

Abstract
Cervical cancer is the fourth most common cancer in women and is almost exclusively caused by human papillomavirus (HPV) infection. HPV is also frequently associated with other cancers arising from mucosal epithelium, including anal and oropharyngeal cancers, which are becoming more common in both men and women. Viral persistence and progression through precancerous lesion stages are prerequisites for HPV-associated cancer and reflect the inability of cell-mediated immune mechanisms to clear infections and eliminate abnormal cells in some individuals. Cell-mediated immune responses are initiated by innate pathogen sensing and subsequent secretion of soluble immune mediators and amplified by the recruitment and activation of effector T lymphocytes. This review discusses early defensive mechanisms of innate responders to natural HPV infection, their influence on response polarization, and the underappreciated role of keratinocytes in this process.
INTRODUCTION
Human papillomavirus (HPV) infects epithelial cells of the skin and mucosal tissues and is best known for its causal role in cervical cancer (1, 2), the fourth most common cancer in women worldwide (3). HPV remains a serious public health problem despite the availability of effective prophylactic vaccines such as Gardasil (Merck, Whitehouse Station, NJ, USA) and Cervarix (GlaxoSmithKline Biologicals, Rixensart, Belgium). In the United States in 2009, cervical cancer represented 53.4% of the newly diagnosed HPV-associated cancers in women and oropharyngeal cancer represented 78.2% of the newly diagnosed HPV-associated cancers in men (4). The incidence rates of HPV-associated anal and oropharyngeal cancers in both men and women increased between 2000 and 2009 (4). In addition, adoption of prophylactic vaccines has been slow. Therefore, understanding of early events that occur upon HPV infection would be important in developing additional modalities for preventing HPV-associated malignancies. This review presents recent advances in our knowledge of the impact of epithelial danger sensing and cytokine responses on the clearance of HPV infections and the emerging role of keratinocytes (KC) as initiators of and partners in the amplification of anti-HPV immunity.
HPV INFECTION AND DISEASE PROGRESSION
The site of infection matters.
HPV can be divided into cutaneous and mucosal types based on their tropism for the epithelium of different tissues (5). Mucosal HPV types are further divided into low- and high-risk categories, depending on oncogenicity. High-risk HPV types cause virtually all cases of cervical cancer (1, 2) and are commonly associated with cancer and high-grade precursor lesions in other mucosal tissues (5–7). HPV16 and -18 cause approximately 70% of cervical cancers, while low-risk HPV6 and -11 cause 90% of anogenital warts. Although HPV broadly infects proliferative cells of cutaneous and mucosal epithelia (2), the risk of HPV-associated disease progression is much higher in the metaplastic transformation zones of the cervix, anus, and oropharynx (8). Active metaplasia of the cervical transformation zone in young women is a significant risk factor for HPV infection and progression to low-grade cervical intraepithelial neoplasia (CIN1, analogous to low-grade squamous intraepithelial lesion [LSIL]) (9, 10). Conversely, the vagina lacks a transformation zone, and HPV-associated cancer of the vagina is much less frequent than cervical cancer, despite common vaginal HPV infections (8).
Danger sensing enables clearance.
The sensing of pathogen-associated molecular patterns via pattern recognition receptors (PRRs) is central to the induction of innate immune responses and identifies the nature of the infection for an adaptive response. Viral nucleic acids can be detected by endosomal Toll-like receptor 3 (TLR3), TLR7, TLR8, and TLR9. Two recent studies by the same group revealed that increased expression of TLR mRNAs, measured by quantitative reverse transcription-PCR (qRT-PCR) in cervical cytobrush specimens, is significantly associated with impending viral clearance. The first study examined women with incident HPV16 infections (11). Greater cervical expression of TLR2, -3, -7, -8, and -9 after incident infection with HPV16 than preinfection was significantly associated with subsequent clearance within 4 months. In women who cleared the infection, expression of TLR1, -3, -7, and -8 was significantly associated with increased secretion of antiviral alpha 2 interferon (IFN-α2). No such associations were found in subjects with HPV51 infections, suggesting type-specific mechanisms by which different HPV types may evade innate immune responses (11). The second study examined the clearance of HPV16 infection following periods of persistence (12). Higher expression of TLR3 or TLR7 was predictive of HPV16 clearance by the following visit. The associations grew stronger and expanded to include TLR3, -7, -8, and -9 in women who produced positive IFN-γ immunospot responses to the HPV16 E6 oncoprotein but not E7. Together, these studies suggest that increased TLR expression is important in the clearance of HPV16 infections. Furthermore, there appears to be a link between the role of TLRs in the clearance of persistent HPV16 infection and an E6-specific effector response.
Soluble immune mediators.
PRR activation induces cytokine secretion both directly and through autocrine and paracrine cytokine signaling. Cytokines and chemokines (chemotactic cytokines) can suppress viral gene expression, create an inflammatory microenvironment, and recruit both innate and adaptive immune cells. Cervicovaginal lavage samples from healthy young women with immature, metaplastic cervical epithelium contain higher levels of interleukin-1α (IL-1α), IL-1β, IL-6, IL-10, IL-12, CCL3, CCL5, CXCL8, tumor necrosis factor alpha (TNF-α), and IFN-γ (measured by Luminex-based multiplex cytokine/chemokine assay) than those from women with mature epithelium, irrespective of HPV status (13). Chemokine nomenclature is described in Table 1. Young women have the highest rate of HPV infection (14), yet it is unclear whether robust cytokine secretion in the healthy state represents a greater readiness to mount an immune response or greater susceptibility to harmful inflammation (13). Two longitudinal studies have shown that IFN-γ mRNA expression, measured by qRT-PCR in cervical cytobrush (15) or cervical biopsy (16) samples, is associated with HPV clearance. IFN-γ expression in natural killer (NK) cells and activated T cells can be stimulated by IFN-α, IL-12, and IL-18 produced by infected cells, activated dendritic cells (DC), or macrophages (17). These studies combined with those linking higher TLR expression and antigen-specific IFN-γ immunospot responses to HPV clearance (11, 12) underscore the importance of cytokine production by innate immune cells to promote an effective T-cell response against HPV.
TABLE 1.
Chemokine nomenclature and recruited cell types
| Chemokine | Other name | Recruited cell type(s) |
|---|---|---|
| CCL2 | MCP-1 | Monocytes, memory T cells, DC/LC, NK cells |
| CCL3 | MIP-1α | Polymorphonuclear leukocytes |
| CCL4 | MIP-1β | Monocytes, NK cells |
| CCL5 | RANTES | T cells, monocytes, other leukocytes |
| CCL20 | MIP-3α | LC precursors, memory T cells |
| CCL27 | CTACK | Memory T cells (homing to skin) |
| CXCL8 | IL-8 | Neutrophils, other phagocytes |
| CXCL9 | MIG | Activated Th1 cells |
| CXCL10 | IP-10 | Activated Th1 cells |
| CXCL11 | I-TAC | Activated Th1 cells |
The fact that 90% of HPV infections are cleared within 2 years (18) speaks to the success of immune effector mechanisms. Nevertheless, some women harbor persistent HPV infections for years. A prospective study by Scott et al. (19) discovered a significant inverse relationship between time to clearance of incident high-risk HPV infections and high levels of IL-12 and TNF-α measured in cervicovaginal lavage samples by a multiplex immune assay. High levels of IL-12 were also associated with significantly longer times to clearance of established infections (defined as clearance after at least two high-risk HPV-positive visits). Given its role as a promoter of IFN-γ secretion and T-helper type 1 (Th1) responses, it is counterintuitive to associate IL-12 with viral persistence, and changes in IFN-γ secretion were not associated with longer times to clearance in this study. However, IL-12 can also stimulate the production of TNF-α by macrophages (20). The inverse association of time to clearance with high levels of both IL-12 and TNF-α may reflect macrophage activation and resultant inflammation. Chronic inflammation is known to cause tissue damage and can promote cellular changes leading to malignancy (21). On the other hand, insufficient immune activation also leads to advanced disease. A previous cross-sectional study (22) of high-risk HPV-associated lesion progression found that women with lower levels of IFN-γ and IL-10 mRNA in cervical cytology brush specimens were significantly more likely to have CIN2/3 than CIN1 or normal histology. These associations were paralleled by significantly higher levels of FoxP3 mRNA with CIN2/3, suggesting larger numbers of regulatory T cells and an immunosuppressive environment. Thus, in this study, reduced Th1 and homeostatic cytokine expression appeared to be permissive for immunosuppression and disease progression.
Additional studies have demonstrated that cytokine responses are increasingly dysregulated during HPV-related disease progression. Azar et al. (23) examined cytokine (TNF-α, IFN-γ, IL-6, and IL-10) levels in cervical cytobrush specimens for a continuum of subject classifications: HPV negative normal, HPV negative inflamed, HPV positive with normal cytology, HPV positive with CIN1, and HPV positive with CIN2/3. All of the cytokine levels were higher in secretions from inflamed cervices and all HPV-positive cervices than in those from HPV-negative normal cervices. An increased level of TNF-α was significantly associated with CIN2/3. The amounts of Th1-promoting cytokines (IFN-γ, TNF) and Th2-promoting cytokines (IL-6, IL-10) were inversely correlated in HPV-positive samples without dysplasia and CIN1, but these correlations decreased with progressive disease grades, suggesting a role for an impaired immune response in disease progression. Mhatre et al. (24) reported that women with negative Papanicolaou test results secreted higher levels of anti-inflammatory markers (IL-1 receptor antagonist, β-defensins 1, 2, and 3) and lower levels of proinflammatory cytokines (IL-1α/β, CCL5, and CXCL8) than did women with CIN1 and CIN3. IL-1 receptor antagonist and β-defensins are normally increased with inflammation and infection (24), so this reciprocal modulation with proinflammatory cytokines suggests dysregulation of the normal cytokine-defensin network.
Defensins can block HPV infection (25, 26) and potentially influence adaptive immunity by recruiting immune cells (27, 28). Greater expression of β-defensins has been reported in HPV-associated genital warts than in uninfected tissue (29) and in anal intraepithelial neoplasia than in nonlesional tissue in men who have sex with men (30). These observations suggest that defensins play a role in the local immune response to HPV. In addition, Hubert et al. (28) noted a regional disparity in the expression of human α-defensin 5, which was readily detectable in ectocervical, vaginal, and vulvar neoplasia but nearly absent from the cervical squamocolumnar junction, and suggested that this absence of α-defensin 5 may contribute to the unique susceptibility of the squamocolumnar junction to HPV infection and progressive disease.
KC AS MEDIATORS OF INNATE IMMUNITY
Pattern recognition.
Undifferentiated basal KC are the target of HPV infection but also the first line of defense. Expression of TLR1 to -7, -9, and -10 has been described in cultured primary human KC (31–35). As mentioned above, TLR3, -7, -8, and -9 detect nucleic acids present in endosomes. TLR3 senses double-stranded RNA (dsRNA) and the synthetic agonist poly(I·C). TLR9 senses unmethylated CpG sequences in DNA and synthetic CpG oligodeoxynucleotides (CpG ODN). TLR7 and -8 sense single-stranded RNA and can be stimulated by synthetic oligoribonucleotides or small imidazoquinoline molecules. The imidazoquinoline drug imiquimod is an immunomodulator and FDA-approved topical therapeutic for skin conditions, including genital warts, and its effectiveness as a treatment for anogenital intraepithelial neoplasias continues to be examined (36–39). KC also constitutively express the cytoplasmic dsRNA sensors protein kinase R (PKR), retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5) (33, 34). Expression of all four dsRNA sensors (TLR3, PKR, RIG-I, and MDA5) is upregulated in KC by type I and II IFNs (40–42) or poly(I·C) (33), further enhancing viral detection capability.
Karim et al. (34) developed a KC culture model by using primary KC from foreskin, vagina, and cervix tissues electroporated with HPV16 or -18 episomes to study the effect of HPV on virus-sensing PRR responses. Genome-wide expression profiling demonstrated that the presence of high-risk HPV episomes downregulated genes involved in innate and adaptive immune responses and upregulated genes involved in the cell cycle, DNA replication, and RNA metabolism. Basal expression of TLR3, PKR, RIG-I, and MDA5 was not altered in KC harboring HPV16 or -18 episomes. However, TICAM1 (encodes a TLR3 adaptor and mediator of type I IFN expression) expression was downregulated and poly(I·C) responses were broadly depressed, suggesting that high-risk HPV suppresses PRR signaling (34). Of the poly(I·C)-stimulated genes most downregulated by HPV, those for IL-1β and IL-6 are the most interconnected to other cytokines and antigen presentation pathways. The gene for CDKN2A, a cell cycle regulator, was the most interconnected of the genes upregulated by high-risk HPV. Reduced secretion of IL-1β and CCL5 in poly(I·C)-stimulated, HPV-positive KC was confirmed by enzyme-linked immunosorbent assay (ELISA), and reduced expression of the inflammasome component NLRP2 was confirmed by qRT-PCR (34). Thus, the ability of HPV16 and -18 to counter PRR-mediated signaling and target central hubs of highly interconnected gene networks favors immune escape and cell proliferation.
Later reanalysis of genome-wide expression profiles (43) revealed a significant upregulation of genes belonging to the protein ubiquitination pathway in HPV-positive KC, and the gene for ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) was the most highly upregulated of these. UCHL1 proved to be a potent negative regulator of PRR-induced immune responses in KC and was exploited by high-risk HPV to prevent the production of IFNs, cytokines, and chemokines and the attraction of professional immune cells. Mechanistic studies showed that UCHL1-mediated suppression of nuclear factor κB (NF-κB) and IFN response factor (IRF) signaling pathways by destabilizing NF-κB essential modulator and preventing polyubiquitination of TNF receptor-associated factor 3, respectively (43). HPV-transformed KC tend to lose expression of UCHL1, suggesting that different mechanisms of immune escape operate in these cells.
In contrast, Hasan et al. (32) demonstrated that HPV16 E6 and E7 downregulate the constitutive expression of the viral DNA sensor TLR9 in undifferentiated KC. HPV16 E6 and E7 also suppressed CCL20 and CXCL8 secretion and inhibited NF-κB reporter activity in CpG ODN-stimulated KC. TLR9 promoter deletions that removed several NF-κB binding sites restored promoter activity to normal levels, suggesting that HPV16 E6 and E7 actively suppress TLR9 promoter activity via the NF-κB binding sites (32). Karim et al. (43) also observed that KC could respond to CpG ODN stimulation by upregulating IFN-β1, IL-8, and CCL20 gene expression and that this response was abrogated by HPV16 and -18. However, findings of the two groups conflict with respect to the differentiation state of TLR9-responsive KC. While Lebre et al. (35) also detected TLR9 expression and CpG ODN-stimulated chemokine secretion in undifferentiated KC, Karim et al. (34, 43) and others (33) failed to detect TLR9 expression in undifferentiated KC. Several groups demonstrated TLR9 expression and responsiveness in in vitro differentiated primary KC (34, 43, 44) and differentiated layers of normal (44) and lesional (34) epithelium. The reason for the conflicting findings is unknown but may reflect differences in culture conditions (media, feeder cells, passage number) and/or tissue sources (embryonic versus adult, foreskin versus cervical).
TLR7/8 are also typically absent in KC. Andersen et al. (31) found that primary cervical epithelial cells mount functional responses to TLR3 and TLR9 activation but fail to respond to several TLR7/8 ligands. It has been thought that in the absence of TLR7/8 responses from cervical epithelial cells, any benefit from imiquimod/resiquimod therapy would arise from stimulation of DC and other TLR7/8-responsive immune cells present in cervical tissue (31). However, a few groups have uncovered circumstances under which TLR7 and -8 are upregulated in KC. One group (33) found that TLR7 was functionally upregulated in primary KC exposed to poly(I·C). They also observed 3-fold higher TLR7 expression in HPV-positive genital wart specimens than in healthy epidermis. As poly(I·C) mimics viral dsRNA, these results suggest that activation of TLR3, PKR, RIG-I, and MDA5 may upregulate TLR7 in HPV-infected tissue and confer lesion-specific sensitivity to the TLR7 agonist imiquimod (33). Another group (45) found that differentiated KC respond to imiquimod in a TLR7-dependent manner by secreting CXCL8 and TNF-α. TLR7 expression was induced 3-fold by KC differentiation and further induced by imiquimod. Interestingly, Hasan et al. (32) discovered that TLR8 was induced in KC lines infected with HPV16 E6/E7-expressing retroviruses and that conditioned medium from these cells could induce TLR8 expression in mock-infected KC.
KC-derived cytokines.
KC constitutively secrete low levels of soluble immune mediators, including cytokines, chemokines, and growth factors (46–48), the production of which can be elevated in response to proinflammatory stimuli, including PRR activation and autocrine or paracrine stimulation with inflammatory cytokines (31, 43, 46, 47, 49). Several cytokines, including type I IFNs, TNF-α, IL-1α, IL-4, IL-13, and transforming growth factor β (TGF-β), share the ability to inhibit HPV early gene expression (50–54). In addition, type I IFNs, TNF-α, and TGF-β suppress the growth of normal KC and nontumorigenic, HPV-transformed KC, although effectiveness can vary with the transforming HPV type and is generally lost with progression to a tumorigenic phenotype (50, 51, 54, 55). The complexity of HPV-host interactions is well reflected by the roles of antiviral type I IFNs and their subversion by HPV.
Type I IFNs are produced by most cells in response to viral infection to induce an antiviral state in both the infected cell and neighboring cells to prevent the spread of viral infection. Initial danger sensing via intracellular nucleic acid-sensing PRRs leads to the activation of constitutively expressed IRF3 and subsequent expression and secretion of IFN-β, which in turn induces the expression of IFN-α-stimulated genes and other IFN-stimulated genes (ISGs) through autocrine and paracrine signaling pathways dependent on signal transducers and activators of transcription (STAT) transcription factors (56, 57). IFN-α/β and IFN-γ exert potent antiproliferative activity on HPV-immortalized KC and reduce the transcription of HPV genes, but IFN sensitivity differs greatly among cell lines (50).
High-risk HPV oncoproteins are effective suppressors of ISG expression (58–60). The E6 and E7 oncoproteins from HPV16 and -31 inhibit both STAT1 (61), an essential mediator of IFN signaling, and PKR (40), which is increased by IFN-α/β/γ to induce translation inhibition. HPV oncoproteins inhibit PKR function by multiple pathways, including inhibition of PKR phosphorylation and alteration of PKR subcellular localization in organotypic raft cultures and CIN lesions (40). STAT1-deficient mice are susceptible to viral infection because of almost complete loss of IFN signaling (62). STAT1 increases during KC differentiation, but to a lesser extent in HPV-positive cells. Suppression of STAT1 expression by viral proteins is required for differentiation-dependent HPV amplification, as well as long-term maintenance of episomal HPV (61). Nees et al. (58) discovered that the HPV16 E6 and E7 oncoproteins downregulate the expression of ISGs, as well as genes involved in NF-κB activation and cell cycle regulation in differentiating but not proliferating KC. E6 was a stronger repressor of IFN and ISG expression than E7 was and strongly reduced IFN-α and STAT1 protein levels and STAT1 binding to DNA. Even so, the combination of E6 and E7 was more effective than E6 alone. In contrast to the group of ISGs, very few of the E6/E7-modulated genes involved in NF-κB activation or cell cycle regulation were modulated by IFN-α/β treatment (58).
IFN-κ is a KC-specific type I IFN that, unlike IFN-β, is constitutively expressed in resting KC (63). Viral infection, dsRNA, IFN-β, and IFN-γ all significantly increase IFN-κ expression. Exogenous IFN-κ signals through type I IFN receptors, activates IRF1 and STAT1 signaling mediators, induces several antiviral effector pathways (PKR, oligoadenylate synthetase, Mx dynamin-like GTPases) common to other type I IFNs, and can protect fibroblasts from viral infection (63). The upregulation of STAT1 by IFN-κ is notable because STAT1-deficient mice are susceptible to viral infection because of almost complete loss of IFN signaling (62). HPV16-, HPV18-, and HPV31-positive KC cell lines downregulate the constitutive expression of IFN-κ and a broad selection of ISGs, including those for STAT1 and dsRNA-sensing PRR, and respond weakly to poly(I·C) (59). However, reexpression of IFN-κ restores ISG and PRR expression in HPV18-positive KC cell lines and HeLa cells (59) and restores p53, IRF-1/7/9, and MxA expression in HPV16-positive SiHa cells (64), highlighting the importance of IFN-κ as a regulator of ISG expression in KC. The E6 oncoprotein is the main driver of IFN-κ repression, which is achieved via promoter hypermethylation (59, 64). Interestingly, promoter demethylation restores IFN-κ expression in CaSki but not SiHa cells (both HPV16 positive), yet forced expression of IFN-κ can protect SiHa cells from viral infection (64).
Sunthamala et al. (60) reported that HPV16 E2 also suppresses many genes associated with innate immunity, including those for IFN-κ and stimulator of IFN genes (STING). Activation of STING by cytosolic DNA sensors induces type I IFN expression via Tank binding kinase 1 and IRF3. This is the first report of a possible involvement of STING in HPV immunity. Furthermore, knockdown of STING moderately reduced IFN-κ expression in primary human KC, and both proteins were reduced in HPV-positive cervical tissue and CIN1 (60). IFN-κ downregulation in HPV-positive precursor lesions and cervical cancer has been observed before (64, 65). Interestingly, De Carlo et al. (65) found that while IFN-β and IFN-γ mRNAs were detectable in HPV16-positive cervical epithelium from LSIL, high-grade squamous intraepithelial lesion, and carcinoma biopsy specimens, IFN-κ mRNA was absent. In contrast, IFN-κ, IFN-β, and IFN-γ mRNAs all increased in stroma associated with higher lesion grades, which was selectively infiltrated by monocytes and DC (65). The authors suggest that these immune cells may be responsible for increased IFN expression in stroma. Thus, the absence of IFN-κ in diseased epithelium and suppression of IFN-κ and STING by E2 may both contribute to viral persistence and immune evasion at an early stage of HPV pathogenesis.
KC COMMUNICATE WITH PROFESSIONAL IMMUNE CELLS
Recruitment.
Leukocytes expressing the appropriate chemokine receptors are recruited to sites of inflammation by gradients of chemokines that are released locally. Several of the studies described above (31, 34, 35, 43) document that KC readily upregulate the production and secretion of chemokines (CCL2, CCL3, CCL4, CCL5, CCL20, CCL27, CXCL8, CXCL9, CXCL10, and CXCL11) in response to danger signals. Chemokine-responsive immune cells are described in Table 1. Importantly, discrimination of the pathogen type by differential PRR activation stimulates the secretion of different patterns of chemokines (31, 35), which may recruit distinct populations of immune cells (66). Lebre et al. (35) examined the secretion of chemokines from primary KC cultures treated with ligands for TLR3 [poly(I·C)], TLR4 (lipopolysaccharide), TLR5 (flagellin), and TLR9 (CpG ODN) and discovered that different TLR ligands induce distinct patterns of chemokine secretion. While CCL2, CCL20, and CXCL8 were inducible by all four TLR ligands, CXCL10 responded to TLR3 and TLR9, CXCL9 responded only to TLR3, and CCL27 responded to TLR3 and TLR5 (35). Another group observed that cervical epithelial cells secrete CXCL8 in response to stimulation by both TLR3 and TLR9 but secrete CCL5 in response to TLR3 but not TLR9 (31). CXCL8 stimulates chemotaxis and phagocytic activity in neutrophils and other phagocytes. CCL5 attracts T cells, monocytes, eosinophils, basophils, NK cells, and DC (67). Langerhans cell (LC) precursors are recruited to the epidermis by CCL20 constitutively expressed by KC (68). KC respond to activated LC and T cells by increased secretion of cytokines and chemokines, including CCL20, which supports the repopulation of the epidermis with LC following the emigration of activated LC (reviewed in reference 66). CCL20 is the most potent inducer of immature LC migration to the skin but also attracts CCR6-positive memory B and T lymphocytes. CXCL9 and CXCL10 are CXCR3 ligands that recruit activated Th1 cells and help establish a local adaptive immune response (69). Several groups have demonstrated that the HPV16 E6 and E7 oncoproteins reduce both the basal and PRR- or cytokine-stimulated expression of chemokines in KC (34, 70–73). Additionally, the reduced ability of conditioned medium from HPV16 E6/E7-positive KC or SiHa cells to support chemotaxis of immune cells has been attributed to reduced secretion of chemokines (34, 72, 73).
Retention.
In addition to recruitment, KC facilitate immune cell retention through the expression of surface receptors. Following recruitment of LC precursors to the epidermis by CCL20 (66, 68), homotypic binding of E-cadherin between KC and LC is critical for epithelial retention (74) and subsequent differentiation of LC (75) and is reduced upon LC activation to permit egress of LC from the epithelium and migration to lymph nodes (76). Forced expression of E-cadherin in normally E-cadherin-deficient SiHa cells greatly enhances LC infiltration of and adhesion to organotypic layers of SiHa cells (77).
Several studies have reported an association between reduced numbers of LC and loss of E-cadherin expression in HPV-positive CIN lesions (77, 78) and noninflamed warts (79). In warts, LC depletion was associated with reduced expression of both CCL20 and E-cadherin in lesional KC. CCL20 was upregulated and LC numbers were increased in inflamed warts associated with massive or diffuse dermal infiltrates of DC and cytotoxic CD8+ T cells (79). Hubert et al. (80) showed that both LC numbers and E-cadherin expression decreased progressively with increasing grades of CIN. Furthermore, reduced numbers of S100-positive LC in CIN suggest that HPV interferes with the recruitment of a functional subset of LC (78, 80). Higher numbers of S100-positive LC were associated with inflammation in CIN lesions (80).
Costimulation and amplification.
In the resting state, KC do not express major histocompatibility complex class II (MHC-II) or costimulatory receptors CD80 and CD86, which are necessary to fully activate T cells, and do not upregulate CD80 or CD86 in response to cytokines or PRR stimulation. Therefore, KC can engage in “nonprofessional” antigen presentation, which may be tolerogenic because of the lack of costimulation. However, KC can express MHC-II in response to IFN-γ, and increased adhesion mediated by intercellular adhesion molecule 1 (ICAM-1) may substitute for CD28 costimulatory interactions (81, 82). Using CD28-deficient mice, it was shown that ICAM-1 can provide necessary costimulation for anti-CD3 antibody-mediated T cell proliferation and IL-2 secretion (81). Grousson et al. (83) found that blocking antibodies to ICAM-1 or its coreceptor CD18 (LFA-1 β-chain) inhibited T cell proliferative responses to superantigen in the presence of IFN-γ-treated primary KC (83). IFN-γ treatment of immortalized human KC increases the surface expression of ICAM-1 and MHC-II. These cells are then able to process protein antigen and prime antigen-specific memory T cells in an ICAM-1/lymphocyte function-associated antigen 1 (LFA-1)-dependent manner, suggesting that ICAM-1 on KC is able to costimulate MHC-II responses (82). Salient features of KC activation responses are summarized in Fig. 1.
FIG 1.
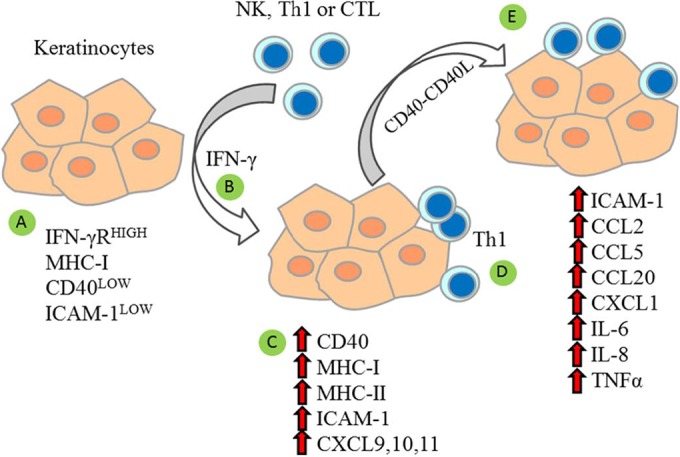
Activated KC amplify cell-mediated immune reactions. Resting KC express low surface levels of CD40 and ICAM-1, moderate levels of MHC-I, and high levels of IFN-γ receptor (IFN-γR) (82, 90, 91) (A). Activation of KC by IFN-γ secreted by NK cells or activated T lymphocytes (B) increases surface expression of MHC-I, MHC-II, CD40, and ICAM-1 (82, 83, 90–92) and induces expression of CXCL9, CXCL10, and CXCL11 (94), chemokines that attract activated Th1 cells (C). KC that present HPV antigens via MHC-II may interact with Th1 cells (D). Higher surface expression of ICAM-1 and CD40 improves adhesion and costimulation (82, 83, 91). Ligation of CD40 on KC by CD40 ligand on Th1 cells further increases ICAM-1 (83, 90) and stimulates further cytokine and chemokine expression (90–94) to activate and recruit additional immune cells (E).
In addition to direct effects on memory T cells, KC may indirectly influence the polarization of naive T cells through their influence on DC. Uncommitted immature DC can acquire a Th1- or Th2-inducing phenotype in response to locally produced inflammatory mediators (84) in a tissue- and pathogen-dependent manner (85). Supernatants from poly(I·C)-treated KC can skew the maturation of DC toward a phenotype that biases the development of naive T cells into Th1 cells (86). KC-secreted IFN-α/β and IL-18 were necessary for this DC-polarizing effect, which was not seen with KC activated by a combination of TNF-α and IL-1β (86). Thus, pathogen-induced KC-derived factors can modulate the functional activation of DC and their subsequent polarizing effects on T cells.
CD40 is another important costimulatory molecule expressed on professional antigen-presenting cells and KC that interacts with CD154 (CD40 ligand) on activated T cells. CD40 has been detected on basal cells of cervical epithelium (87–89) and is increased in HPV-related CIN and cervical cancer (87–89). CD40 can also be upregulated by IFN-γ in normal KC (90–92) and HPV-transformed cell lines (87). CD40 ligation was found to directly influence the susceptibility of cervical carcinoma cells to cytotoxic T lymphocyte (CTL)-mediated killing (88, 89). Importantly, the level of ICAM-1 stimulated by CD40 ligation of IFN-γ-stimulated KC is higher than that produced by IFN-γ alone (83, 90). Increased adhesion between cervical carcinoma cells and activated T cells may promote antigen-specific lysis. Hill et al. (89) discovered that CD40 ligation on cervical carcinoma cells increased the production of transporter associated with antigen processing 1 and antigen-specific lysis by CTL, which was dependent on an endogenously processed, transporter associated with antigen processing 1-dependent HPV16 E6 antigen.
CD40 ligation on both normal KC and cervical carcinoma cells also amplifies inflammatory reactions by increasing chemokine secretion to recruit leukocytes (90–94). However, chemokine secretion stimulated by CD40 ligation is depressed by HPV (87, 94). Altenburg et al. (87) reported that cervical carcinoma cells secrete much less CCL2 in response to CD40 ligation than do nontumorigenic HPV-positive cells. Yet, IFN-γ combined with CD40 ligation was able to synergistically increase CCL2 secretion in both cell types, and the synergistic upregulation was more pronounced for CXCL10. The authors concluded that the combined signals are adequate to induce large changes in chemokine secretion, despite lower levels overall. Tummers et al. (94) made similar observations by using genome-wide expression analysis of IFN-γ-prestimulated, CD40-ligated primary epithelial cells from foreskin, vagina, and cervix tissues. CD40 ligation produced defined networks of gene expression coordinated by early high expression of IL-8 and TNF. Genes involved in immunity, inflammation, cell adhesion, and leukocyte migration were upregulated. HPV-positive epithelial cells produced a similar network of gene expression following CD40 ligation, but gene expression amplitude was reduced. In uninfected epithelial cells, CD40 ligation did not boost the gains in CXCL9, CXCL10, and CXCL11 gene expression achieved with IFN-γ alone. However, CD40 ligation did increase their expression further in HPV-positive cells than in uninfected epithelial cells (94), indicating that CD40 ligation is able to partially reverse gene expression deficits caused by HPV. Similarly, lower levels of CXCL8, CXCL9, CXCL10, and RANTES (measured by ELISA) were secreted by HPV-positive rather than uninfected epithelial cells in response to IFN-γ stimulation and CD40 ligation, and only supernatants of uninfected epithelial cells were able to increase peripheral blood mononuclear cell migration in response to IFN-γ stimulation and CD40 ligation (94). The in vivo balance of these immune response-promoting and evasive mechanisms in epithelial cells may significantly impact HPV persistence and disease progression.
CONCLUDING REMARKS
Danger sensing is the first step toward the clearance of natural HPV infections. While the danger-activated responses of LC and DC have garnered much attention, the role of KC in HPV clearance is less well explored. Even so, it is becoming increasingly clear that KC have the capability to be active participants in the immune response to HPV, and additional strategies to harness the innate immune mechanisms of KC are needed. The ability of KC-derived cytokines to affect DC phenotype and T cell polarization is particularly encouraging and may be exploited to develop new therapeutic modalities for treating HPV-associated diseases.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01CA143130.
We have no conflicts of interest to declare.
REFERENCES
- 1.Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. 2002. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol 55:244–265. doi: 10.1136/jcp.55.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 3.International Agency for Research on Cancer. 2012. Globocan 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012. World Health Organization, Geneva, Switzerland: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. [Google Scholar]
- 4.Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, Cronin KA, Watson M, Schiffman M, Henley SJ, Schymura MJ, Anderson RN, Yankey D, Edwards BK. 2013. Annual report to the nation on the status of cancer, 1975-2009, featuring the burden and trends in human papillomavirus (HPV)-associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst 105:175–201. doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leto M, Santos Junior GF, Porro AM, Tomimori J. 2011. Human papillomavirus infection: etiopathogenesis, molecular biology and clinical manifestations. An Bras Dermatol 86:306–317. doi: 10.1590/S0365-05962011000200014. [DOI] [PubMed] [Google Scholar]
- 6.Frisch M, Fenger C, van den Brule AJ, Sorensen P, Meijer CJ, Walboomers JM, Adami HO, Melbye M, Glimelius B. 1999. Variants of squamous cell carcinoma of the anal canal and perianal skin and their relation to human papillomaviruses. Cancer Res 59:753–757. [PubMed] [Google Scholar]
- 7.Tota JE, Chevarie-Davis M, Richardson LA, Devries M, Franco EL. 2011. Epidemiology and burden of HPV infection and related diseases: implications for prevention strategies. Prev Med 53(Suppl 1):S12–S21. doi: 10.1016/j.ypmed.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Schiffman M, Kjaer SK. 2003. Chapter 2: natural history of anogenital human papillomavirus infection and neoplasia. J Natl Cancer Inst Monogr 31:14–19. [DOI] [PubMed] [Google Scholar]
- 9.Moscicki AB, Burt VG, Kanowitz S, Darragh T, Shiboski S. 1999. The significance of squamous metaplasia in the development of low grade squamous intraepithelial lesions in young women. Cancer 85:1139–1144. doi:. [DOI] [PubMed] [Google Scholar]
- 10.Hwang LY, Ma Y, Shiboski SC, Farhat S, Jonte J, Moscicki AB. 2012. Active squamous metaplasia of the cervical epithelium is associated with subsequent acquisition of human papillomavirus 16 infection among healthy young women. J Infect Dis 206:504–511. doi: 10.1093/infdis/jis398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daud II, Scott ME, Ma Y, Shiboski S, Farhat S, Moscicki AB. 2011. Association between Toll-like receptor expression and human papillomavirus type 16 persistence. Int J Cancer 128:879–886. doi: 10.1002/ijc.25400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott ME, Ma Y, Farhat S, Moscicki AB. 2015. Expression of nucleic acid-sensing Toll-like receptors predicts HPV16 clearance associated with an E6-directed cell-mediated response. Int J Cancer 15:2402–2408. doi: 10.1002/ijc.29283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang LY, Scott ME, Ma Y, Moscicki AB. 2011. Higher levels of cervicovaginal inflammatory and regulatory cytokines and chemokines in healthy young women with immature cervical epithelium. J Reprod Immunol 88:66–71. doi: 10.1016/j.jri.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castle PE, Schiffman M, Herrero R, Hildesheim A, Rodriguez AC, Bratti MC, Sherman ME, Wacholder S, Tarone R, Burk RD. 2005. A prospective study of age trends in cervical human papillomavirus acquisition and persistence in Guanacaste, Costa Rica. J Infect Dis 191:1808–1816. doi: 10.1086/428779. [DOI] [PubMed] [Google Scholar]
- 15.Scott M, Stites DP, Moscicki AB. 1999. Th1 cytokine patterns in cervical human papillomavirus infection. Clin Diagn Lab Immunol 6:751–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song SH, Lee JK, Lee NW, Saw HS, Kang JS, Lee KW. 2008. Interferon-gamma (IFN-gamma): a possible prognostic marker for clearance of high-risk human papillomavirus (HPV). Gynecol Oncol 108:543–548. doi: 10.1016/j.ygyno.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, Sareneva T. 2001. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol 31:2236–2245. doi:. [DOI] [PubMed] [Google Scholar]
- 18.Moscicki AB, Schiffman M, Kjaer S, Villa LL. 2006. Chapter 5: updating the natural history of HPV and anogenital cancer. Vaccine 24(Suppl 3):42–51. doi: 10.1016/j.vaccine.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Scott ME, Shvetsov YB, Thompson PJ, Hernandez BY, Zhu X, Wilkens LR, Killeen J, Vo DD, Moscicki AB, Goodman MT. 2013. Cervical cytokines and clearance of incident human papillomavirus infection: Hawaii HPV cohort study. Int J Cancer 133:1187–1196. doi: 10.1002/ijc.28119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. 1998. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol 16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 21.Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 22.Scott ME, Ma Y, Kuzmich L, Moscicki AB. 2009. Diminished IFN-gamma and IL-10 and elevated Foxp3 mRNA expression in the cervix are associated with CIN 2 or 3. Int J Cancer 124:1379–1383. doi: 10.1002/ijc.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azar KK, Tani M, Yasuda H, Sakai A, Inoue M, Sasagawa T. 2004. Increased secretion patterns of interleukin-10 and tumor necrosis factor-alpha in cervical squamous intraepithelial lesions. Hum Pathol 35:1376–1384. doi: 10.1016/j.humpath.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Mhatre M, McAndrew T, Carpenter C, Burk RD, Einstein MH, Herold BC. 2012. Cervical intraepithelial neoplasia is associated with genital tract mucosal inflammation. Sex Transm Dis 39:591–597. doi: 10.1097/OLQ.0b013e318255aeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. 2006. Human alpha-defensins block papillomavirus infection. Proc Natl Acad Sci U S A 103:1516–1521. doi: 10.1073/pnas.0508033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiens ME, Smith JG. 2015. Alpha-defensin HD5 inhibits furin cleavage of human papillomavirus 16 L2 to block infection. J Virol 89:2866–2874. doi: 10.1128/JVI.02901-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubert P, Herman L, Maillard C, Caberg JH, Nikkels A, Pierard G, Foidart JM, Noel A, Boniver J, Delvenne P. 2007. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed in vitro and transplanted in vivo. FASEB J 21:2765–2775. doi: 10.1096/fj.06-7646com. [DOI] [PubMed] [Google Scholar]
- 28.Hubert P, Herman L, Roncarati P, Maillard C, Renoux V, Demoulin S, Erpicum C, Foidart JM, Boniver J, Noel A, Delvenne P, Herfs M. 2014. Altered alpha-defensin 5 expression in cervical squamocolumnar junction: implication in the formation of a viral/tumour-permissive microenvironment. J Pathol 234:464–477. doi: 10.1002/path.4435. [DOI] [PubMed] [Google Scholar]
- 29.Erhart W, Alkasi O, Brunke G, Wegener F, Maass N, Arnold N, Arlt A, Meinhold-Heerlein I. 2011. Induction of human beta-defensins and psoriasin in vulvovaginal human papillomavirus-associated lesions. J Infect Dis 204:391–399. doi: 10.1093/infdis/jir079. [DOI] [PubMed] [Google Scholar]
- 30.Kreuter A, Skrygan M, Gambichler T, Brockmeyer NH, Stucker M, Herzler C, Potthoff A, Altmeyer P, Pfister H, Wieland U. 2009. Human papillomavirus-associated induction of human beta-defensins in anal intraepithelial neoplasia. Br J Dermatol 160:1197–1205. doi: 10.1111/j.1365-2133.2009.09090.x. [DOI] [PubMed] [Google Scholar]
- 31.Andersen JM, Al-Khairy D, Ingalls RR. 2006. Innate immunity at the mucosal surface: role of Toll-like receptor 3 and Toll-like receptor 9 in cervical epithelial cell responses to microbial pathogens. Biol Reprod 74:824–831. doi: 10.1095/biolreprod.105.048629. [DOI] [PubMed] [Google Scholar]
- 32.Hasan UA, Bates E, Takeshita F, Biliato A, Accardi R, Bouvard V, Mansour M, Vincent I, Gissmann L, Iftner T, Sideri M, Stubenrauch F, Tommasino M. 2007. TLR9 expression and function is [sic] abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol 178:3186–3197. doi: 10.4049/jimmunol.178.5.3186. [DOI] [PubMed] [Google Scholar]
- 33.Kalali BN, Kollisch G, Mages J, Muller T, Bauer S, Wagner H, Ring J, Lang R, Mempel M, Ollert M. 2008. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J Immunol 181:2694–2704. doi: 10.4049/jimmunol.181.4.2694. [DOI] [PubMed] [Google Scholar]
- 34.Karim R, Meyers C, Backendorf C, Ludigs K, Offringa R, van Ommen GJ, Melief CJ, van der Burg SH, Boer JM. 2011. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One 6:e17848 doi: 10.1371/journal.pone.0017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebre MC, van der Aar AM, van Baarsen L, van Capel TM, Schuitemaker JH, Kapsenberg ML, de Jong EC. 2007. Human keratinocytes express functional Toll-like receptor [sic] 3, 4, 5, and 9. J Investig Dermatol 127:331–341. doi: 10.1038/sj.jid.5700530. [DOI] [PubMed] [Google Scholar]
- 36.Tristram A, Hurt CN, Madden T, Powell N, Man S, Hibbitts S, Dutton P, Jones S, Nordin AJ, Naik R, Fiander A, Griffiths G. 2014. Activity, safety, and feasibility of cidofovir and imiquimod for treatment of vulval intraepithelial neoplasia (RT3VIN): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 15:1361–1368. doi: 10.1016/S1470-2045(14)70456-5. [DOI] [PubMed] [Google Scholar]
- 37.Grimm C, Polterauer S, Natter C, Rahhal J, Hefler L, Tempfer CB, Heinze G, Stary G, Reinthaller A, Speiser P. 2012. Treatment of cervical intraepithelial neoplasia with topical imiquimod: a randomized controlled trial. Obstet Gynecol 120:152–159. doi: 10.1097/AOG.0b013e31825bc6e8. [DOI] [PubMed] [Google Scholar]
- 38.Daayana S, Elkord E, Winters U, Pawlita M, Roden R, Stern PL, Kitchener HC. 2010. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer 102:1129–1136. doi: 10.1038/sj.bjc.6605611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stern PL, van der Burg SH, Hampson IN, Broker TR, Fiander A, Lacey CJ, Kitchener HC, Einstein MH. 2012. Therapy of human papillomavirus-related disease. Vaccine 30(Suppl 5):F71–F82. doi: 10.1016/j.vaccine.2012.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. 2006. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J Gen Virol 87:3183–3193. doi: 10.1099/vir.0.82098-0. [DOI] [PubMed] [Google Scholar]
- 41.Rácz E, Prens EP, Kant M, Florencia E, Jaspers NG, Laman JD, de Ridder D, van der Fits L. 2011. Narrowband ultraviolet B inhibits innate cytosolic double-stranded RNA receptors in psoriatic skin and keratinocytes. Br J Dermatol 164:838–847. doi: 10.1111/j.1365-2133.2010.10169.x. [DOI] [PubMed] [Google Scholar]
- 42.Prens EP, Kant M, van Dijk G, van der Wel LI, Mourits S, van der Fits L. 2008. IFN-alpha enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: relevance for psoriasis. J Investig Dermatol 128:932–938. doi: 10.1038/sj.jid.5701087. [DOI] [PubMed] [Google Scholar]
- 43.Karim R, Tummers B, Meyers C, Biryukov JL, Alam S, Backendorf C, Jha V, Offringa R, van Ommen GJ, Melief CJ, Guardavaccaro D, Boer JM, van der Burg SH. 2013. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte's innate immune response. PLoS Pathog 9:e1003384 doi: 10.1371/journal.ppat.1003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller LS, Sorensen OE, Liu PT, Jalian HR, Eshtiaghpour D, Behmanesh BE, Chung W, Starner TD, Kim J, Sieling PA, Ganz T, Modlin RL. 2005. TGF-alpha regulates TLR expression and function on epidermal keratinocytes. J Immunol 174:6137–6143. doi: 10.4049/jimmunol.174.10.6137. [DOI] [PubMed] [Google Scholar]
- 45.Li ZJ, Sohn KC, Choi DK, Shi G, Hong D, Lee HE, Whang KU, Lee YH, Im M, Lee Y, Seo YJ, Kim CD, Lee JH. 2013. Roles of TLR7 in activation of NF-kappaB signaling of keratinocytes by imiquimod. PLoS One 8:e77159 doi: 10.1371/journal.pone.0077159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stadnyk AW. 1994. Cytokine production by epithelial cells. FASEB J 8:1041–1047. [DOI] [PubMed] [Google Scholar]
- 47.Woodworth CD, Simpson S. 1993. Comparative lymphokine secretion by cultured normal human cervical keratinocytes, papillomavirus-immortalized, and carcinoma cell lines. Am J Pathol 142:1544–1555. [PMC free article] [PubMed] [Google Scholar]
- 48.Feliciani C, Gupta AK, Sauder DN. 1996. Keratinocytes and cytokine/growth factors. Crit Rev Oral Biol Med 7:300–318. doi: 10.1177/10454411960070040101. [DOI] [PubMed] [Google Scholar]
- 49.Barker JN, Mitra RS, Griffiths CE, Dixit VM, Nickoloff BJ. 1991. Keratinocytes as initiators of inflammation. Lancet 337:211–214. doi: 10.1016/0140-6736(91)92168-2. [DOI] [PubMed] [Google Scholar]
- 50.Scott M, Nakagawa M, Moscicki AB. 2001. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol 8:209–220. doi: 10.1128/CDLI.8.2.209-220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woodworth CD, Notario V, DiPaolo JA. 1990. Transforming growth factors beta 1 and 2 transcriptionally regulate human papillomavirus (HPV) type 16 early gene expression in HPV-immortalized human genital epithelial cells. J Virol 64:4767–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kyo S, Inoue M, Hayasaka N, Inoue T, Yutsudo M, Tanizawa O, Hakura A. 1994. Regulation of early gene expression of human papillomavirus type 16 by inflammatory cytokines. Virology 200:130–139. doi: 10.1006/viro.1994.1171. [DOI] [PubMed] [Google Scholar]
- 53.Lembo D, Donalisio M, De Andrea M, Cornaglia M, Scutera S, Musso T, Landolfo S. 2006. A cell-based high-throughput assay for screening inhibitors of human papillomavirus-16 long control region activity. FASEB J 20:148–150. doi: 10.1096/fj.05-3904fje. [DOI] [PubMed] [Google Scholar]
- 54.Braun L, Durst M, Mikumo R, Gruppuso P. 1990. Differential response of nontumorigenic and tumorigenic human papillomavirus type 16-positive epithelial cells to transforming growth factor beta 1. Cancer Res 50:7324–7332. [PubMed] [Google Scholar]
- 55.Kowli S, Velidandla R, Creek KE, Pirisi L. 2013. TGF-beta regulation of gene expression at early and late stages of HPV16-mediated transformation of human keratinocytes. Virology 447:63–73. doi: 10.1016/j.virol.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 57.Fensterl V, Sen GC. 2009. Interferons and viral infections. Biofactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 58.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. 2001. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol 75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reiser J, Hurst J, Voges M, Krauss P, Munch P, Iftner T, Stubenrauch F. 2011. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol 85:11372–11380. doi: 10.1128/JVI.05279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, Sangkomkamhang U, Ekalaksananan T. 2014. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-kappa transcription in keratinocytes. PLoS One 9:e91473 doi: 10.1371/journal.pone.0091473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84:431–442. doi: 10.1016/S0092-8674(00)81288-X. [DOI] [PubMed] [Google Scholar]
- 63.LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V, Chen G, Ruben SM, Pitha PM, Coleman TA, Moore PA. 2001. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem 276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 64.Rincon-Orozco B, Halec G, Rosenberger S, Muschik D, Nindl I, Bachmann A, Ritter TM, Dondog B, Ly R, Bosch FX, Zawatzky R, Rosl F. 2009. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res 69:8718–8725. doi: 10.1158/0008-5472.CAN-09-0550. [DOI] [PubMed] [Google Scholar]
- 65.DeCarlo CA, Severini A, Edler L, Escott NG, Lambert PF, Ulanova M, Zehbe I. 2010. IFN-kappa, a novel type I IFN, is undetectable in HPV-positive human cervical keratinocytes. Lab Invest 90:1482–1491. doi: 10.1038/labinvest.2010.95. [DOI] [PubMed] [Google Scholar]
- 66.Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, Homey B, Massacrier C, Vanbervliet B, Zlotnik A, Vicari A. 2000. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin Immunopathol 22:345–369. doi: 10.1007/s002810000053. [DOI] [PubMed] [Google Scholar]
- 67.Appay V, Rowland-Jones SL. 2001. RANTES: a versatile and controversial chemokine. Trends Immunol 22:83–87. doi: 10.1016/S1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- 68.Charbonnier AS, Kohrgruber N, Kriehuber E, Stingl G, Rot A, Maurer D. 1999. Macrophage inflammatory protein 3alpha is involved in the constitutive trafficking of epidermal Langerhans cells. J Exp Med 190:1755–1768. doi: 10.1084/jem.190.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang SM, McCance DJ. 2002. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol 76:8710–8721. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kleine-Lowinski K, Rheinwald JG, Fichorova RN, Anderson DJ, Basile J, Munger K, Daly CM, Rosl F, Rollins BJ. 2003. Selective suppression of monocyte chemoattractant protein-1 expression by human papillomavirus E6 and E7 oncoproteins in human cervical epithelial and epidermal cells. Int J Cancer 107:407–415. doi: 10.1002/ijc.11411. [DOI] [PubMed] [Google Scholar]
- 72.Guess JC, McCance DJ. 2005. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J Virol 79:14852–14862. doi: 10.1128/JVI.79.23.14852-14862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caberg JH, Hubert P, Herman L, Herfs M, Roncarati P, Boniver J, Delvenne P. 2009. Increased migration of Langerhans cells in response to HPV16 E6 and E7 oncogene silencing: role of CCL20. Cancer Immunol Immunother 58:39–47. doi: 10.1007/s00262-008-0522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang A, Amagai M, Granger LG, Stanley JR, Udey MC. 1993. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 361:82–85. doi: 10.1038/361082a0. [DOI] [PubMed] [Google Scholar]
- 75.Mayumi N, Watanabe E, Norose Y, Watari E, Kawana S, Geijtenbeek TB, Takahashi H. 2013. E-cadherin interactions are required for Langerhans cell differentiation. Eur J Immunol 43:270–280. doi: 10.1002/eji.201242654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jakob T, Ring J, Udey MC. 2001. Multistep navigation of Langerhans/dendritic cells in and out of the skin. J Allergy Clin Immunol 108:688–696. doi: 10.1067/mai.2001.118797. [DOI] [PubMed] [Google Scholar]
- 77.Hubert P, Caberg JH, Gilles C, Bousarghin L, Franzen-Detrooz E, Boniver J, Delvenne P. 2005. E-cadherin-dependent adhesion of dendritic and Langerhans cells to keratinocytes is defective in cervical human papillomavirus-associated (pre)neoplastic lesions. J Pathol 206:346–355. doi: 10.1002/path.1771. [DOI] [PubMed] [Google Scholar]
- 78.Matthews K, Leong CM, Baxter L, Inglis E, Yun K, Backstrom BT, Doorbar J, Hibma M. 2003. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated down regulation of E-cadherin. J Virol 77:8378–8385. doi: 10.1128/JVI.77.15.8378-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakayama Y, Asagoe K, Yamauchi A, Yamamoto T, Shirafuji Y, Morizane S, Nakanishi G, Iwatsuki K. 2011. Dendritic cell subsets and immunological milieu in inflammatory human papilloma virus-related skin lesions. J Dermatol Sci 63:173–183. doi: 10.1016/j.jdermsci.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 80.Connor JP, Ferrer K, Kane JP, Goldberg JM. 1999. Evaluation of Langerhans' cells in the cervical epithelium of women with cervical intraepithelial neoplasia. Gynecol Oncol 75:130–135. doi: 10.1006/gyno.1999.5559. [DOI] [PubMed] [Google Scholar]
- 81.Gaglia JL, Greenfield EA, Mattoo A, Sharpe AH, Freeman GJ, Kuchroo VK. 2000. Intercellular adhesion molecule 1 is critical for activation of CD28-deficient T cells. J Immunol 165:6091–6098. doi: 10.4049/jimmunol.165.11.6091. [DOI] [PubMed] [Google Scholar]
- 82.Black AP, Ardern-Jones MR, Kasprowicz V, Bowness P, Jones L, Bailey AS, Ogg GS. 2007. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur J Immunol 37:1485–1493. doi: 10.1002/eji.200636915. [DOI] [PubMed] [Google Scholar]
- 83.Grousson J, Concha M, Schmitt D, Péguet-Navarro J. 1998. Effects of CD40 ligation on human keratinocyte accessory function. Arch Dermatol Res 290:325–330. doi: 10.1007/s004030050312. [DOI] [PubMed] [Google Scholar]
- 84.Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kaliński P. 2000. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol 164:4507–4512. doi: 10.4049/jimmunol.164.9.4507. [DOI] [PubMed] [Google Scholar]
- 85.Kaliński P, Hilkens CM, Wierenga EA, Kapsenberg ML. 1999. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today 20:561–567. doi: 10.1016/S0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 86.Lebre MC, Antons JC, Kaliński P, Schuitemaker JH, van Capel TM, Kapsenberg ML, De Jong EC. 2003. Double-stranded RNA-exposed human keratinocytes promote Th1 responses by inducing a type-1 polarized phenotype in dendritic cells: role of keratinocyte-derived tumor necrosis factor alpha, type I interferons, and interleukin-18. J Investig Dermatol 120:990–997. doi: 10.1046/j.1523-1747.2003.12245.x. [DOI] [PubMed] [Google Scholar]
- 87.Altenburg A, Baldus SE, Smola H, Pfister H, Hess S. 1999. CD40 ligand-CD40 interaction induces chemokines in cervical carcinoma cells in synergism with IFN-gamma. J Immunol 162:4140–4147. [PubMed] [Google Scholar]
- 88.King AE, Kelly RW, Critchley HO, Malmstrom A, Sennstrom M, Phipps RP. 2001. Cd40 expression in uterine tissues: a key regulator of cytokine expression by fibroblasts. J Clin Endocrinol Metab 86:405–412. doi: 10.1210/jcem.86.1.7133. [DOI] [PubMed] [Google Scholar]
- 89.Hill SC, Youde SJ, Man S, Teale GR, Baxendale AJ, Hislop A, Davies CC, Luesley DM, Blom AM, Rickinson AB, Young LS, Eliopoulos AG. 2005. Activation of CD40 in cervical carcinoma cells facilitates CTL responses and augments chemotherapy-induced apoptosis. J Immunol 174:41–50. doi: 10.4049/jimmunol.174.1.41. [DOI] [PubMed] [Google Scholar]
- 90.Denfeld RW, Hollenbaugh D, Fehrenbach A, Weiss JM, von Leoprechting A, Mai B, Voith U, Schopf E, Aruffo A, Simon JC. 1996. CD40 is functionally expressed on human keratinocytes. Eur J Immunol 26:2329–2334. doi: 10.1002/eji.1830261009. [DOI] [PubMed] [Google Scholar]
- 91.Gaspari AA, Sempowski GD, Chess P, Gish J, Phipps RP. 1996. Human epidermal keratinocytes are induced to secrete interleukin-6 and co-stimulate T lymphocyte proliferation by a CD40-dependent mechanism. Eur J Immunol 26:1371–1377. doi: 10.1002/eji.1830260629. [DOI] [PubMed] [Google Scholar]
- 92.Péguet-Navarro J, Dalbiez-Gauthier C, Moulon C, Berthier O, Reano A, Gaucherand M, Banchereau J, Rousset F, Schmitt D. 1997. CD40 ligation of human keratinocytes inhibits their proliferation and induces their differentiation. J Immunol 158:144–152. [PubMed] [Google Scholar]
- 93.Pasch MC, Timar KK, van Meurs M, Heydendael VM, Bos JD, Laman JD, Asghar SS. 2004. In situ demonstration of CD40- and CD154-positive cells in psoriatic lesions and keratinocyte production of chemokines by CD40 ligation in vitro. J Pathol 203:839–848. doi: 10.1002/path.1581. [DOI] [PubMed] [Google Scholar]
- 94.Tummers B, Goedemans R, Jha V, Meyers C, Melief CJ, van der Burg SH, Boer JM. 2014. CD40-mediated amplification of local immunity by epithelial cells is impaired by HPV. J Investig Dermatol 134:2918–2927. doi: 10.1038/jid.2014.262. [DOI] [PMC free article] [PubMed] [Google Scholar]