10.Dalmasso AP, He T, Benson BA. Human IgM xenoreactive natural antibodies can induce resistance of porcine endothelial cells to complement-mediated injury. Xenotransplantation. 1996;3:54–62. [Google Scholar]
- 11.Dalmasso AP, Benson BA. Pore siz of lesions induced by complement on red cell membranes and its relation to C5b-8, C5b-9 and poly C9. In: Podack ER, editor. Cytolytic lympphocytes and complement: Effectors of the immune system. CRC Press; Boca Raton FL: 1988. pp. 207–19. [Google Scholar]
- 12.Muller-Eberhard HJ. The membrane attack complex of complement. Springer Sem Immunopath. 1984;7:227–75. doi: 10.1007/BF01893017. [DOI] [PubMed] [Google Scholar]
- 13.Laine RO, Morgan BP, Esser AF. Comparison between complement and melittin hemolysis: anti-melittin antibodies inhibit complement lysis. Biochemistry. 1988;27:5308–14. doi: 10.1021/bi00414a054. [DOI] [PubMed] [Google Scholar]
- 14.Walev I, Hombach M, Bobkiewicz W, et al. Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J. 2002;16:237–9. doi: 10.1096/fj.01-0572fje. [DOI] [PubMed] [Google Scholar]
- 15.Hadders MA, Beringer DX, Gros P. Structure of C8alpha-MACPF reveals mechanism of membrane attack in complement immune defense. Science. 2007;317:1552–4. doi: 10.1126/science.1147103. [DOI] [PubMed] [Google Scholar]
- 16.Rosado CJ, Buckle AM, Law RH, et al. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–51. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 17.Contreras JL, Bilbao G, Smyth C, et al. Gene transfer of the Bcl-2 gene confers cytoprotection to isolated adult porcine pancreatic islets exposed to xenoreactive antibodies and complement. Surgery. 2001;130:166–74. doi: 10.1067/msy.2001.115828. [DOI] [PubMed] [Google Scholar]
- 18.Heckl-Ostreicher B, Binder R, Kirschfink M. Functional activity of the membrane-associated complement inhibitor CD59 in a pig-to-human in vitro model for hyperacute xenograft rejection. Clin Exp Immunol. 1995;102:589–95. doi: 10.1111/j.1365-2249.1995.tb03857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Denderen BJ, Pearse MJ, Katerelos M, et al. Expression of functional decay-accelerating factor (CD55) in transgenic mice protects against human complement-mediated attack. Transplantation. 1996;61:582–8. doi: 10.1097/00007890-199602270-00012. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto A, Ikeda K, Wang D, et al. Trial using pig cells with the H-D antigen knocked down. Surg Today. 2013;43:782–6. doi: 10.1007/s00595-012-0274-x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang C, Wang L, Zhong S, et al. Over-expression of heme oxygenase-1 does not protect porcine endothelial cells from human xenoantibodies and complement-mediated lysis. J Huazhong Univ Sci Technolog Med Sci. 2013;33:102–6. doi: 10.1007/s11596-013-1079-x. [DOI] [PubMed] [Google Scholar]
- 22.Black SM, Benson BA, Idossa D, et al. Protection of porcine endothelial cells against apoptosis with interleukin-4. Xenotransplantation. 2011;18:343–54. doi: 10.1111/j.1399-3089.2011.00678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]