

Abstract
POLR3-related (or 4H) leukodystrophy is an autosomal recessive disorder caused by mutations in POLR3A or POLR3B and is characterized by neurological and non-neurological features. In a small proportion of patients, no mutation in either gene or only one mutation is found. Analysis of the POLR3B cDNA revealed a large deletion of exons 21–22 in one case and of exons 26–27 in another case. These are the first reports of long deletions causing POLR3-related leukodystrophy, suggesting that deletions and duplications in POLR3A or POLR3B should be investigated in patients with a compatible phenotype, especially if one pathogenic variant has been identified.
Keywords: 4H leukodystrophy, Pol III (POLR3)-related leukodystrophy, POLR3A, POLR3B, Deletion
Findings
POLR3-related leukodystrophy or 4H (Hypomyelination, Hypodontia, Hypogonadotropic Hypogonadism) leukodystrophy (MIM#607694) is a hypomyelinating leukodystrophy with typical onset in early childhood [1–3]. Neurological features include motor delay or regression, cerebellar and pyramidal features, and, later in the course of the disease, cognitive regression. Non-neurological features include a variety of dental and hormonal abnormalities, and myopia [1, 3]. Brain MRI shows hypomyelination, i.e. variable (hypo-, hyper- or iso- intense) signal on T1-weighted images and hyperintense signal of the white matter on T2-weighted images compared to grey matter structures [4], with relative preservation of myelination (i.e. T2-hypointensity), of specific structures, with or without cerebellar atrophy and thinning of the corpus callosum [3, 5, 6]. 4H is caused by recessive mutations in POLR3A (MIM#614258) [7–10] or POLR3B (MIM#614366), encoding respectively the largest and second largest subunits of the DNA-directed RNA polymerase III (POLR3). POLR3 is responsible for the transcription of transfer RNAs and other small RNAs essential for cellular processes [11]. Amino acid changes in the protein domains of POLR3A or POLR3B suggest a direct interference with DNA binding, a modification of the catalytic cleft structure, or a change in protein interactions of POLR3 subunits [9, 10]. Current knowledge suggests that mutations are uncovered in the vast majority of 4H cases. A small percentage of cases remain negative or have only one mutation after sequencing analysis [3, 8]. To date, no large deletion or duplication has been reported in 4H cases [3, 9].
We here report the first large exonic deletions in patients with clinical and radiological diagnosis of 4H. The first index patient is a 15-year-old girl, first evaluated by a child neurologist at the age of 19 months for unstable stance and delayed walking. She was born from healthy non-consanguineous parents after a normal pregnancy and delivery. At initial evaluation, the patient had normal height, weight and head circumference. Neurological examination was significant for mild hypotonia as well as truncal ataxia. Patient evolution and annual examinations revealed delayed decidual teeth eruption, slowly progressive gait ataxia and halted pubertal development at Tanner Stage IV with primary amenorrhea. At the time, LHRH stimulation showed lack of LH/FSH pulsatility. The second patient, a boy, was born at term by elective caesarean section after a pregnancy sustained by progesterone in the context of having had three prior miscarriages of unknown cause. Congenital hip dislocation was first treated conservatively, and then corrected surgically. In the second year of life, delay of gross and fine motor skills became obvious. He developed frank ataxia and a mild pyramidal syndrome. Eruption of upper medial incisors was delayed. He developed myopia and short stature. Neurological deterioration led to death at age 8 years. MRI of both patients were compatible with 4H leukodystrophy.
Both patients underwent sequencing of all exons and intron-exon boundaries of POLR3A and POLR3B. Patient 1 was heterozygous for a maternally-inherited variant, c.1568 T > A (p.Val523Glu), located in exon 15 of POLR3B (Figs. 1a and 2c). The Val523Glu have been reported several times in the literature as pathogenic [1]. Patient 2 was apparently homozygous for a missense variant, c.3008A > G (p.Tyr1003Cys), located in exon 26 of POLR3B (Figs. 1b and 2c). Parental testing failed to uncover the Tyr1003Cys in the mother suggesting the presence of a large deletion on the other allele or uniparental isodisomy. No mutation in POLR3A was uncovered in either patient.
Fig. 1.
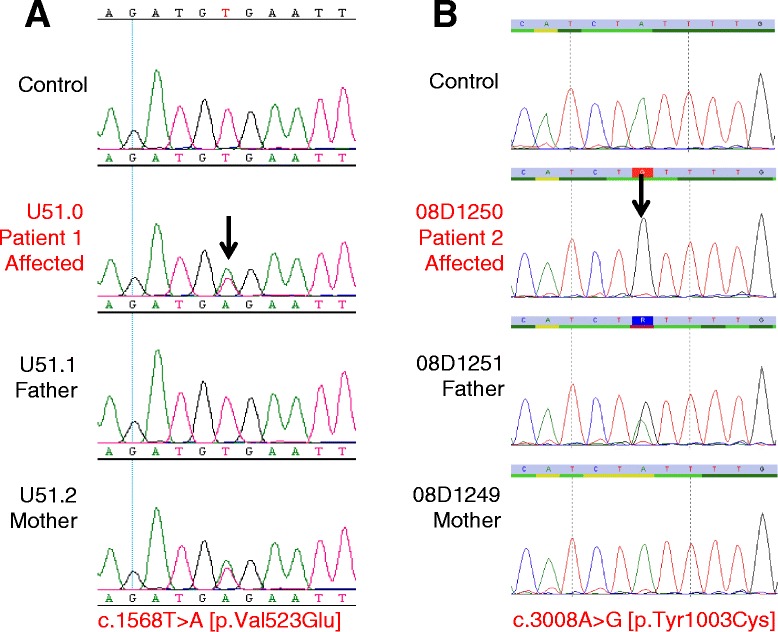
Sanger sequencing of POLR3B genomic coding sequence and segregation analysis in families. a Patient 1 was heterozygous for a maternally-inherited pathogenic variant, c.1568 T > A (p.Val523Glu), located in exon 15 of POLR3B gene. Father was negative for the variant. The Val523Glu variant has not been reported in the general population databases. While not validated for clinical use, multiple in silico analyses predict that this variant is disease-causing. b Patient 2 was found apparently homozygous for a paternally-inherited pathogenic variant, c.3008G > A (p.Tyr1003Cys), located in exon 26 of POLR3B gene. Father was a heterozygous carrier but mother was negative for the variant, suggesting a deletion on the other allele. The Tyr1003Cys variant has not been reported in the general population databases. While not validated for clinical use, multiple in silico analyses predict that this variant is disease-causing
Fig. 2.
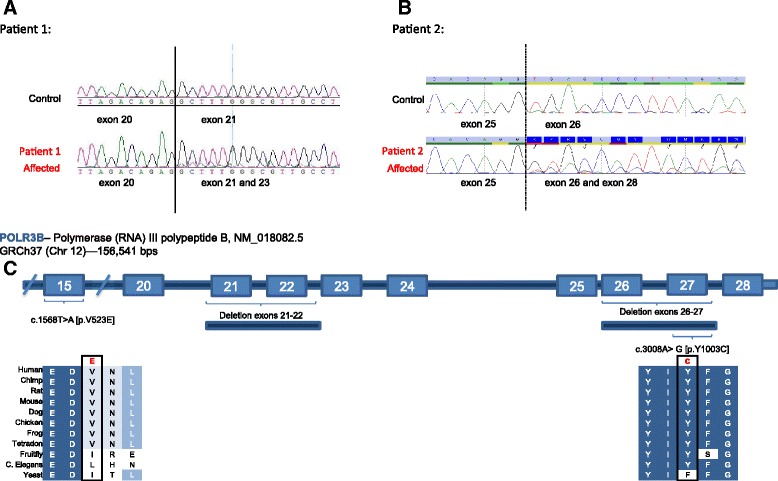
cDNA amplification and sequencing of fragments encompassing the putative POLR3B exonic deletions. a Patient 1: RNA isolation was performed using standard method and followed by reverse transcription-polymerase chain reaction (RT-PCR). Amplification of the fragment of complement DNA (cDNA) encompassing exons 20 to 23 revealed the presence of a putative deletion. The sequence in the overlapping reading frame were showing a normal cDNA PCR product of 630 bp, covering exons 20 to 23. The second sequence of a reduced product of 353 bp was encompassing exons 20 to 23, suggesting the complete or partial deletion of exons 21 and 22. b Patient 2: RNA isolation was performed using standard method and followed by reverse transcription-polymerase chain reaction (RT-PCR). Amplification of the fragment of complement DNA (cDNA) encompassing the exon 25 to 28 revealed the presence of a putative deletion. The sequence in the overlapping reading frame were showing a normal cDNA PCR product of 634 bp, covering exons 25 to 28. The second sequence of a reduced product of 346 bp was encompassing exons 25 and 28, suggesting the complete or partial deletion of exons 26 and 27. c Scheme depicts the large deletions in POLR3B gene characterized in our index patients with classical clinical and radiological findings of 4H
Further studies were conducted in order to uncover exonic deletion in our index patients. In patient 1, RNA analysis revealed the presence of a heterozygous deletion encompassing exons 21 and 22 (Fig. 2a and c). Subsequently, long-range genomic PCR using exon-specific primers for exons 20 and 23, confirmed a ~6Kb paternally-inherited deletion in patient 1 (Figs. 2c and 3a). In patient 2, RNA analysis revealed the presence of a heterozygous deletion encompassing exons 26 and 27 (Fig. 2b and c). Subsequently, long-range genomic PCR using exon-specific primers for exons 25 and 27 respectively, confirmed a ~4Kb maternally-inherited deletion. Since no homology was found at the deletion breakpoints, the POLR3B exonic deletions appear to have arisen by the simple rejoining of non-homologous DNA ends during double stranded break repair.
Fig. 3.
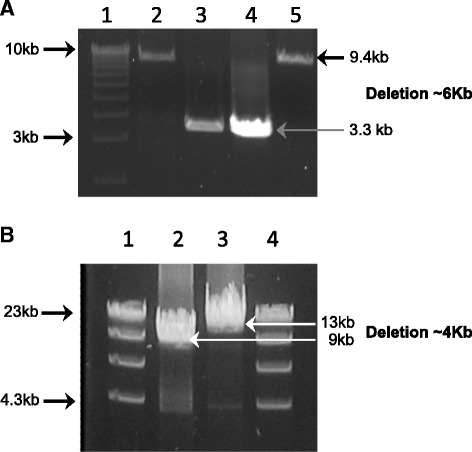
Long-Range PCR of the putative POLR3B exonic deletions and sequencing of the breakpoints. a. Patient 1: Long-Range (LR- PCR) was performed on genomic DNA using exon-specific forward and reverse primers for exons 20 and 23 respectively. The expected fragment size of this region was estimated at 10 kb (Control DNA; Lane 2 and U51.2; Lane 5), however the proband LR-PCR amplification generated a smaller DNA fragment with average product length of approximately 3 kb, as indicated by the arrow (U51.0; Lane 3). This 3 kb DNA fragment was also seen in the LR-PCR amplification of paternal DNA (U51.1; Lane 4), but not of maternal DNA (U51.2; Lane 5). Lane 1 is the molecular size marker. Sanger sequencing of the abnormal fragment identified in both the proband and the unaffected father allowed mapping of the breakpoint on the 5’ at position 106, 751, 658 on chromosome 12, and the breakpoint at the 3’ was at position 106, 857, 267 (data not shown). No homology was found at either of the deletion breakpoints. Tandem Repeats finder, QuadParser and Repeat Masker databases were used for sequence motif analysis. These tools failed to identify motif, suggesting that homologous recombination events were unlikely. The exonic deletions of POLR3B appear to have arisen by the simple rejoining of non-homologous DNA ends during double stranded break repair. b. Patient 2: Long-Range (LR- PCR) was performed on genomic DNA using intron-specific forward and reverse primers for introns 25 and 27 respectively. The expected fragment size of this region was estimated at 13 kb (Control DNA; Lane 3), however the proband LR-PCR amplification generated a smaller DNA fragment with average product length of approximately 9 kb, as indicated by the arrow (08D1250; Lane 2). Lane 1 and 4 is the molecular size marker
The clinical picture in these patients is typical of 4H. The presence of a deletion together with a missense mutation reiterates the idea that complete loss of POLR3B is lethal [1]. Patients with compound heterozygous mutations have shown no significant differences in the clinical course of deterioration as compared to homozygous affected patients, except for the patients homozygous for the common POLR3B mutation (c.1568 T > A, p.Val523Glu), whose clinical symptoms are significantly milder [3, 10]. This could be, at least partly, attributed to the debilitating effect of the missense mutation on the formation of multiplex complexes [9]. In summary, our findings highlight that multi-modal approaches in mutation screening of POLR3A and POLR3B genes may be required for clinically suspected 4H cases when Sanger sequencing of coding elements is negative or reveals only one mutation.
Ethics and consent statement
The project was approved by the research ethics committee of the Montreal Children Hospital (11–105-PED) and the institutional review board of the VU University Medical Center, Neuroscience Campus Amsterdam, Netherlands. Written informed consent was obtained from the patients’ legal guardians.
Acknowledgments
We are grateful to the patients and their families. Dr. Bernard has received a Research Scholar Junior 1 of the Fonds de Recherche du Québec en Santé (FRQS). She wishes to thank the Canadian Institutes of Health Research (CIHR) [MOP-G-287547]. Dr. Thiffault has received a scholarship from the RMGA (Réseau de Médecine Génétique Appliquée du FRQS). We thank the McGill University–Genome Quebec Innovation Center for their technical assistance.
Bioinformatics
The accession numbers for the reference sequences are listed as follows: POLR3B NM_018082, NC_000012.12, NP_060552.4
UCSC web
Primer3
Exome Variant Database (EVS)
Abbreviations
- 4H
Hypomyelination Hypodontia Hypogonadotropic Hypogonadism
- DNA
Deoxyribonucleic Acid
- FSH
Follicle stimulating hormone
- LH
Luteinizing hormone
- LHRH
Luteinizing hormone-releasing hormone
- LR-PCR
Long-Range Polymerase Chain Reaction
- MRI
Magnetic Resonance Imaging
- PCR
Polymerase Chain Reaction
- POLR3
DNA-directed RNA polymerase III
- POLR3A
DNA-Directed RNA Polymerase III Subunit A
- POLR3B
DNA-Directed RNA Polymerase III Subunit B
- RNA
Ribonucleic Acid
Footnotes
Nicole I. Wolf and Geneviève Bernard dual senior authors.
Mariana Gutierrez and Isabelle Thiffault contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The authors have made the following declarations about their contributions: Conceived and designed the experiments: KG, IT, RvS, NIW, GB. Performed the experiments: KG, RvS. Analyzed the data: KG, IT, RvS, NIW, GB. Contributed reagents/materials/analysis tools: GB, NIW, KG, IT, RvS. Wrote the paper: MG, IT. Reviewed the manuscript: LT, KG, GAMM, RvS, MSvdK, NIW, GB. Contributed to the recruitment of the patients for the study: LT, GAMM, MSvdK, NIW, GB. All authors read and approved the final manuscript.
Contributor Information
Mariana Gutierrez, Email: mariana.gutierrez@mail.mcgill.ca.
Isabelle Thiffault, Email: isabelle.thiffault@mail.mcgill.ca.
Kether Guerrero, Email: kether.guerrero@mail.mcgill.ca.
Gabriel Á. Martos-Moreno, Email: gabrielangelmartos@yahoo.es
Luan T. Tran, Email: luan.tran@mail.mcgill.ca
William Benko, Email: wbenko@wellspan.org.
Marjo S. van der Knaap, Email: ms.vanderknaap@vumc.nl
Rosalina M. L. van Spaendonk, Email: r.vervenne@vumc.nl
Nicole I. Wolf, Email: n.wolf@vumc.nl
Geneviève Bernard, Email: genevieve.bernard@mcgill.ca.
References
- 1.Bernard G, Vanderver A. Pol III-related leukodystrophies (august 2012) in: GeneReviews at GeneTests: medical genetics information resource [database online] Seattle: Copyright, University of Washington; 1997. [Google Scholar]
- 2.Potic A, Brais B, Choquet K, Schiffmann R, Bernard G. 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol. 2012;69.7:920–923. doi: 10.1001/archneurol.2011.1963. [DOI] [PubMed] [Google Scholar]
- 3.Wolf NI, Vanderver A, van Spaendonk RML, Schiffmann R, Brais B, Bugiani M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83(21):1898–1905. doi: 10.1212/WNL.0000000000001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72(8):750–9. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van der Knaap MS, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133(10):2971–82. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.La Piana R, Tonduti D, Dressman HG, Schmidt JL, Murnick J, Brais B, Vanderver A, et al. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol. 2014;29(2):214–20. doi: 10.1177/0883073813503902. [DOI] [PubMed] [Google Scholar]
- 7.Saitsu H, Osaka H, Sasaki M, Takanashi JI, Hamada K, Yamashita A, Matsumoto N, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89(5):644–651. doi: 10.1016/j.ajhg.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, Brais B, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89(3):415–423. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tétreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, Bernard G, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III. Cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89(5):652–655. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daoud H, Tétreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, Bernard G, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet. 2013;50(3):194–197. doi: 10.1136/jmedgenet-2012-101357. [DOI] [PubMed] [Google Scholar]
- 11.Dumay-Odelot H, Durrieu-Gaillard S, Da Silva D, Roeder RG, Teichmann M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle. 2010;9(18):3711–3723. doi: 10.4161/cc.9.18.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]