

Abstract
Patient-derived induced pluripotent stem cells (iPSCs) are valuable tools for the study of developmental biology and disease modeling. In both applications, genetic correction of patient iPSCs is a powerful method to understand the specific contribution of a gene(s) in development or diseased state(s). Here, we describe a protocol for the targeted integration of a doxycycline-inducible transgene expression system in a safe harbor site in iPSCs. Our gene targeting strategy uses zinc finger nucleases (ZFNs) to enhance homologous recombination at the AAVS1 safe harbor locus, thus increasing the efficiency of the site-specific integration of the two targeting vectors that make up the doxycycline-inducible system. Importantly, the use of dual-drug selection in our system increases the efficiency of positive selection for double-targeted clones to >50 %, permitting a less laborious screening process. If desired, this protocol can also be adapted to allow the use of tissue-specific promoters to drive gene expression instead of the doxycycline-inducible promoter (TRE). Additionally, this protocol is also compatible with the use of Transcription-Activator-Like Effector Nucleases (TALENs) or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 system in place of ZFNs.
Keywords: Genetic correction, Disease modeling, Homologous recombination, Doxycycline-inducible expression, Gene targeting, Zinc finger nucleases
1 Introduction
In the characterization of iPSC disease models, a commonly used strategy is to genetically manipulate the patient-derived iPSCs to correct the disease phenotype, thus ascertaining the role of a specific gene in the disease. One way to genetically correct defects in iPSC-based disease models is by the site-specific targeting of transgenes into a safe harbor locus. In human iPSCs, the AAVS1 gene has been identified as a putative safe harbor locus (1–4), where we have expressed a number of transgenes (5, 6).
This chapter describes a protocol for the generation of iPSC lines with a doxycycline-inducible transgene expression system targeted into both alleles of the AAVS1 safe harbor locus. To enhance homologous recombination (7, 8), a pair of ZFNs are used to induce a double-stranded break specifically in the AAVS1 locus. The two targeting vectors with arms of homology to AAVS1 express reverse tet activator (rtTA) driven by a constitutive promoter and a tet-response element (TRE) driving the gene of interest (Fig. 1). If knock down of a gene is desired, short hairpins directed against the gene of interest can be cloned downstream of the TRE in the targeting construct. The two targeting vectors are designed to confer dual-drug resistance to double-targeted clones. Clones are screened using PCR to confirm the integration of the targeting vectors and Southern blotting to identify the presence of off-target integration. With the use of dual drug selection, on average >50 % of selected clones are correctly double-targeted (Table 1). The entire process takes about 3 weeks, excluding the time needed for the clones to expand (Fig. 2). This protocol can also be adapted to accommodate the use of TALENs (8, 9) or (CRISPR)-Cas9 system (8, 10–12) in place of ZFNs.
Fig. 1.

Design of targeting vectors. ZFNs specific to the AAVS1 locus will cut the DNA in intron 1 of AAVS1. Integration of both targeting vectors will confer dual drug resistance to ESCs/iPSCs. The location of PCR screening primers and Southern blot probe is indicated
Table 1. Percentage of selected clones that are correctly double-targeted from three independent transfections.
| Transfection attempt | Number of clones screened | Number of double-targeted clones | Percentage of double-targeted clones (%) |
|---|---|---|---|
| 1 | 12 | 8 | 67 |
| 2 | 6 | 5 | 83 |
| 3 | 10 | 5 | 50 |
| Overall | 28 | 18 | 64 |
Fig. 2. Schematic outlining the processes involved in the generation of a doxycycline-inducible transgene expression system in hESCs/iPSCs.

As a proof of concept, we have generated a doxycycline-inducible human embryonic stem cell (hESC) line expressing GFP. This line was generated using an earlier version of this protocol, which utilized single drug selection. We have differentiated this line into hematopoietic progenitors or pancreatic beta cells and observed that GFP expression is visible 2 days after the addition of doxycycline and is maintained as long as doxycycline is present in the system. Importantly, the expression of GFP is proportional to the amount of doxycycline added and the length of time doxycycline is present in the medium (Fig. 3). This implies that we can induce a suitable, physiological level of transgene expression by titrating different doses of doxycycline. Since then, we have developed an improved protocol described here, which uses dual drug selection to increase the specificity of selection and decrease the need to screen large numbers of clones.
Fig. 3.
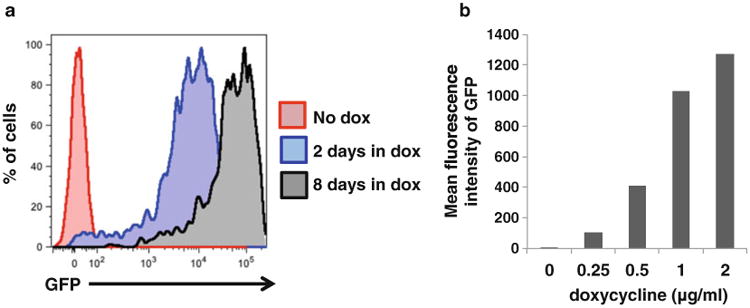
GFP expression in hematopoietic progenitors differentiated from hESC line with doxycycline-inducible GFP expression under different conditions. (a) GFP expression in hematopoietic progenitors treated with doxycycline for different lengths of time. Untreated hematopoietic progenitors (red) do not express GFP. Treatment with doxycycline for 2 days during differentiation is sufficient to induce GFP expression in hematopoietic progenitors (blue). When doxycycline was given for 8 days, the induction of GFP expression is stronger (black), suggesting that transgene expression does not get silenced when hESCs undergo hematopoietic differentiation. (b) The level of GFP expression in hematopoietic progenitors increases with the concentration of doxycycline added
2 Materials
2.1 Reagents and Supplies
2.1.1 Vectors Encoding Zinc Finger Nucleases Specific to the AAVS1 Locus
PGK-AAVS1-ZFN-Left (addgene cat. no. #60915).
PGK-AAVS1-ZFN-right (addgene cat. no. #60916).
2.1.2 Targeting Vectors To Be Integrated into the AAVS1 Locus
AAVS1-SA-2A-NEO-CAG-RTTA3 (addgene cat. no. #60431).
AAVS1-SA-2A-PURO-TRE-eGFP (addgene cat. no. #22074) (GFP can be replaced by any gene of interest).
2.1.3 Transfection Reagent
Lipid transfection reagent: X-tremeGENE 9 DNA transfection reagent (Roche Cat. no. 06 365 787 001).
2.1.4 Cell Culture Media and Reagents
Dulbecco's Modified Eagle's Medium/Ham's F12 50/50 mix (DMEM/F12 50/50) (Corning Cellgro Cat. no. 10-092-CV).
Knockout™ Serum Replacement (Knockout™ SR) (Life Technologies Cat. no. 10828-028).
Penicillin Streptomycin Solution (Pen-Strep), 100× (Corning Cellgro Cat. no. 30-002-Cl).
l-Glutamine Solution, 100× (Corning Cellgro Cat. no. 25-005-Cl).
MEM Nonessential Amino Acids (NEAA), 100× (Life Technologies Cat. no. 11140-050).
2-Mercaptoethanol (55 mM) (Life Technologies Cat. no. 21985-023).
Iscove's Modification of DMEM (IMDM) (Corning Cellgro Cat. no. 10-016-CV).
PES Sterilizing 0.22 μm Filter System, Low Protein binding (250 ml) (Corning Cat. no. 431096).
Dimethyl sulfoxide (DMSO) (Sigma Cat. no. D2650).
Fetal Bovine Serum (Tissue Culture Biologicals Cat. no. 101).
TrypLE™ Express (1×), phenol red (Life Technologies Cat. no. 12605-010).
Trypsin-EDTA (0.25 %), phenol red (Life Technologies Cat. no. 25200-056).
Rock inhibitor Y-27632 dihydrochloride (TOCRIS Cat. no. 1254).
Basic Fibroblast Growth Factor (bFGF) (R&D Systems Cat no. 233-FB-025).
Matrigel Matrix, Growth Factor Reduced (Corning Cat. no. 354230).
Irradiated drug resistant (DR4) mouse embryonic fibroblasts (MEFs) (1.2 × 106/vial) (ATCC® SCRC-1045.1) (see Note 1).
Irradiated CF-1™ MEFs (1.2 × 106/vial) (ATCC® SCRC-1040.1).
Geneticin®/Neomycin (G418) (Life Technologies Cat. no. 10131035).
Puromycin dihydrochloride (Sigma Cat. no. P8833-25MG).
2.1.5 Molecular Biology Reagents
PureLink® Genomic DNA Mini Kit (Invitrogen Cat. no. K1820-01).
Platinum® Blue PCR SuperMix (Invitrogen Cat. no. 12580-015).
-
Screening primers reconstituted to 100 μM.
- Primer set WT
- WT-F: 5′ CCC CTA TGT CCA CTT CAG GA 3′
- WT-R: 5′ CAG CTC AGG TTC TGG GAG AG 3′
- Primer set AAVS1-CAG
- AAVS1-CAG-F: 5′ GAG CAT CTG ACT TCT GGC TAA TA 3′
- AAVS1-CAG-R: 5′ GAA GGA TGC AGG ACG AGA AA 3′
- Primer set AAVS1-TRE
- AAVS1-TRE-F: 5′ GCA ATA GCA TCA CAA ATT TCA C 3′
- AAVS1-TRE-R: Same as AAVS1-CAG-R
Ultrapure Agarose (Invitrogen Cat. no. 16500-500).
1 kb Plus DNA ladder (Invitrogen Cat. no. 10787-018).
2.1.6 Southern Blot Reagents
Probe: 480 bp fragment from the digestion of AAVS1-SA-2A-PURO plasmid (addgene cat. no. #22075) with BamHI restriction enzyme.
- Restriction enzymes:
- SphI-HF® (New England Biolabs Inc. Cat. no. R3182S).
- BamHI (New England Biolabs Inc. Cat. no. R0136S).
Standard Southern blotting reagents.
2.2 Cell Culture Media
hESC medium—DMEM/F12 50/50, 1 % l-Glutamine, 1 % Pen-Strep, 1 % NEAA, 15 % Knockout™ SR, 100 μM 2-ME. Add reagents together in a PES Sterilizing 0.22 μm Filter System. Filter sterilize. Add bFGF to a final concentration of 5 ng/ml. Store at 4 °C for up to 10 days.
MEF medium—IMDM, 10 % serum, 1 % Pen-Strep, 1 % l-Glutamine. Add reagents together in a PES Sterilizing 0.22 μm Filter System. Filter sterilize. Store at 4 °C for up to a month.
3 Methods
Two days prior to transfection.
3.1 Preparation of 1:3 Matrigel/DR4 MEF Plates
– Thaw Matrigel on ice or overnight at 4 °C. Dilute 1:3 in coldIMDM in pre-cooled tubes on ice. Extra 1:3 Matrigel can bealiquoted and stored at —20 °C (see Note 2).
– Pre-cool a 6-well plate and 2 ml pipettes in the —20 °C freezerfor 10 min.
– Coat the pre-cooled 6-well plate with diluted 1:3 Matrigel onice using cold 2 ml pipettes. The whole surface should becompletely covered with a thin coat of Matrigel.
– Incubate the plate on ice for 20 min.
– Aspirate excess 1:3 Matrigel from the wells.
– Incubate the 1:3 Matrigel-coated plate at 37 °C for at least30 min, up to 4 h.
– Thaw one vial of irradiated DR4 MEFs (1.2 × 106/vial) in the37 °C water bath.
– Add to 10 ml of IMDM in a 50 ml tube. Centrifuge 335 × g for 3 min. Aspirate and resuspend the cell pellet in 12 ml of MEF medium.
– Plate MEFs on the 1:3 Matrigel-coated 6-well plate at2 ml/well.
One day prior to transfection.
3.2 Passaging iPSCs onto 1:3 Matrigel/DR4 MEFs for Transfection
– Aspirate hESC medium from wells of iPSCs to be split for transfection (see Note 3).
– Add 1 ml/well of TrypLE (warmed to room temperature), incubate for 3–4 min in the hood, and aspirate.
– Add 1 ml IMDM/well, incubate for 1 min, and repeat wash step.
– Add 1 ml/well hESC medium containing rock inhibitor (10 μM).
– Remove iPSCs using a cell scraper.
– Wash 1:3 Matrigel/DR4 MEF plates two times using 1 ml/well IMDM per well, and plate cells at a split ratio of 1:4 in hESC medium containing rock inhibitor (10 μM) (see Note 4).
Day of transfection.
3.3 Medium Change to Remove Rock Inhibitor
– Add 2 ml/well hESC media without rock inhibitor to cells first thing in the morning.
3.4 Transfection
– Add ZFN, TRE, and rtTA vectors to 1.5 ml Eppendorf tube bringing the total volume to 100 μl per tube using IMDM.
– Add XtremeGENE 9 DNA transfection reagent to IMDM/DNA mixture using 3 μl for every 1 μg of DNA.
– Mix the reagents by gently flicking the tube and incubate atroom temperature for 20 min.
– Add the 100 μl of IMDM/DNA/lipid mixture to cellsdropwise.
– Gently shake the plate to distribute the mixture uniformly inthe well.
– Incubate for 16 h at 37 °C 5 % CO2, 5 % O2, and 90 % N2.
– Aspirate transfection mixture and add 2 ml/well hESCmedium.
– See Notes 5 and 6 for transfection troubleshooting.
3.5 Selection of Transfected Colonies
Start puromycin selection (0.5 μg/ml in hESC medium) 48 h after transfection. Feed cells with fresh hESC medium containing puromycin (2 ml/well) every day for 2 days.
Feed cells with fresh hESC medium without drugs (2 ml/well) for 2 days.
Start neomycin selection (40 μg/ml in hESC medium). Feed cells with hESC medium containing neomycin (2 ml/well) every day for 8–10 days (see Notes 7–9)
The day before picking clones, prepare 12-well plates containing 1:3 Matrigel/CF-1 MEFs.
Pick colonies into 12-well plates containing 1:3 Matrigel/CF-1 MEFs and allow colonies to grow until 80–90 % confluent (see Notes 10 and 11).
Passage the clones when 80–90 % confluent. Plate 25 % of the cells into 1 well of a 5-well plate and use the remaining 75 % for genomic DNA extraction.
Expand and freeze individual clones (see Note 12).
3.6 PCR Screening of Double-Targeted Clones
|
| |
| Reagent | Amount/volume |
|
| |
| Platinum® Blue PCR SuperMix | 20 μl |
|
| |
| Forward primer (10 μM) | 1 μl |
|
| |
| Reverse primer (10 μM) | 1 μl |
|
| |
| Genomic DNA | 100 ng (up to 5 μl) |
|
| |
Perform PCRs using primer sets WT, AAVS1-CAG, and AAVS1-TRE listed in Sect. 2.1.5, item 3.
- PCR cycle:
- – Step 1—95 °C for 10 min
- – Step 2—95 °C for 30 s
- – Step 3—55 °C for 30 s
- – Step 4—72 °C for 2 min
- – Repeat steps 2-4 for 35 cycles
- Analyze PCR products on an ethidium bromide (EtBr) agarosegel for the following predicted band sizes:
- – WT (AAVS1 locus)—500 bp
- Presence of a WT band indicates that the clone is heterozygous, thus not a double-targeted clone.
- – AAVS1-CAG-rtTA—1,920 bp
- Presence of an AAVS1-CAG-rtTA band indicates CAG-RTTA plasmid integration at the AAVS1 locus
- – AAVS1-TRE—1,380 bp
- Presence of an AAVS1-TRE band indicates TRE-GFP plasmid integration at the AAVS1 locus
3.7 Southern Blotting of Double-Targeted Clones to Reveal Off-Target Integrations
Digest genomic DNA isolated from double-targeted clones with SphI-HF® restriction enzyme.
Run digested DNA on a 0.7 % agarose gel.
Transfer to membrane and probe using DNA fragment from AAVS1 vector backbone as described in Sect. 2.1.6.
- Southern blot results:
- – Nontargeted cells should result in a 6.5 kb fragment.
- – TRE target allele should result in a 3.4 kb fragment and CAG-rtTA in a 3.7 kb fragment.
3.8 Addition of Doxycycline to Induce Transgene Expression
As a proof-of-principle study, we generated a hESC line expressing doxycycline-inducible GFP. We differentiated the hESC line into hematopoietic progenitors, adding doxycycline (2 μg/ml) 2 days or 8 days before the emergence of the hematopoietic progenitors. The longer doxycycline was present in the media, the stronger the induction of GFP expression in the hematopoietic progenitors (Fig. 3a). The induction of GFP expression increases with the amount of doxycycline added (Fig. 3b). In addition, this proof-of-principle study establishes that the doxycycline system of inducing transgene expression does not get silenced through epigenetic mechanisms as iPSCs undergo hematopoietic differentiation, and thus is suitable for use in the study of blood disorders.
4 Notes
Irradiated drug resistant and normal MEFs are available commercially. Alternatively, MEFs can be expanded, irradiated, and frozen down at 1.2 × 106 cells/vial. Each vial is sufficient for one 6-well plate.
Keep tubes, plates, and pipette tips cold to prevent Matrigel from clumping. Frozen 1:3 Matrigel aliquots will thaw in a couple of hours or overnight at 4 °C.
iPSCs are ready to be split for transfection when they are 80-90 % confluent, with good morphology.
Passage cells the day before transfection and adjust split ratios to obtain small colonies that are 25–30 % confluent. Individual colonies should be small, as large colonies do not transfect well. Cells should be transfected the day after splitting as cells that were plated for longer than a day do not transfect well even if the confluency is right. We found a split ratio of 1:4 works best for our cell lines.
We recommend starting the transfection in the evening as prolonged incubation of cells with transfection reagent (>16 h) leads to less efficient transfection.
If you encounter the problem of one targeting vector integrating preferentially over the other, adjust the ratio of the two targeting vectors (see Table 2). If there are very few surviving clones after dual drug selection, we recommend transfecting more wells and/or increasing the amount of targeting vectors.
The doses for puromycin and neomycin work for several lines that we have tested. If there are problems with selection, we recommend doing a kill curve to titrate concentrations suitable for different iPSC lines.
We recommend a recovery period of 2 days after puromycin selection before starting neomycin selection. The colonies should be very small when you begin neomycin selection as neomycin takes 8–10 days to kill. If colonies are too large or growing too fast, the colonies will overgrow before neomycin takes effect. Decrease recovery period to a day for fast growing cell lines.
Some DR4 MEFs may die during the whole process of drug selection. Supplement MEFs to the plate if necessary.
Clones are ready to be picked when the colony occupies half the view when viewed under the 10× magnification.
To increase the growth rate of hESC/iPSCs, increase the concentration of bFGF in the media up to 20 ng/ml.
Freeze down one 90 % confluent well of a 6-well plate. To freeze iPSCs, split cells with TrypLE as described above in Sect. 3.2. Instead of scraping cells into hESC media, scrape cells into pre-cooled freezing media: 50 % fetal bovine serum, 40 % hESC media, 10 % DMSO. Cells in freezing media can be immediately transferred to the —80 °C freezer. For long-term storage, keep frozen cells in liquid nitrogen.
Table 2. Recommended concentrations of reagents and vectors for transfection.
| Reagent/vector | Well #1 | Well #2 | Well #3 |
|---|---|---|---|
| AAVS1-ZFN-Left | 0.2 μg | 0.2 μg | 0.2 μg |
| AAVS1-ZFN-Right | 0.2 μg | 0.2 μg | 0.2 μg |
| AAVS1-SA-2A-PURO-TRE-GFP | 1.5 μg | 1 μg | 3 μg |
| AAVS1-SA-2A-NEO-CAG-RTTA | 1.5 μg | 3 μg | 1 μg |
| IMDM | Top up to 100 μl total volume | Top up to 100 μl total volume | Top up to 100 μl total volume |
| XtremeGENE 9 | 10.2 μl | 13.2 μl | 13.2 μl |
| TRE-GFP:CAG-RTTA ratio | 1:1 | 1:3 | 3:1 |
| Total DNA | 3.4 μg | 4.4 μg | 4.4 μg |
Acknowledgments
This work was supported by NIH grant U01 HL099656.
References
- 1.DeKelver RC, Choi VM, Moehle E, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lombardo A, Cesana D, Genovese P, et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 4.Smith JR, Maguire S, Davis L, et al. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem cells. 2008;26:496–504. doi: 10.1634/stemcells.2007-0039. [DOI] [PubMed] [Google Scholar]
- 5.Tiyaboonchai A, Mac H, Shamsedeen R, et al. Utilization of the AAVS1 safe harbor locus for hematopoietic specific transgene expression and gene knockdown in human ES cells. Stem Cell Res. 2014;12:630–637. doi: 10.1016/j.scr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan SK, Mills JA, Koukouritaki SB, et al. High-level transgene expression in induced pluripotent stem cell-derived megakaryocytes: correction of Glanzmann thrombasthenia. Blood. 2014;123:753–757. doi: 10.1182/blood-2013-10-530725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian K, Huang C, Chen H, et al. A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cells. 2014;32:1230–1238. doi: 10.1002/stem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Liang D, Wang Y, et al. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Cho SW, Kim S, Kim JM, et al. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 12.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]