

Abstract
Candida glabrata is reported as the second most prevalent human opportunistic fungal pathogen in the United States. Over the last decades, its incidence increased, whereas that of Candida albicans decreased slightly. One of the main reasons for this shift is attributed to the inherent tolerance of C. glabrata toward the commonly used azole antifungal drugs. Despite a close phylogenetic distance to Saccharomyces cerevisiae, homologous recombination works with poor efficiency in C. glabrata compared to baker's yeast, in fact limiting targeted genetic alterations of the pathogen's genome. It has been shown that nonhomologous DNA end joining is dominant over specific gene targeting in C. glabrata. To improve the homologous recombination efficiency, we have generated a strain in which the LIG4 gene has been deleted, which resulted in a significant increase in correct gene targeting. The very specific function of Lig4 in mediating nonhomologous end joining is the reason for the absence of clear side effects, some of which affect the ku80 mutant, another mutant with reduced nonhomologous end joining. We also generated a LIG4 reintegration cassette. Our results show that the lig4 mutant strain may be a valuable tool for the C. glabrata research community.
INTRODUCTION
Candida spp. are part of the commensal microbiota in most humans. In healthy individuals they do not cause disease, but in immunocompromised patients they can result in systemic infections with a high mortality rate (1–3). The most commonly isolated Candida species is C. albicans, but whereas its incidence is slightly decreasing, the incidence of another species, C. glabrata, is on the rise (4). One of the underlying causes is that C. glabrata is inherently tolerant to azole antifungals, the most commonly used family of drugs against Candida spp. (4–7). Because of this, more and more research groups have started to work on this pathogen. To identify virulence factors or the genetic information that explains why this species is tolerant toward azoles, it would be important to have a genome-wide deletion strain collection. For the closely related species Saccharomyces cerevisiae, such a collection already has been available to the research community for more than 1 decade, and it has contributed to increased understanding of the cell biology of this species (8). A major problem is that homologous recombination in C. glabrata is far less efficient than in S. cerevisiae. In S. cerevisiae, efficient gene targeting requires typically 40 bp of homologous flanking sequence at each end, and gene disruption cassettes can easily be made by PCR using 60- to 80-bp oligonucleotides. However, like many other Candida species, in C. glabrata, much longer flanking sequences of up to 400 bp on each side are needed. This can also be performed by PCR, using a two-step PCR approach, in which 5′ and 3′ flanking sequences are first amplified and the products obtained are then used as primers to amplify a marker gene to replace one's favorite gene in the genome. This method was first developed for C. albicans (9) and was recently used to generate a collection of 619 deletion strains in the C. glabrata ATCC 2001 background (10). Compared to C. albicans, however, this method still results in many false positives when performed with C. glabrata, i.e., many transformants carrying nonspecific genomic integrations of the disruption cassette are obtained. Integration of foreign DNA into the genome requires either functional nonhomologous end-joining (NHEJ) or homologous recombination (HR), the two mechanisms to repair the double-stranded DNA breaks (11, 12). HR targets DNA by homologous sequences, while NHEJ does not (11, 13). The low gene-targeting efficiency of C. glabrata is due to a very efficient NHEJ and less efficient HR.
The process of NHEJ is less complicated in yeasts than in mammalian cells, where it requires many proteins. In yeast, NHEJ depends on the combined action of KU genes and DNA ligase IV (13, 14). Deletions of genes homologous to the human genes KU70 and KU80 were shown to decrease the number of NHEJ events, resulting in an increase in correct gene targeting, in Neurospora crassa (15), Kluyveromyces lactis (16), Cryptococcus neoformans (17), and some Aspergillus spp. (18–20). Transcriptional repression of KU80 in C. glabrata also resulted in an increased gene-targeting efficiency (21). However, KU genes are also involved in telomere length maintenance and regulation of gene silencing at telomeres. Telomere sequences are enriched for genes involved in adhesion to biotic and abiotic substrates, which is one of the main virulence factors of C. glabrata (22). One family of adhesins, the EPA gene family, plays a major role in this process, and it is well established that Ku70/Ku80 are involved in the regulation of subtelomeric silencing of some EPA genes, so that modification of this part of the NHEJ mechanism may have undesired consequences affecting the physiology of C. glabrata (23). Another component involved in the NHEJ process is ligase IV (Lig4). This enzyme was previously shown, in other organisms, to be more specific for the NHEJ process, so its deletion may result in fewer side effects (12, 13, 24–27). To test whether deletion of LIG4 in C. glabrata results in strongly improved gene targeting without any unwanted phenotype, we generated a deletion in this gene (CAGL0E02695g), and we show that the corresponding mutants display a strong increase in correct gene targeting. Differently from the ku80 mutants, no detectable side effects could be observed on growth, DNA stress tolerance, or antifungal drug resistance. Lig4 mutants show apparently no phenotype that should be of concern to the research community, other than an increase in the gene-targeting efficiency. Despite this, we also generated a LIG4 reintegration construct for reconstitution of LIG4 in its original locus once the desired genetic alterations have been performed. Thus, we provide a useful strain and plasmid that can serve as a basis for targeted engineering in this human fungal pathogen.
MATERIALS AND METHODS
Yeast strains, primers, and media.
The C. glabrata strains used in this study are listed in Table 1. C. glabrata cells were grown in either liquid YPD (1% yeast extract, 2% peptone, 2% dextrose) or synthetic complete (SC; 1.7 g/liter Difco yeast nitrogen base without ammonium sulfate, 0.79 g/liter complete supplement mixture [CSM; MP Biomedicals], 5 g/liter ammonium sulfate, supplemented with 2% dextrose) medium. For selection of transformants, l-histidine, adenine, or l-arginine was omitted from the SC medium. For solid media, 1.5 g/100 ml Difco agar granulated was supplemented to the liquid medium mentioned.
TABLE 1.
Strains used in this study
| Strain | Genotype or description | Reference or source |
|---|---|---|
| 2001HT | his3Δ trp1Δ (made from CBS138) | 28 |
| AFG1 (lig4-A) | lig4::HIS3 trp1Δ isolate A (made from 2001HT) | This study |
| AFG2 (lig4-B) | lig4::HIS3 trp1Δ isolate B (made from 2001HT) | This study |
| CYC001 | ade2::SAT1 his3Δ trp1Δ isolate A | This study |
| CYC002 | ade2::SAT1 his3Δ trp1Δ isolate B | This study |
| CYC003 | ade2::SAT1 lig4::HIS3 trp1Δ isolate A | This study |
| CYC004 | ade2::SAT1 lig4::HIS3 trp1Δ isolate B | This study |
| CYC005 | arg8::SAT1 his3Δ trp1Δ isolate A | This study |
| CYC006 | arg8::SAT1 lig4::HIS3 trp1Δ isolate A | This study |
| CYC007 | arg8::SAT1 lig4::HIS3 trp1Δ isolate B | This study |
| CYC008 | cna1::SAT1 his3Δ trp1Δ isolate A | This study |
| CYC009 | 2001HT ku80::SAT1 isolate A | This study |
| CYC010 | 2001HT ku80::SAT1 isolate B | This study |
| CYC011 | 2001HT ku80::SAT1 isolate C | This study |
| KUE200 | 2001HT ku80::SAT1 | 21 |
Full-length gene sequences were obtained from the Candida Genome Database (http://www.candidagenome.org/). The primers used in this study are listed in Table S1 in the supplemental material. These sequences were compared to those in the Candida Genome Database using BLAST in order to determine their specificity.
Construction of LIG4 deletion strains and KU80 deletion strains.
We used the fusion PCR method to generate the deletion of LIG4 (9). The 500-bp 5′ flanking region was amplified from genomic DNA using primers Cglig4-a and Cglig4-b. The 500-bp 3′ flanking site was amplified using primes Cglig4-c and Cglig4-d. The ScHIS3 marker was amplified using primers Schis3-for and Schis3-rev with plasmid pRS423 (GenBank accession number U03454) as the template. The whole deletion cassette was amplified by fusion PCR using the three amplified fragments due to the complementary tails present in Schis3-for and Cglig4-b and in Schis3-rev and Cglig4-c, respectively. The cassette was transformed in C. glabrata strain 2001HT (28) by electroporation (0.2-cm cuvette, 1.5 kV) as previously described (29). Genomic DNA from selected transformants was isolated using the FastPrep method. For the identification of correct lig4Δ strains, genomic DNA, prepared using the FastPrep method, was used as the template for PCR to check for correct deletion of LIG4 as well as for its correct replacement by the HIS3 marker. Primers cglig4-ko-check-for/Schis3-rev and cglig4-ko-check-rev/Schis3-for were used to test the replacement of LIG4 by ScHIS3 gene. Primers cglig4-ko-check-for/cglig4-check-inrev and primers cglig4-ko-check-rev/cglig4-check-infor were used to test the loss of LIG4 from the genome. Genomic DNA from the wild-type strain was used as a control.
We have used the same approach to generate the KU80 deletion strain. The 500-bp 5′ flanking region was amplified using primers Ku80-a and Ku80-b. The 500-bp 3′ flanking side was amplified using primers Ku80-c and Ku80-d. The SAT1 marker was amplified using primers KU80-SAT-for and KU80-SAT-rev from plasmid pSFS2 (GenBank accession number AY524979). The whole deletion cassette was amplified by fusion PCR using the three amplified fragments due to the complementary tails present in KU80-SAT-for and Ku80-b and in KU80-SAT-rev and Ku80-c, respectively. Primers KU80-CHECK-FOR/L4-REV-3 and KU80-CHECK-REV/L4-FOR-9 were used to test the replacement of KU80 by the SAT1 gene. Primers KU80-CHECK-FOR/KU80-CHECK-INREV and KU80-CHECK-REV/KU80-CHECK-INFOR were used to test the loss of KU80 from the genome. Apart from our own ku80 mutants, we also used a deletion strain (KUE200) that was previously generated and kindly provided to us by H. Chibana (Japan) (21).
Construction of ADE2, ARG8, and CNA1 deletion strains.
To delete the ADE2 gene, we used 40-bp and 100-bp flanking regions fused with the SAT1 marker. The SAT1 marker deletion cassettes were amplified either with cgade2-ko-sat-for and cgade2-ko-sat-rev or with 100ADE2-ko-sat-for and 100ADE2-ko-sat-rev from plasmid pSFS2. Amplified DNA fragments were then transformed with electroporation as described above. Putative correct transformants were identified by color and adenine auxotrophy and by colony PCR (30). Primers cgade2-ko-check-for/cgade2-check-inrev and cgade2-ko-check-rev/cgade2-check-infor were used to test the loss of ADE2. Primers cgade2-ko-check-for/Cgade2-ko-sat-rev and cgade2-ko-check-rev/Cgade2-ko-sat-for were used to test for the correct replacement of ADE2 by the SAT1 marker gene.
A similar approach was used to generate ARG8 and CNA1 deletion strains. We used 40-bp flanking sites to generate the deletion cassettes. Primers ARG-KO-FOR and ARG-KO-REV and primers CNA1-KO-FOR and CNA1-KO-REV were used to amplify the SAT1 marker from plasmid pSFS2 to generate the ARG8 and CNA1 deletion cassettes, respectively.
Primers ARG-CHECK-FOR/ARG-CHECK-INREV and ARG-CHECK-REV/ARG-CHECK-INFOR were used to test the loss of ARG8. Primers ARG-CHECK-FOR/L4-REV-3 and ARG-CHECK-REV/L4-FOR-9 were used to test for the correct replacement of ARG8 by the SAT1 marker gene.
Primers CNA1-CHECK-FOR/CNA1-CHECK-INREV and CNA1-CHECK-REV/CNA1-CHECK-INFOR were used to test the loss of CNA1. Primers CNA1-CHECK-FOR/L4-REV-3 and CNA1-CHECK-REV/L4-FOR-9 were used to test for the correct replacement of CNA1 by the SAT1 marker gene.
Generation of the LIG4 reintegration construct.
The LIG4 reintegration construct was made using the Gibson Assembly master mix (provided by New England BioLabs) and assembled in the bacterial vector pUC18 (GenBank accession number L09136). The LIG4 open reading frame (ORF) together with a 500-bp promoter sequence was amplified by PCR using primers Plig4-for-EcoRI-1 and Plig4-rev-2. The ScADH1 terminator was inserted downstream of the LIG4 fragment and amplified by primers ADH1-ter-for and ADH1-ter-rev from plasmid pBEVY-T (GenBank accession number AF069723). The nourseothricin marker gene SAT1 was amplified by primers Plig4-SAT1-for and Plig4-SAT1-rev from plasmid pSFS2, while the 500-bp LIG4 terminator was amplified by primers PLIG4-TER-FOR and PLIG4-TER-EcoRI-REV. Plasmid pUC18 was digested with restriction enzymes BamHI and EcoRI. The five linear fragments were assembled using the Gibson Assembly kit, and the resulting plasmid was checked by sequence analysis.
To further increase the efficiency of the reintegration cassette, we inserted a fragment in the EcoRI site that contains the recognition sequence of the CspCI restriction endonuclease, which is a double digesting enzyme, and that was amplified using the oligonucleotides LIG4-REIN-CSPCI-FOR and LIG4-REIN-CSPCI-REV and pUC18 as the template. Digestion by CspCI will result in the complete removal of nonhomologous DNA (EcoRI restriction site), leaving homologous flanking sites.
Southern blotting.
Southern blotting was performed to determine the level of ectopic integration. Briefly, genomic DNA from overnight cultures of the ade2, arg8, cna1, and ku80 mutant strains as well as the wild-type strain was isolated and digested for 14 h with EcoRI, EcoRV, EcoRI, and KpnI, respectively. A 0.8% agarose gel was run overnight at 40 V, and the DNA was transferred to a Hybond-NX membrane (Amersham Biosciences). The different probes were generated by PCR using ADE-sb-for and ADE-sb-rev, ARG8-sb-for and ARG8-sb-rev, CNA1-sb-for and CNA1-sb-rev, and KU80-sb-for and KU80-sb-rev, respectively. The probes (200 bp to 300 bp) were located in the flanking region of the deleted genes (ADE2, ARG8, CNA1, and KU80, respectively). Primers L4-FOR-8 and L4-REV-2 were used to amplify the probe for detection of the SAT1 marker. Generation of the probe and hybridization were performed using the AlkPhos direct-labeling reagents (Amersham GE Healthcare) according to the instructions in the manual. The signals were detected using the CDP-Star detection reagent (Amersham, GE Healthcare).
Real-time PCR analysis.
For expression analysis, overnight cultures were diluted to an optical density at 600 nm (OD600) of 0.2 in YPD and were harvested after either 4 h (exponential phase) or 24 h (stationary phase) of incubation at 37°C. RNA was extracted by the TRIzol RNA isolation reagent (Life technologies). The amount of RNA was quantified by a NanoDrop spectrophotometer (ND-1000; Life Science). After DNase treatment, the RNA concentration was adjusted to a final concentration of 1 μg/μl. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed in 96-well plates using the Step One Plus real-time PCR system (Applied Biosystems) and the Go Taq qPCR Master Mix (Promega). Five microliters of 0.4 ng/μl cDNA samples and 15 μl of Master Mix (containing the primers) were added to the plates. Real-time PCRs were performed at 95°C for 2 min, followed by 40 cycles of 3 s at 95°C and 30 s at 60°C. Samples were then kept at 95°C for 15 s and 60°C for 1 min. Finally, samples were subjected to incubations at increasing temperatures from 60°C to 95°C and kept 15 s at 95°C. ACT1 was used as a control gene, and normalized data were then used to calculate the relative gene expression levels. Data for each target gene were calculated as the fold change in comparison with the reference gene ACT1 using the ΔΔCT quantification method (where CT is threshold cycle) (31).
The sequences of the primers are given in Table S1 in the supplemental material. The quantitative PCR results were obtained from 3 independent biological repeats.
Growth and stress experiments.
To determine whether the absence of Lig4 had no effect on growth and stress tolerance (especially DNA stress), we tested two independent lig4 mutants under different growth conditions, including different media (YPD, SC, and RPMI [RPMI 1640 from Sigma]), as well as upon applying different types of stress to the cells such as different temperatures (23°C, 30°C, 37°C, and 42°C), different pHs (pH 2, 5, or 8), different concentrations of SDS (0.02% and 0.04%), calcofluor white (CFW; 0.5 mg/ml and 1 mg/ml), Congo red (3 mg/ml and 6 mg/ml), amphotericin B (1 μg/ml), caspofungin (0.04 μg/ml and 0.16 μg/ml), sodium chloride (1 M), fetal bovine serum (40% and 50%), ethyl methanesulfonate (EMS; 0.01%, 0.05%, 0.1%, 0.2%, and 0.4%), H2O2 (5 mM, 10 mM, and 25 mM), and hydroxyurea (HOU; 65 mM), and different doses of UV, and we compared (some of) the phenotypes with those obtained for a wild-type strain as well as with those obtained for two independent ku80 mutants and the published KUE200 strain. The stress tolerance assays were done by spot assays with a starting OD600 of 1 and plating 10-fold serial dilutions using YPD agar media under different conditions or mixed with corresponding concentrations of stress reagents. Different pH YPD agar media were buffered by 150 mM HEPES. For UV stress assays, overnight cultures were diluted to an OD600 of 0.2 in YPD medium. Further 10-fold dilutions were applied using YPD. One hundred microliters of the suspensions was spread onto YPD agar plates, and different UV energy doses were applied (using the Bio-Rad GS Gene Linker UV Chamber, C-L program), while the control plates were kept unexposed to UV light. Plates were incubated for 24 h at 37°C, and the numbers of surviving cells were determined by counting the CFU.
MICs using the broth dilution assay and Etest strips.
The MIC was determined by broth dilution assay according to the CLSI standard reference method M27-S4 (32). Briefly, cells were harvested from overnight YPD culture and diluted to around 2,500 CFU/ml (verified by plating serial dilution on YPD plates) with RPMI medium (RPMI 1640). One hundred microliters of the diluted culture was added to each well. MIC plates were incubated at 37°C for 24 h. The MIC50 values were defined as the lowest concentration of the antifungal (either fluconazole, caspofungin, or amphotericin B) that caused a ≥50% decrease in OD600. The broth dilution assays were repeated 3 times independently.
Fluconazole MICs were also determined using Etest strips (AB Biodisk) on RPMI-glucose plates. Cells were harvested from overnight cultures in YPD, diluted to an OD600 of 0.2, and plated using sterile swabs. Plates were incubated at 37°C for 48 h.
RESULTS AND DISCUSSION
To obtain a LIG4 deletion strain, we used a deletion construct consisting of a 500-bp promoter and 500-bp terminator sequence flanking the ScHIS3 gene. Among the ∼1,000 histidine prototrophic colonies generated by transformation of this construct and verified by colony PCR, two clones showing correct integration of the disruption cassette were found. This confirms that in C. glabrata (at least in the standard strain 2001HT), there is an extremely low HR and a high NHEJ efficiency, as in nearly all transformants the deletion cassette was integrated in a wrong place in the genome.
As previously used approaches to improve homologous recombination, such as deletion of KU70/KU80, result in side effects such as changes in subtelomeric silencing (23), we tested the two independent lig4Δ mutants (lig4-A and lig4-B) under different growth and stress conditions using spot assays (Fig. 1). Deletion of LIG4 has no effect on growth at different temperatures, at different pHs, or in different growth media. Similarly, the lig4Δ mutants show no altered sensitivity to cell wall and salt stress or to antifungals when added in the medium (Fig. 1). We also determined the MIC values for amphotericin B, caspofungin, and fluconazole, and they were also not changed in the two independent lig4 mutants compared to the wild-type strain and were 1 μg/ml, 0.1 μg/ml, and 16 μg/ml, respectively. As NHEJ is crucial for cell response to DNA damage, we also tested the susceptibility of the mutants to a number of conditions such as UV light, EMS, H2O2, and HOU (Fig. 2). The two independent LIG4 deletion strains show the same sensitivity toward all these stresses as the wild-type strain, suggesting that absence of Lig4 does not interfere with cell growth and stress tolerance, at least phenotypically under all tested stress conditions. The expression of adhesion-encoding genes is an important virulence factor of C. glabrata, and it was previously shown that the ku80 mutants resulted in differences in the expression of some of these genes (23). EPA6 and EPA7 are induced in ku80 mutants, whereas the expression levels of EPA1 and EPA2 are unaltered. In order to compare the expression of the EPA genes in our lig4 mutants with that in ku80 mutants in the same background, we have generated our own set of ku80 mutants. Expression of the four EPA genes in exponential or stationary phase was not significantly different between the wild type and the lig4 mutant, and also not in the ku80 mutant (see Fig. S1 in the supplemental material). A possible explanation for the normal EPA gene expression in our ku80 mutants compared to the previously published data is the strain background. Our ku80 mutants were generated in ATCC 2001, whereas the published data were obtained in the BG2 background. Apart from differences in expression of EPA genes, no other phenotypes of the ku80 mutants were previously described. In our effort to validate the lig4 mutants as much as possible, we identified environmental conditions that resulted in phenotypes for the ku80 mutants but not for the lig4 mutants. The ku80 mutants showed a clear phenotype when growing on media containing membrane or cell wall stress reagents (Fig. 3). Compared to the wild type and the lig4 mutants, the three independent ku80 mutants as well as the published ku80 mutant (KUE200) (21) showed an increased sensitivity toward SDS and CFW and an increased tolerance against fluconazole, not against caspofungin. MIC analysis for caspofungin confirmed these data. For fluconazole, however, the difference in sensitivity was confirmed using two different approaches. MIC determinations using the broth dilution method showed that the ku80 mutants had an increased MIC for fluconazole (32 μg/ml) compared to all other strains and that there was no difference in MIC between the wild-type strain and the lig4 mutants. This was confirmed using E-strips with fluconazole. The wild-type strain and the lig4 mutant showed an MIC of 8 μg/ml, whereas the ku80 (a) and KUE200 mutant strains have an MIC of 16 μg/ml (see Fig. S2 in the supplemental material).
FIG 1.

Deletion of LIG4 does not affect growth or stress tolerance of C. glabrata cells. (A) Growth curve of wild-type strain and the two lig4 mutants (AFG1 and AFG2) in YPD medium at 37°C. (B) The growth and stress tolerance of the wild type and two independent lig4 mutants were tested by spot assays under different conditions/stresses. Cells were assayed in different media (YPD, RPMI, SC) at 37°C and at different temperatures and pHs and on YPD supplemented with the indicated compounds at 37°C. Spot assays were done by 10-fold serial dilutions.
FIG 2.

DNA damage conditions affect the lig4 mutant in the same way as the wild-type strain. (A) Spot assays (10× serial dilutions) of wild-type and lig4 mutant cells on YPD plates containing different concentrations of mutagenic chemicals. Plates were incubated for 15 h at 37°C. (B) Survival of C. glabrata wild-type and lig4 mutant cells upon exposure to UV light. To calculate the survival rates, the number of CFU on the exposed plates was divided by the number of CFU on the unexposed plates and multiplied by 100. Cell viability was calculated based on three independent experiments.
FIG 3.
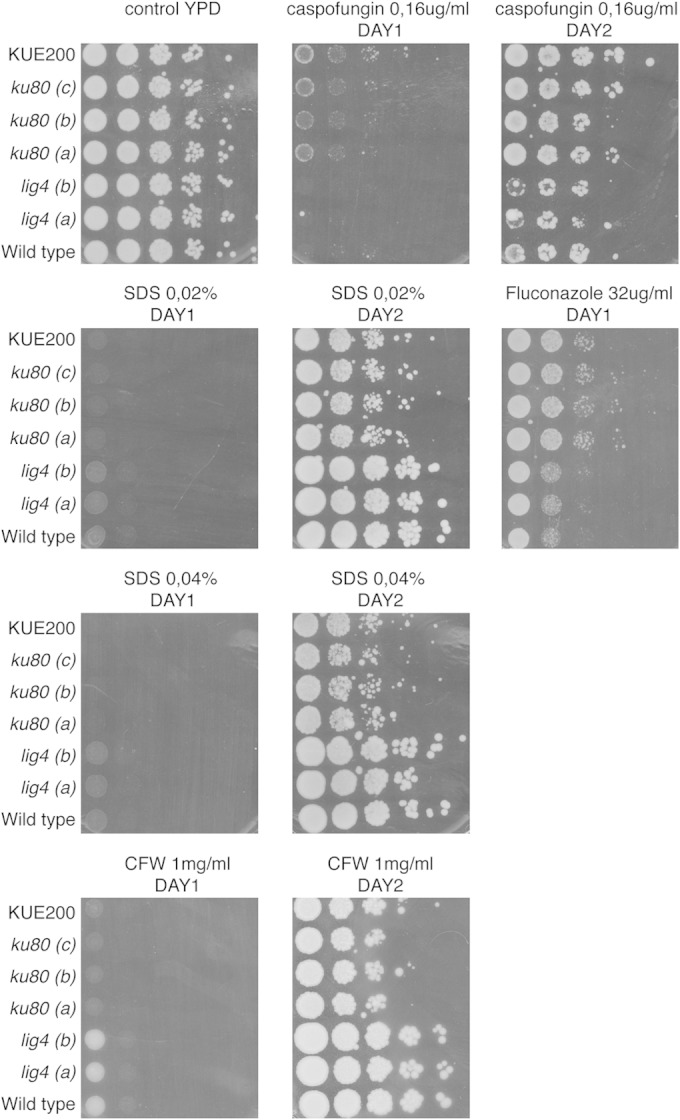
Deletion of KU80 affects cell membrane and cell wall stress tolerance of C. glabrata cells, whereas deletion of LIG4 has no effect. Two independent lig4 mutants (a and b), three independent ku80 mutants (a, b, and c), and the published ku80 mutant (21) were grown in overnight cultures, diluted using serial 10-fold dilutions, and plated on YPD medium containing caspofungin, fluconazole, SDS, and calcofluor white (CFW) at the indicated concentrations. Plates were incubated at 37°C for the indicated times.
Our phenotypic analysis shows that the lig4 mutant strain behaves exactly as does the wild type for all conditions that we have tested and therefore may be a very good strain to use for gene targeting, as it may strongly decrease the number of transformants that need to be checked to obtain the correct desired strain.
To determine whether the absence of Lig4 results in an improved gene-targeting efficiency, the wild-type strain and the two lig4Δ mutants were transformed with the SAT1 marker flanked by either 40 bp or 100 bp of promoter and terminator sequences of the target genes. As a proof of principle, we have chosen to delete the ADE2 gene. Deletion of ADE2 results in pink to red colonies because of the accumulation of a red compound in the cells (33). All transformants were screened for colony color as well as for adenine auxotrophy. For the constructs with the 100-bp flanking sequence, we obtained 120 transformants in the wild-type background and 22 in the lig4Δ background. Interestingly, only 2 transformants were found to be correct (based on color and adenine auxotrophy) in the wild-type background whereas 5 were correct in the lig4Δ mutant. These seven colonies together with 20 randomly picked white colonies from each transformation were checked with colony PCR for loss of ADE2 and replacement by the marker gene SAT1. Only the seven red colonies turned out to be the correct ones. Similar results were obtained when the flanking sequences were further reduced to 40 bp, which is the typical size of flanking sequences used in S. cerevisiae for gene-targeting approaches. In this case, 1 of the 368 transformants in the wild-type background and 3 of 32 transformants in the lig4Δ strain were correct. These results show that the efficiency of correct targeting is about 35 times higher in the lig4Δ strain than in the wild-type strain, when using 40-bp flanking sequences (Fig. 4). Using 100-bp flanking regions, the gene targeting efficiency was even higher in the lig4Δ mutant (Fig. 4).
FIG 4.

Gene-targeting efficiency is strongly increased in the lig4 mutant. The wild type and the lig4 (a) mutant were transformed with DNA fragments containing either 40- or 100-bp flanking sequences to target a deletion cassette to the ADE2 locus. The percentage of correct ADE2 deletion strains to the total number of transformants is shown.
To validate the use of the lig4Δ mutant for improved gene targeting, we repeated the experiment with ADE2 several independent times and also included two other genes, ARG8 and CNA1, which are located on different chromosomes. ARG8 encodes an enzyme involved in arginine biosynthesis, so the deletion of ARG8 results in auxotrophy for arginine. Cna1 is a subunit of the calcineurin phosphatase, and deletion of CNA1 has no easy detectible phenotype. Therefore, all CNA1 transformants were screened by colony PCR using primers CNA1-CHECK-FOR and CNA1-CHECK-INREV (see Table S1 in the supplemental material). Correct gene targeting for the three genes was further tested by colony PCR for loss of the target genes and their replacement by the SAT1 marker gene. Table 2 lists the numbers of transformants that we obtained and the numbers of correct deletion strains we selected and verified. The number of overall transformants clearly shows that the lig4 mutants resulted in a strongly reduced nonhomologous integration in comparison to the wild-type strain. The number of correct deletion strains remains more or less the same for the wild type and for the lig4 mutants. This also implies that Lig4 does not interfere with the homologous recombination pathway and is specific for the NHEJ pathway. To better demonstrate the increased gene targeting efficiency in the lig4Δ background compared to the wild-type strain, we calculated the relative aspecific integration (number of false-positive transformants of each batch/number of false-positive transformants of the wild-type strain of the batch) (Fig. 5). Finally, we confirmed correct deletion of the target genes using Southern blot analysis (see Fig. S3 in the supplemental material). The Southern blot also shows that there is no ectopic integration in any of the checked deletion strains, either in the wild-type background or in the lig4 mutant background.
TABLE 2.
Correct transformation efficiency in wild-type C. glabrata and two independent lig4 mutant strainsa
| Gene deletion tested and batch no.b | No. of correct gene deletions/total no. of transformants |
||
|---|---|---|---|
| Wild type | lig4 (a) | lig4 (b) | |
| ADE2 | |||
| Batch1 | 1/105 | 1/3 | 0/1 |
| Batch2 | 0/71 | 0/0 | 0/2 |
| Batch3 | 0/31 | 0/2 | 0/2 |
| Batch4 | 0/29 | 0/6 | 1/5 |
| Overall | 1/236 | 1/11 | 1/10 |
| ARG8 | |||
| Batch1 | 1/332 | 0/1 | 1/4 |
| Batch2 | 0/17 | 0/2 | 0/0 |
| Batch3 | 0/40 | 0/0 | 0/1 |
| Batch4 | 0/45 | 0/0 | 1/2 |
| Overall | 1/434 | 0/3 | 2/7 |
| CNA1 | |||
| Batch1 | 0/37 | 0/0 | 0/0 |
| Batch2 | 0/33 | 0/0 | 0/0 |
| Batch3 | 0/39 | 0/1 | 0/0 |
| Batch4 | 0/120 | 0/0 | 0/0 |
| Batch5 | 0/196 | 0/1 | 1/3 |
| Batch6 | 0/121 | 0/0 | 0/1 |
| Overall | 0/546 | 0/2 | 1/4 |
For each experiment, the wild-type strain and the two lig4 mutants were incubated in 50 ml YPD medium at an OD600 of 0.2 and grown at 37°C to an OD600 of 1.5. Competent cells harvested from one such culture were defined as one batch.
Data are from at least four independent transformation rounds in the wild-type background and in the two independent lig4 mutants.
FIG 5.

Relative aspecific integration is much lower in the lig4 mutants than in the wild-type strain. Relative aspecific integration values, calculated as the number of false-positive transformants of each batch/number of false-positive transformants of the wild-type strain of the batch, for deletion cassettes targeting ADE2, ARG8, or CNA1 are shown. P < 0.05 for comparison of the wild type to the lig4 mutants.
To avoid any phenotype caused by the absence of LIG4, we also generated a reintegration cassette that can be used to reintegrate the LIG4 gene at its original locus. We have generated this cassette using the Gibson Assembly kit as described in Materials and Methods (Fig. 6). To target the LIG4 gene to its original locus, an EcoRI or CspCI digestion (the latter is a double-digester enzyme and will result in a complete homologous DNA fragment, possibly further increasing efficiency) of this plasmid followed by transformation and selection on nourseothricin will restore proper expression of this ligase gene at its original locus. Both the strain and the reintegration plasmid will be available for the Candida research community.
FIG 6.

Map of the LIG4 reintegration plasmid. Digestion of this plasmid with EcoRI or CspC1 will target the LIG4 gene to its original locus in the genome.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Taiga Miyazaki and Hironobu Nakayama for their generous gift of C. glabrata strain 2001HT and Hiroji Chibana for kindly providing us the C. glabrata strain KUE200. We thank Ilse Palmans and Cindy Colombo for excellent technical assistance and Nico Vangoethem for help with the preparation of figures.
This work was supported by a Ph.D. scholarship (file no. 2010833102) from the China Scholarship Council to Y.C. and by the Fund for Scientific Research Flanders (FWO project VS.036.14N).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00281-14.
REFERENCES
- 1.Rubin RH. 2002. Overview: pathogenesis of fungal infections in the organ transplant recipient. Transpl Infect Dis 4:12–17. doi: 10.1034/j.1399-3062.4.s3.2.x. [DOI] [PubMed] [Google Scholar]
- 2.Eggimann P, Garbino J, Pittet D. 2003. Epidemiology of Candida species infections in critically ill non-immunosuppressed patients. Lancet Infect Dis 3:685–702. doi: 10.1016/S1473-3099(03)00801-6. [DOI] [PubMed] [Google Scholar]
- 3.Lass-Flörl C. 2009. The changing face of epidemiology of invasive fungal disease in Europe. Mycoses 52:197–205. doi: 10.1111/j.1439-0507.2009.01691.x. [DOI] [PubMed] [Google Scholar]
- 4.Pfaller MA, Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- 5.Hitchcock CA, Pye GW, Troke PF, Johnson EM, Warnock DW. 1993. Fluconazole resistance in Candida glabrata. Antimicrob Agents Chemother 37:1962–1965. doi: 10.1128/AAC.37.9.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfaller MA, Messer SA, Moet GJ, Jones RN, Castanheira M. 2011. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in intensive care unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program. Int J Antimicrob Agents 38:65–69. doi: 10.1016/j.ijantimicag.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Pfaller MA, Castanheira M, Lockhart SR, Ahlquist AM, Messer SA, Jones RN. 2012. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J Clin Microbiol 50:1199–1203. doi: 10.1128/JCM.06112-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 9.Noble SM, Johnson AD. 2005. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell 4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarzmüller T, Ma B, Hiller E, Istel F, Tscherner M, Brunke S, Ames L, Firon A, Green B, Cabral V, Marcet-Houben M, Jacobsen ID, Quintin J, Seider K, Frohner IE, Glaser W, Jungwirth H, Bachellier-Bassi S, Chauvel M, Zeidler U, Ferrandon D, Gabaldón T, Hube B, d' Enfert C, Rupp S, Cormack BP, Haynes K, Kuchler K. 2014. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog 10:e1004211. doi: 10.1371/journal.ppat.1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paques F, Haber JE. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63:349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schorsch C, Köhler T, Boles E. 2009. Knockout of the DNA ligase IV homolog gene in the sphingoid base producing yeast Pichia ciferrii significantly increases gene targeting efficiency. Curr Genet 55:381–389. doi: 10.1007/s00294-009-0252-z. [DOI] [PubMed] [Google Scholar]
- 13.Daley JM, Palmbos PL, Wu D, Wilson TE. 2005. Nonhomologous end joining in yeast. Annu Rev Genet 39:431–451. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 14.Dudásová Z, Dudás A, Chovanec M. 2004. Non-homologous end-joining factors of Saccharomyces cerevisiae. FEMS Microbiol Rev 28:581–601. doi: 10.1016/j.femsre.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Ninomiya Y, Suzuki K, Ishii C, Inoue H. 2004. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci U S A 101:12248–12253. doi: 10.1073/pnas.0402780101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kooistra R, Hooykaas PJ, Steensma HY. 2004. Efficient gene targeting in Kluyveromyces lactis. Yeast 21:781–792. doi: 10.1002/yea.1131. [DOI] [PubMed] [Google Scholar]
- 17.Goins CL, Gerik KJ, Lodge JK. 2006. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet Biol 43:531–544. doi: 10.1016/j.fgb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi T, Masuda T, Koyama Y. 2006. Enhanced gene targeting frequency in ku70 and ku80 disruption mutants of Aspergillus sojae and Aspergillus oryzae. Mol Genet Genomics 275:460–470. doi: 10.1007/s00438-006-0104-1. [DOI] [PubMed] [Google Scholar]
- 19.Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, Van den Hondel CA, Ram AF. 2007. Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775. doi: 10.1016/j.jbiotec.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 20.da Silva Ferreira ME, Kress MR, Savoldi M, Goldman MH, Hartl A, Heinekamp T, Brakhage AA, Goldman GH. 2006. The akuB (KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot Cell 5:207–211. doi: 10.1128/EC.5.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueno K, Uno J, Nakayama H, Sasamoto K, Mikami Y, Chibana H. 2007. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata. Eukaryot Cell 6:1239–1247. doi: 10.1128/EC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kucharíková S, Neirinck B, Sharma N, Vleugels J, Lagrou K, Van Dijck P. 2015. In vivo Candida glabrata biofilm development on foreign bodies in a rat subcutaneous model. J Antimicrob Chemother 70:846–856. doi: 10.1093/jac/dku447. [DOI] [PubMed] [Google Scholar]
- 23.Rosas-Hernández LL, Juárez-Reyes A, Arroyo-Helguera OE, De Las Peñas A, Pan SJ, Cormack BP, Castaño I. 2008. yKu70/yKu80 and Rif1 regulate silencing differentially at telomeres in Candida glabrata. Eukaryot Cell 7:2168–2178. doi: 10.1128/EC.00228-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE. 2003. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 17:2384–2395. doi: 10.1101/gad.1125903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myung K, Ghosh G, Fattah FJ, Li G, Kim H, Dutia A, Pak E, Smith S, Hendrickson EA. 2004. Regulation of telomere length and suppression of genomic instability in human somatic cells by Ku86. Mol Cell Biol 24:5050–5059. doi: 10.1128/MCB.24.11.5050-5059.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher TS, Taggart AK, Zakian VA. 2004. Cell cycle-dependent regulation of yeast telomerase by Ku. Nat Struct Mol Biol 11:1198–1205. doi: 10.1038/nsmb854. [DOI] [PubMed] [Google Scholar]
- 27.Boulton SJ, Jackson SP. 1996. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazaki T, Yamauchi S, Inamine T, Nagayoshi Y, Saijo T, Izumikawa K, Seki M, Kakeya H, Yamamoto Y, Yanagihara K, Miyazaki Y, Kohno S. 2010. Roles of calcineurin and Crz1 in antifungal susceptibility and virulence of Candida glabrata. Antimicrob Agents Chemother 54:1639–1643. doi: 10.1128/AAC.01364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reuß O, Vik A, Kolter R, Morschhäuser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Akada R, Murakane T, Nishikawa Y. 2000. DNA extraction method for screening yeast clones by PCR. Biotechniques 28:668–670. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Clinical and Laboratory Standards Institute.2012. CLSI M27-S4: reference method for broth dilution antifungal susceptibility testing of yeasts, 4th informational supplement, M27S4 CLSI, Wayne, PA. [Google Scholar]
- 33.Reaume SE, Tatum EL. 1949. Spontaneous and nitrogen mustard-induced nutritional deficiencies in S. cerevisiae. Arch Biochem 22:331–338. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.