

Abstract
Nodal peripheral T-cell lymphomas are a rare group of neoplasms derived from post-thymic and activated T lymphocytes. A review of scientific articles listed in PubMed, Lilacs, and the Cochrane Library databases was performed using the term “peripheral T-cell lymphomas”. According to the World Health Organization classification of hematopoietic tissue tumors, this group of neoplasms consists of peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), angioimmunoblastic T-cell lymphoma (AITL), anaplastic large cell lymphoma-anaplastic lymphoma kinase positive (ALCL-ALK+), and a provisional entity called anaplastic large cell lymphoma-anaplastic lymphoma kinase negative (ALCL-ALK−). Because the treatment and prognoses of these neoplasms involve different principles, it is essential to distinguish each one by its clinical, immunophenotypic, genetic, and molecular features. Except for anaplastic large cell lymphoma-anaplastic lymphoma kinase positive, which has no adverse international prognostic index, the prognosis of nodal peripheral T-cell lymphomas is worse than that of aggressive B-cell lymphomas. Chemotherapy based on anthracyclines provides poor outcomes because these neoplasms frequently have multidrug-resistant phenotypes. Based on this, the current tendency is to use intensified cyclophosphamide, doxorubicin, vincristine, prednisolone (CHOP) regimens with the addition of new drugs, and autologous hematopoietic stem cell transplantation. This paper describes the clinical features and diagnostic methods, and proposes a therapeutic algorithm for nodal peripheral T-cell lymphoma patients.
Keywords: T-cell Lymphoma, Diseases classification, Immunophenotyping, World Health Organization, Antineoplastic combined chemotherapy protocols
Introduction
Non-Hodgkin lymphomas (NHLs) are a heterogeneous group of malignancies of the immune system, encompassing more than 40 entities with specific clinical, morphologic, immunophenotypic, and molecular characteristics. In the Western World, NHLs originate from B lymphocytes in 85–90% of cases and from lymphoid T cells and natural killer cells (NK) in 10–15% of cases, while T-cell lymphomas represent a higher proportion of NHL cases in Asian countries, accounting for up to 25% of these neoplasms.1
T-cell malignancies derived from precursor or immature T cells originate from leukemia-lymphoblastic T-cell lymphoma. Otherwise, the NHLs that originate from mature T lymphocytes or NK cells are recognized as peripheral T-cell lymphomas (PTCL). The latter represent 12–15% of all NHLs and, according to the World Health Organization (WHO) Classification of Tumors, comprise 22 different entities categorized according to clinical presentation as primary nodal, primary extranodal, primary cutaneous, and disseminated or leukemic forms as summarized in Table 1.1–3
Table 1.
World Health Organization classification of peripheral natural killer (NK)/T-cell lymphomas.
| Primary cutaneous lymphomas |
| Mycosis fungoides and Sézary syndrome |
| Primary cutaneous CD30+T-cell lymphoproliferative disease |
| - Primary cutaneous anaplastic large-cell lymphoma (C-ALCL) |
| - Lymphomatoid papulosis (LYP) |
| Primary cutaneous peripheral T-cell lymphomas (PTCLs) |
| - Gama-delta T-cell lymphoma |
| - CD8+ aggressive epidermotropic cytotoxic |
| - CD4+ small-medium |
| Nodal peripheral T-cell lymphomas (PTCL) |
| Peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) |
| Angioimmunoblastic T-cell lymphoma (AITL) |
| Anaplastic large-cell lymphoma (ALCL) anaplastic lymphoma kinase (ALK) positive |
| Anaplastic large-cell lymphoma (ALCL) ALK negative (provisional) |
| Extranodal peripheral T-cell lymphomas |
| Extranodal NK-T-cell lymphoma, nasal type |
| Enteropathy-associated T-cell lymphoma (EATL) |
| Hepatosplenic T-cell lymphoma (HSTL) |
| Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) |
| Epstein–Barr virus (EBV)-positive T-cell lymphoproliferative childhood disorders |
| - EBV-positive T-cell lymphoproliferative childhood disease |
| - Hydroa vacciniforme-like |
| Widespread or leukemic |
| T-cell prolymphocytic leukemia (T-PLL) |
| T-cell large granular lymphocytic leukemia (T-LGL) |
| Chronic lymphoproliferative disorders of NK cells (provisional) |
| Aggressive NK-cell leukemia |
| Adult T-cell leukemia/lymphoma |
This review discusses the main clinical and therapeutic aspects of primary nodal PTCLs such as peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), angioimmunoblastic T-cell lymphoma (AITL), anaplastic large-cell lymphoma-anaplastic lymphoma kinase (ALK) positive (ALCL-ALK+), and anaplastic large-cell lymphoma-ALK negative (ALCL-ALK−).
Methods
A systematic literature search using PubMed, Lilacs (Literatura Latino-Americana e do Caribe em Ciências da Saúde), and the Cochrane Library databases was conducted to identify scientific papers related to the search term “peripheral T-cell lymphomas”. Only papers focused on human subjects were included.
Clinical and epidemiological aspects
PTCL-NOS are represented by nodal or extranodal T-cell lymphomas not otherwise categorized as any specific entity in the current WHO classification. These tumors represent 60–70% of all PTCLs, and 5–7% of all NHLs. They occur more commonly in adults with a median age of 60 years and have a slight male predominance.4–6 Although lymph node involvement predominates in the majority of cases, these lymphomas often disseminate to the bone marrow, liver, spleen, and other extranodal sites such as the skin and lung. They rarely have a leukemic presentation.5,6 Patients often present with unfavorable clinical characteristics including B symptoms, elevated lactate dehydrogenase (LDH) levels, high tumor burden, advanced disease (Stage III or IV), and poor performance status.5,6
The AITL subtype is characterized by a specific clinical syndrome and polymorphic infiltrate involving lymph nodes, with proliferation of high endothelial venules in a tree-like pattern and irregular proliferation of follicular dendritic cells (FDC) with predominantly perivascular distribution. AITL represents the second most common subtype of PTCL (15–20% of cases) with its prevalence only being exceeded by PTCL-NOS. It occurs most commonly in elderly patients (aged 60–65 years), with a slight male predominance.7
Typical cases of AITL show acute or subacute systemic features that may mimic drug reactions or systemic infections. Clinically, AITL presents as small and generalized lymphadenopathy, hepatosplenomegaly, and constitutional symptoms. Maculopapular rash occurs in 50% of cases and paraneoplastic manifestations such as arthritis, vasculitis, serous effusions, and neurological manifestations are not uncommon. Laboratory features include eosinophilia, polyclonal hypergammaglobulinemia, elevated serum LDH and erythrocyte sedimentation rate (ESR), as well as circulating autoantibodies (cryoagglutinins, cryoglobulins, immune complexes, positive direct Coombs test) and paraneoplastic phenomena of an immune nature (hemolytic anemia, leukocytoclastic vasculitis, rheumatoid arthritis, and autoimmune thyroid disease).1,4,7
ALCL is a PTCL characterized by large polymorphic lymphoid CD30+ cells with abundant cytoplasm and horseshoe- or kidney-shaped nuclei. The three recognized subtypes are ALCL-ALK+, ALCL-ALK−, and primary cutaneous anaplastic lymphoma.8 ALCL-ALK+ predominates in younger patients, with a median age of 30 years, and represents 3–5% of NHLs in adults and 30% of NHLs in children, with a male predominance. Extranodal involvement is frequent, and 50% of cases are diagnosed at an advanced stage (III or IV) and in patients with B symptoms. The most commonly involved sites include the skin, subcutaneous tissue, bone, lung, and liver. Bone marrow involvement occurs in 10–30% of cases, but the leukemic phase is unusual and occurs more frequently in the small-cell morphological variant. It rarely presents with gastrointestinal and central nervous system involvement. Despite the advanced stage involving multiple extranodal sites, most patients present with low-risk or low-intermediate risk disease according to the International Prognostic Index (IPI), since performance status is frequently preserved and most patients are younger, with normal LDH levels at diagnosis.4,9
ALCL-ALK− is morphologically indistinct from ALCL-ALK+, but does not show chromosomal translocations involving the ALK gene or expression of the ALK protein. The disease predominates in the elderly; extranodal involvement occurs in up to half of cases and is usually diagnosed in patients with advanced stage disease who have B symptoms and aggressive disease. Recently, an unusual variant associated with prosthetic breast implants that has an indolent behavior has been described.8–10
Histopathological and molecular diagnostic criteria
An excisional tumor biopsy is essential for correct diagnosis. Needle biopsy guided by imaging is not recommended.1 It should be highlighted that for the correct diagnosis and classification of PTCL, histopathological analysis by an experienced hematopathologist using a large sample with a considerable amount of tissue that allows visualization of the broad pattern of lymph node infiltration is necessary, as well as applying a wide immunohistochemical panel.
In PTCL-NOS, the compromised lymph node shows paracortical or diffuse infiltration with loss of the normal lymph node architectural pattern. Although large tumor cells predominate, a few cases exhibit small neoplastic cells. The morphologic spectrum varies from monotonous to pleomorphic cells with an excess of mitotic cells (Figure 1). The WHO classification recognizes histological variants such as lymphoepitheliod lymphoma (Lennert lymphoma), follicular, and T-like zone. Phenotypically, it exhibits aberrant antigen expression characterized by a lack of T-cell markers such as CD5 and CD7. Most cases express the CD4 antigen, although some others may be CD8+, particularly the lymphoepitheliod variant. About 30% of PTCL-NOS express the CD30 marker, but the phenotypic profile and morphologic aspect allow it to be distinguished from ALCL-ALK− since the latter expresses epithelial membrane antigens (EMA) and contains cytotoxic granules. Distinguishing it from classical Hodgkin's lymphoma (HL) can be achieved by observing the expression of CD15, PAX-5, and Epstein–Barr virus (EBV). Also, PTCL-NOS present with a high proliferative rate, with Ki67 higher than 70%, and strong and homogeneous expression of CD3.1,4,11 Cases of PTCL CD30+/ALK− with no histologic similarity to classic ALCL should be categorized as PTCL-NOS.1 A monoclonal rearrangement of the T-cell receptor (TCR) gene is usually seen in PTCL-NOS by polymerase chain reaction (PCR); a cytogenetic abnormality with complex karyotype is the rule. In the subtype with a follicular growth pattern, it is common to observe the recurrent aberration t(5;9)(q33;q22) that produces the oncogenic protein ITK-SYK.11
Figure 1.

Morphologic aspects of Peripheral T-cell lymphoma not otherwise specified (PTCL-NOS). (A) Diffuse proliferation of atypical medium sized lymphoid cells. Note some intermingled eosinophils [hematoxylin and eosin (H&E)-stained section]. (B) Diffuse expression of CD3 by the neoplastic lymphoid cells. (C) These cells also express CD4. (D) CD7 antigen is only partially expressed by neoplastic T cells.
The morphological aspects of classical AITL include a diffuse pattern of infiltration by polymorphic cells and variable proportions of atypical neoplastic T-cells together with small lymphocytes, histiocytes, immunoblasts, eosinophils, and plasma cells with peri-lymph node infiltration into subcortical sinuses. Likewise, there is prominent vascular proliferation with a tree-like pattern of endothelial venules and irregular proliferation of FDC. B immunoblasts are usually present in the paracortical region and may mimic HL (Figure 2). Neoplastic cells express a pattern of follicular T helper cells such as CD3+, CD4+, CD10+, BCL-6+, CXCL13+, PD1+, and ICOS+ cells. Indeed, vascular expansion can be demonstrated by immunohistochemistry for CD31 and CD34. Irregular proliferation of FDC can be defined by CD21, CD23, and CD35 staining that are often found with vessels. The B-cell-associated antigen markers, such as CD20 and CD79a, may show immunoblasts in the interfollicular areas. These cells are infected by EBV and are Reed-Sternberg-like cells mimicking classical HL.7,11–14 Most of the AITL cases contain a monoclonal population of T cells. The presence of B lymphocyte clones can be detected in up to 30% of cases and the EBV genome is detected in nearly 100%. Karyotypic aberrations occur in 70–80% of cases, mainly trisomy of chromosome 3 and 5, and an additional X chromosome.11,15
Figure 2.
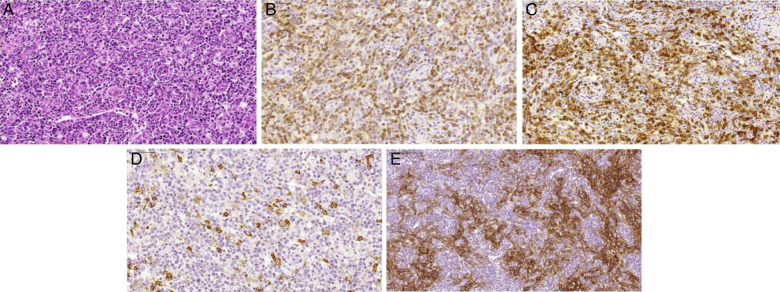
Morphologic aspects of angioimmunoblastic T-cell lymphoma. (A) Proliferation of small to medium sized lymphoid cells with mild atypia surrounding prominent high endothelial venules. Occasional large cells are observed, as well as some plasmocytes (H&E-stained section). (B) The majority of neoplastic lymphoid cells express membrane CD3. (C) Neoplastic lymphoid cells also express CD10. (D) CD20 antibody highlights the large B immunoblasts. (E) Follicular dendritic cell meshwork expansion surrounding high endothelial venules is evident with CD21 immunostain.
Although ALCL-ALK+ presents with a broad morphological spectrum, all cases contain a variable proportion of cells with eccentric nuclei (kidney-like or horseshoe-shaped) with an eosinophilic perinuclear area corresponding to the Golgi, and abundant cytoplasm; these cells are called ‘hallmark cells’. The morphological findings of this disease vary from small to very large malignant cells (Figure 3). In addition, five distinct morphological patterns are recognized including classical, lymphohistiocytic, small-cell, ‘Hodgkin-like’ variant, and mixed pattern.9,11 The classical variants represent 60–70% of all cases and their malignant cells are almost exclusively large and sometimes Reed-Sternberg-like. When the lymph node architecture is partially compromised, the cells grow within sinusoids and may simulate metastatic tumors. The cell phenotype exhibits diffuse positivity for CD30 and sometimes presents as a ‘perinuclear dot’ that corresponds to Golgi. Expression of the CD2 antigen is common, but CD45RO, CD5, and CD7 are usually negative. Most patients present with a CD4+/CD8− phenotype and show co-expression of cytotoxic antigens such as granzyme B, TIA-1, perforin and the EMA antigen.4,9,11 Most cases of ALCL-ALK+ have t(2;5)(p23;q35), resulting in the hybrid product NPM1-ALK. In these cases, the ALK protein can be found in both the cytoplasm and nucleus. About 90% of these lymphomas have a clonal rearrangement of the TCR gene. In addition to the classic t(2;5), a variant chromosome translocation involving the ALK gene and other genes located on chromosomes 1, 2, 3, 17, 19, 22, and X may occur. The morphological and phenotypic aspects of ALCL-ALK− cases are identical to those observed in ALCL-ALK+ cases.4,11,16
Figure 3.
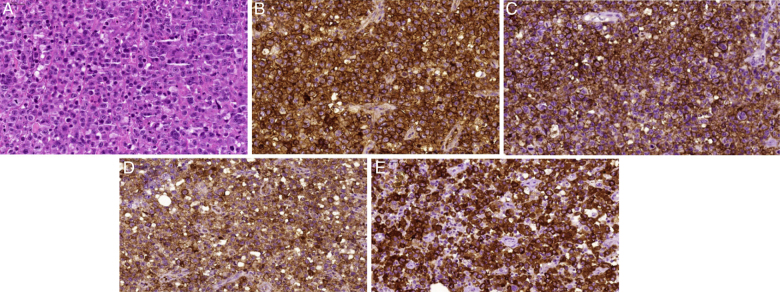
Morphologic aspects of anaplastic large cell lymphoma-anaplastic lymphoma kinase (ALK) positive. (A) Diffuse proliferation of large, highly atypical cells, with prominent pleomorphism (H&E-stained section). Some hallmark cells can be observed; they present large multilobulated ‘horseshoe-like’ nuclei. Neoplastic cells show diffuse expression of (B) CD30; (C) CD43; (D) epithelial membrane antigens and (E) ALK-1. In this case, ALK-1 positivity present exclusive cytoplasmic pattern.
Prognostic factors
Different studies have shown that the phenotype of T-cell lymphomas can be effectively stratified using the IPI.17,18 However, a Prognostic Index for T-cell Lymphomas (PIT) was developed in an attempt to produce more accurate prognostic indicators. This prognostic index identified four risk factors; age, serum LDH, performance status, and histopathological involvement of the bone marrow. Patients were stratified into four risk groups with 0, 1, 2, or more than 2 risk factors with overall survival (OS) of 62%; 53%, 33%, and 18% for each risk group, respectively.19
Another index is the modified Prognostic Index for T-cell Lymphoma (mPIT), which incorporates a biological variable (Ki67 expression) and clinical aspects such as age, LDH, and performance status. The mPIT distinguishes three different risk groups: good risk prognosis, intermediate risk, and poor risk.20 The International Peripheral T-cell Lymphoma Project (IPTCLP) proposed another prognostic score that includes age, performance status, and platelet count.18,21
Therapeutic considerations
Conventional multidrug regimens used for aggressive B cell NHL such as CHOP are routinely considered for PTCL treatment. However, the outcomes with anthracycline-based regimens have been disappointing in patients with ALCL-ALK+ with low risk IPI.21–23 The worse outcome of PTCL is the result of many adverse factors such as high expression of P-glycoprotein, which is associated with the anthracycline resistance phenotype (MDR phenotype).3 Usually, the overall response rate is low and there is a high rate of early death during first-line treatment. Relapses are common and often occur in the first two years after treatment. Because of the lack of more effective treatment for PTCL that is not ALK positive, the CHOP regimen remains the most commonly used first-line therapy for these malignancies. Several studies demonstrated complete response (CR) rates of 50% and OS at 5 years of 30% with the CHOP regimen.23
These disappointing results led to the investigation of new therapeutic strategies such as intensified chemotherapy and alternative protocols such as high-dose therapy followed by rescue with autologous hematopoietic stem cell transplantation (aHSCT). Although the results between different studies were discordant, an intensified CHOP regimen with the addition of etoposide has shown better progression-free survival (PFS), especially in under 60-year-old patients with favorable risk factors.23–25
Although spontaneous remissions have been described, AITL often presents with an aggressive clinical course. Occasionally, asymptomatic patients may be observed or treated with corticosteroids or immunomodulators and antiangiogenic drugs such as cyclosporine, thalidomide, and bevacizumab. In general, patients with AITL requiring therapy should be allocated to clinical studies. Of those, CHOP or fludarabine and cyclophosphamide (FC) regimens have been used the most often.7,22,23
On the other hand, patients with ALCL-ALK+ without adverse IPI risk factors have very chemosensitive disease that responds with comparable or superior outcomes to diffuse large B cell lymphoma (DLBCL) when treated with anthracycline-based therapy. Several studies have demonstrated overall response rate (ORR) of 90% with CHOP and 5-year OS of 70–80%. ALCL-ALK− and ALCL-ALK+ high risk groups have an intermediate prognosis between ALCL-ALK+ without adverse risk factors and PTCL-NOS, with a 5-year OS of 49% for ALCL-ALK− and 19% for PTCL-NOS. Currently, the management of these patients is similar to those positive for the variant ALK with low IPI, but because the outcome is worse, the same approach used for PTCL-NOS is recommended, with intensification of CHOP by adding etoposide or allocating patients to clinical trials or consolidation with aHSCT.6,16,22,23
Usually, the therapeutic approach to PTCL does not involve irradiation, which is reserved for bulky disease (tumor greater than 10 cm) or a residual mass after chemotherapy. However, there is insufficient evidence of clinical benefits using this approach. Many groups have evaluated the role of aHSCT in PTCL that is not ALK positive for first CR or partial response (PR). Although controversial, outside of the clinical trial setting patients with AITL, ALCL-ALK− and PTCL-NOS should be treated with CHOP-like therapy and consolidated with aHSCT after the first CR or PR.22,23,26–29
Patients with relapsed or refractory PTCL, acceptable performance status, and no comorbidities should be treated with platinum-based rescue chemotherapy followed by aHSCT. There is still no consensus regarding allogeneic bone marrow transplantation in these patients, with mortality rates as high as 30–50% when using myeloablative conditioning regimens. This fact has promoted the use of reduced intensity conditioning regimens in some cases.23,30,31
For many patients with PTCL, the available treatment strategies are ineffective and new therapies should be explored. Gemcitabine is a nucleoside analog that overcomes the p-glycoprotein system. Bendamustine and antifolate agents such as pralatrexate are being tested in these lymphomas.32,33 Therapy with monoclonal antibodies such as brentuximab vedotin (anti CD30), alemtuzumab (anti CD52), and zanolimumab (anti CD4) have also been evaluated.
For the subgroup of PTCL patients whose tumors express the CD30 antigen, particularly systemic ALCL and CD30+ PTCL-NOS, brentuximab vedotin has emerged as an interesting therapy. The antibody-drug conjugate combines a monoclonal antibody directed against CD30 with a potent antimicrotubule agent named monomethylauristatin E (MMAE). CD30 is an ideal therapeutic target because of its strong and uniform expression in anaplastic lymphoma. After binding to its target molecule, brentuximab vedotin is internalized and MMAE is cleaved from the antibody molecule, exerting its action through inhibition of microtubule formation. In a phase II multicenter trial of brentuximab vedotin in relapsed and refractory ALCL, the overall response rate was 86%, with a CR rate of 53%. Significant adverse events were peripheral neuropathy, lung toxicity, neutropenia, and thrombocytopenia.34
Another new interesting drug for the treatment of these neoplasms is alisertib (MLN8237). This drug is a selective inhibitor of Aurora A kinase (AAK), and shows preclinical activity in PTCL cell lines and patient samples. Recently, Friedberg et al. reported the interim results of a phase II clinical trial of alisertib in patients with aggressive B and T-cell NHL. ORR for the whole study population was 32%, but when divided by subtype, patients with PTCL had an ORR of 57%.35
A better understanding of the pathophysiology of these tumors is allowing the use of targeted therapies, such as the ALK inhibitor crizotinib, modulators of epigenetic phenomenon drugs able to restore the transcriptional status of tumor suppressor genes (histone deacetylase inhibitors), and immunomodulating agents that have greater antitumor activity in AITL. Disease progression is associated with more dysregulation of the immune system than tumor growth phenomena.31,36–40
Table 2 shows a comparative analysis of the major epidemiological and clinical characteristics, pathological and therapeutic approaches, prognosis, and new therapeutic agents that can be employed in patients with each of the four main entities discussed in this article.
Table 2.
Main characteristics of nodal peripheral T-cell lymphomas.
| PTCL-NOS | AITL | ALCL-ALK+ | ALCL-ALK- | |
|---|---|---|---|---|
| Age Gender predominance |
>60 years Male |
>60 years Male |
<40 years Male |
>60 years Male |
| Clinical-laboratory markers and prognosis | Frequent extranodal involvement; Very aggressive course |
Nodal predominance; Splenomegaly; Autoimmune disorders; Cutaneous rash; Eosinophilia; Hypergammaglobulinemia |
Aggressive course; Low and low-intermediate IPI; Good prognosis, particularly if low IPI |
Aggressive course, but better than PTCL-NOS; Intermediate prognosis between ALCL-ALK+ and PTCL-NOS |
| Histopathology | Monomorphic infiltrate by small and medium lymphoid cells | Polymorphic infiltrate; Vascular proliferation; Follicular dendritic cell proliferation; Large immunoblasts EBV+ |
Hallmark cells; Mimic metastatic carcinoma |
Hallmark cells; Mimic metastatic carcinoma |
| Immunohistochemistry | CD3+ (75%), CD4+, CD7−, CD30+ or− | CD10+, BCL6+, CXCL13+, PD1+, ICOS+, EBV+ | CD3+ (25%), CD4+, CD30+ high, ALK1+ | CD3+ (25%), CD4+, CD30+ high, ALK– |
| Cytogenetics | t(5;9) ITK-SYK | +3, +5, +X | t(2;5) NPM-ALK | Non-specific |
| 5-year overall survival with CHOP | 19% | 18% | 70–80% | 49% |
| Treatment option | 6–8 CHO(E)P + aHSCT | 6–8 (R)CHO(E)P + aHSCT | 6–8 CHO(E)P | 6–8 CHO(E)P + aHSCT |
| Potential new therapeutic agents | Pralatrexate Vorinostat |
Bevacizumab Lenalidomide |
Crizotinib Brentuximab vedotin |
Brentuximab vedotin |
PTCL-NOS: peripheral T-cell lymphoma not otherwise specified; AITL: angioimmunoblastic T-cell lymphoma; ALCL-ALK+: anaplastic large cell lymphoma-anaplastic lymphoma kinase positive; ALCL-ALK−: anaplastic large cell lymphoma-anaplastic lymphoma kinase negative; IPI: International Prognostic Index; EBV: Epstein–Barr virus; CHO(E)P: cyclophosphamide, doxorubicin, vincristine, prednisolone; aHSCT: autologous hematopoietic stem cell transplantation.
Conclusion
Primary nodal peripheral T-cell lymphomas comprise a heterogeneous group of neoplasms arising from the proliferation of activated lymphoid T-cells. Clinical, morphologic, immunophenotypic, genetic, and molecular criteria are used to classify these neoplasms, which are frequently difficult to diagnose. Correct identification of each entity is critical to establish appropriate therapeutic interventions, which currently still offer poor response rates. Intensification of treatment using new drugs or specific monoclonal antibodies, as well as HSCT, may change this reality in the near future. More studies are necessary to define the best treatment approach for these entities.
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Jaffe E.S., Nicolae A., Pittaluga S. Peripheral T-cell NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(Suppl. 1):71–87. doi: 10.1038/modpathol.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaffe E.S. Pathobiology of peripheral T-cell lymphomas. Hematol Am Soc Hematol Educ Prog. 2006:317–322. doi: 10.1182/asheducation-2006.1.317. [DOI] [PubMed] [Google Scholar]
- 3.Vose J., Armitage J., Weisenburger D. International peripheral T-cell and natural killer lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 4.Dogan A., Feldman A F. T- and NK-cell lymphoma update. Diagn Histopathol. 2009;16(2):99–110. [Google Scholar]
- 5.Rizvi M.A., Evens A.M., Tallman M.S., Nelson B.P., Rosen S.T. T-cell non-Hodgkin lymphoma. Blood. 2006;107(4):1255–1264. doi: 10.1182/blood-2005-03-1306. [DOI] [PubMed] [Google Scholar]
- 6.Tang T., Tay K., Quek R., Tao M., Tan S.Y., Tan L. Peripheral T-cell lymphoma: review and updates of current management strategies. Adv Hematol. 2010;2010:624040. doi: 10.1155/2010/624040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leval L., Gisselbrecht C., Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol. 2010;148(5):673–689. doi: 10.1111/j.1365-2141.2009.08003.x. [DOI] [PubMed] [Google Scholar]
- 8.Medeiros L.J., Elenitoba-Johnson K.S. Anaplastic large cell lymphoma. Am J Clin Pathol. 2007;127(5):707–722. doi: 10.1309/r2q9ccuvtlrycf3h. [DOI] [PubMed] [Google Scholar]
- 9.Fornari A., Piva R., Chiarle R., Novero D., Inghirami G. Anaplastic large cell lymphoma: one or more entities among T-cell lymphoma. Hematol Oncol. 2009;27(4):161–170. doi: 10.1002/hon.897. [DOI] [PubMed] [Google Scholar]
- 10.Ten Berge R.L., Oudejans J.J., Ossenkoppele G.J., Meijer C.J. ALK-negative systemic anaplastic large cell lymphoma: differential diagnostic and prognostic aspects – a review. J Pathol. 2003;200(1):4–15. doi: 10.1002/path.1331. [DOI] [PubMed] [Google Scholar]
- 11.Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H. IARC Press; Lyon: 2008. World Health Organization classification: tumors of hematopoietic and lymphoid tissues. [Google Scholar]
- 12.Attygalle A.D., Kyriakou C., Dupuis J., Grogg K.L., Diss T.C., Wotherspoon A.C. Histologic evolution of angioimmunoblastic T-cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression. Am J Surg Pathol. 2007;31(7):1077–1088. doi: 10.1097/PAS.0b013e31802d68e9. [DOI] [PubMed] [Google Scholar]
- 13.Attygalle A.D., Chuang S.S., Diss T.C., Du M.Q., Isaacson P.G., Dogan A. Distinguishing angioimmunoblastic T-cell lymphoma, unspecified, using morphology, immunophenotype and molecular genetics. Histopathology. 2007;50(4):498–508. doi: 10.1111/j.1365-2559.2007.02632.x. [DOI] [PubMed] [Google Scholar]
- 14.Konstantinou K., Yamamoto K., Ishibashi F., Mizoguchi Y., Kurata M., Nakagawa Y. Angiogenic mediators of the angiopoietin system are highly expressed by CD10-positive lymphoma cells in angioimmunoblastic T-cell lymphoma. Br J Haematol. 2009;144(5):696–704. doi: 10.1111/j.1365-2141.2008.07534.x. [DOI] [PubMed] [Google Scholar]
- 15.Nelson M., Horsman D.E., Weisenburger D.D., Gascoyne R.D., Dave B.J., Loberiza F.R. Cytogenetic abnormalities and clinical correlations in peripheral T-cell lymphoma. Br J Haematol. 2008;141(4):461–469. doi: 10.1111/j.1365-2141.2008.07042.x. [DOI] [PubMed] [Google Scholar]
- 16.Ferreri A.J., Govi S., Pileri S.A., Savage K.J. Anaplastic large cell lymphoma ALK-positive. Crit Rev Oncol Hematol. 2012;83(2):293–302. doi: 10.1016/j.critrevonc.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Gutiérrez-García G1, García-Herrera A., Cardesa T., Martínez A., Villamor N., Ghita G. Comparison of four prognostic scores in peripheral T-cell lymphoma. Ann Oncol. 2011;22(2):397–404. doi: 10.1093/annonc/mdq359. [DOI] [PubMed] [Google Scholar]
- 18.López-Guillermo A., Cid J., Salar A., López A., Montalbán C., Castrillo J.M. Peripheral T-cell lymphomas: initial features, natural history, and prognostic factors in a series of 174 patients diagnosed according to the REAL classification. Ann Oncol. 1998;9(8):849–855. doi: 10.1023/a:1008418727472. [DOI] [PubMed] [Google Scholar]
- 19.Gallamini A., Stelitano C., Calvi R., Bellei M., Mattei D., Vitolo U. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood. 2004;103(7):2474–2479. doi: 10.1182/blood-2003-09-3080. [DOI] [PubMed] [Google Scholar]
- 20.Went P., Agostinelli C., Gallamini A., Piccaluga P.P., Ascani S., Sabattini E. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J Clin Orthod. 2006;24(16):2472–2479. doi: 10.1200/JCO.2005.03.6327. [DOI] [PubMed] [Google Scholar]
- 21.Vose J. International peripheral T-cell lymphoma (PTCL) clinical and pathology review project: poor outcome by prognostic indices and lack of efficacy with anthracyclines. Blood. 2005;106 Abstract 811a. [Google Scholar]
- 22.Armitage J.O. The aggressive peripheral T-cell lymphomas: 2012 update on diagnosis, risk stratification and management. Am J Hematol. 2012;87(5):511–519. doi: 10.1002/ajh.23144. [DOI] [PubMed] [Google Scholar]
- 23.Dearden C.E., Johnson R., Pettengell R., Devereux S., Cwynarski K., Whittaker S. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphomas) Br J Haematol. 2011;153(4):451–485. doi: 10.1111/j.1365-2141.2011.08651.x. [DOI] [PubMed] [Google Scholar]
- 24.Karakas T., Bergmann L., Stutte H.J., Jäger E., Knuth A., Weidmann E. Peripheral T-cell lymphomas respond well to vincristine, adriamycin, cyclophosphamide, prednisone and etoposide (VACPE) and have a similar outcome as high grade B-cell lymphomas. Leuk Lymphoma. 1996;24(1–2):121–129. doi: 10.3109/10428199609045720. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz N., Trümper L., Ziepert M., Nickelsen M., Ho A.D., Metzner B. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin's Lymphoma Study Group. Blood. 2010;116(18):3418–3425. doi: 10.1182/blood-2010-02-270785. [DOI] [PubMed] [Google Scholar]
- 26.Eyre T.A., Collins G.P. Current treatment and future prospects for peripheral T-cell lymphoma. Drugs Today (Barc) 2013;49(10):631–646. doi: 10.1358/dot.2013.49.10.2025391. [DOI] [PubMed] [Google Scholar]
- 27.Yin J., Wei J., Xu J.H., Xiao Y., Zhang Y.C. Autologous stem cell transplantation as the first line treatment for peripheral T-cell lymphoma: results of a comprehensive meta-analysis. Acta Haematol. 2014;131(2):114–125. doi: 10.1159/000353778. [DOI] [PubMed] [Google Scholar]
- 28.d’Amore F., Relander T., Lauritzsen G.F., Jantunen E., Hagberg H., Anderson H. Up front autologous stem cell transplantation in peripheral T-cell lymphoma: NGL-T-01. J Clin Orthod. 2012;30(25):3093–3099. doi: 10.1200/JCO.2011.40.2719. [DOI] [PubMed] [Google Scholar]
- 29.Yared J., Kimball A. The role of high dose chemotherapy and autologous stem cell transplantation in peripheral T-cell lymphoma: a review of the literature and new perspectives. Cancer Treat Rev. 2013;39(1):51–59. doi: 10.1016/j.ctrv.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 30.Savage K.J. Peripheral T-cell Lymphomas. Blood Rev. 2007;21(4):201–216. doi: 10.1016/j.blre.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Foss F.M. Treatment strategies for peripheral T-cell lymphomas. Best Pract Res Clin Haematol. 2013;26(1):43–56. doi: 10.1016/j.beha.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Zinzani P.L., Venturini F., Stefoni V., Fina M., Pellegrini C., Derenzini E. Gemcitabine as single agent in pretreated T-cell lymphoma patients: evaluation of the long-term outcome. Ann Oncol. 2010;21(4):860–863. doi: 10.1093/annonc/mdp508. [DOI] [PubMed] [Google Scholar]
- 33.Bergman A.M., Pinedo H.M., Talianidis I., Veerman G., Loves W.J., van der Wilt C.L. Increased sensivity to gemcitabine of p-glycoproteinand multidrug resistance-associated protein-overexpressing human cancer cell lines. Br J Cancer. 2003;88(12):1963–1970. doi: 10.1038/sj.bjc.6601011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pro B., Advani R., Brice P., Bartlett N.L., Rosenblatt J.D., Illidge T. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Orthod. 2012;30(18):2190–2196. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 35.Friedberg J., Mahadevan D., Jung J., Persky D., Lossos I., Danaee H. Phase II trial of alisertib (MLN8237), an investigational, potent inhibitor of aurora A kinase (AAK), in patients with aggressive B- and T-cell non-Hodgkin lymphoma. ASH Annual Meeting Abstracts, vol. 118; San Diego; 2011. p. 95. [Google Scholar]
- 36.Dearden C.E. Role of single agent analogues in therapy of peripheral T-cell lymphomas. Semin Hematol. 2006;43(2 Suppl. 2):S22–S26. doi: 10.1053/j.seminhematol.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 37.O’Connor O.A., Pro B., Pinter-Brown L., Bartlett N., Popplewell L., Coiffier B. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Orthod. 2011;29(9):1182–1189. doi: 10.1200/JCO.2010.29.9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchi E., Bongero D.C., Kalac M., Scotto L., O’Connor O.A. The combination of histone deacetylase inhibitors and hypomethylating agents exhibits marked synergy in preclinical models of T-cell lymphomas. Blood (ASH Annu Meet Abstract) 2013;116(21):3937. [Google Scholar]
- 39.Pierkarz R., Wright J., Frye R., Allen S.L., Joske D., Kirschbaum M. Final results of a phase 2 NCI multicenter study of romidepsin in patients with relapsed peripheral T-cell lymphoma (PTCL) Blood (ASH Annu Meet Abstr) 2009;114(22):1657. [Google Scholar]
- 40.Ansell S.M., Horwitz S.M., Engert A., Khan K.D., Lin T., Strair R. Phase I-II study of an anti-CD30 antibody (MDX-060) in Hodgkin's lymphoma and anaplastic large-cell lymphoma. J Clin Orthod. 2007;25(19):2764–2769. doi: 10.1200/JCO.2006.07.8972. [DOI] [PubMed] [Google Scholar]