

Abstract
Angiotensin II is an important mediator of CKD of diverse etiology. A common pathologic feature of CKD is glomerular fibrosis, a central mediator of which is the profibrotic cytokine TGF-β. The mechanisms underlying the induction of TGF-β and matrix by angiotensin II are not completely understood. Recent studies showed that overexpression of the transcription factor SREBP-1 induces glomerular sclerosis and that angiotensin II can activate SREBP-1 in tubular cells. We thus studied whether SREBP-1 is activated by angiotensin II and mediates angiotensin II–induced profibrogenic responses in primary rat mesangial cells. Treatment of cells with angiotensin II induced the upregulation and activation of SREBP-1. Angiotensin II–induced activation of SREBP-1 required signaling through the angiotensin II type I receptor and activation of PI3K/Akt in addition to the chaperone SCAP and protease S1P. Notably, angiotensin II-induced endoplasmic reticulum stress was identified as a key mediator of Akt-SREBP-1 activation, and inhibition of endoplasmic reticulum stress or SREBP-1 prevented angiotensin II–induced SREBP-1 binding to the TGF-β promoter, TGF-β upregulation, and downstream fibronectin upregulation. Endoplasmic reticulum stress alone, however, did not induce TGF-β upregulation despite activating SREBP-1. Although not required for SREBP-1 activation by angiotensin II, EGF receptor signaling was necessary for activation of the SREBP-1 cotranscription factor Sp1, which provided a required second signal for TGF-β upregulation. In vivo, endoplasmic reticulum stress and SREBP-1-dependent effects were induced in glomeruli of angiotensin II-infused mice, and administration of the SREBP inhibitor fatostatin prevented angiotensin II–induced TGF-β upregulation and matrix accumulation. SREBP-1 and endoplasmic reticulum stress thus provide potential novel therapeutic targets for the treatment of CKD.
Keywords: cell signaling, chronic kidney disease, TGF-β, angiotensin, fibrosis
Angiotensin II (AngII) is a major pathogenic factor in the progression of both diabetic and nondiabetic CKD. Glomerular sclerosis is a characteristic pathologic feature marked by extracellular matrix accumulation. Mesangial cells (MCs) play a key role in this process. AngII induces MCs to produce the profibrogenic cytokine TGF-β and matrix proteins, such as fibronectin and collagen. AngII inhibitors, however, are not completely effective in preventing disease progression and are not tolerated by all patients. Improved understanding of how AngII mediates kidney fibrosis will facilitate the development of new therapeutic agents.
Sterol regulatory element-binding proteins (SREBPs) are transcription factors most extensively studied in lipid homeostasis, but data suggest an additional important role in matrix regulation. Three isoforms exist: SREBP-1a/1c, generated from alternate transcription start sites, and SREBP-2.1 In its inactive precursor form, SREBP resides in the endoplasmic reticulum (ER) membrane. When intracellular cholesterol is low, SREBP cleavage-activating protein (SCAP) escorts SREBP to the Golgi for sequential cleavage by proteases S1P and S2P to release the N-terminus. This mature transcription factor translocates to the nucleus as a dimer and binds promoters of target genes at sterol regulatory element sequences.1,2 Non–sterol-mediated SREBP activation through the same pathways has also been described, such as with shear stress, glucose, and growth factors.3–6
Renal overexpression of active SREBP-1a or -1c induced glomerular sclerosis with upregulation of TGF-β, fibronectin, and collagen.7,8 Although SREBP-1 activation was recently observed in tubular epithelium of AngII-infused rats,9 whether it contributes to glomerular fibrosis remains unknown. AngII effects are primarily mediated by the type 1 receptor (AT1R) through coupling to G proteins.10 Downstream signaling molecules include PI3K and Akt, which positively regulate SREBP activation.1 AngII also cross-talks with EGFR,10 which we showed mediates glucose-induced SREBP-1 activation.4 In MCs, both PI3K/Akt and EGFR signaling regulate TGF-β1 synthesis by AngII,11,12 but whether SREBP-1 is also required is unknown.
ER stress is emerging as an important pathophysiologic factor in CKD of various causes.13 A response to disruption of ER homeostasis, it is characterized by activation of three signaling proteins regulated by the master chaperone GRP78, namely ATF6, IRE1, and PERK. These upregulate chaperones, attenuate translation through phosphorylation and inhibition of the translation-initiating factor eIF2-α, and, if prolonged, activate proapoptotic signaling mediators.14,15 Interestingly, SREBP-1 contains an internal ribosome entry site, allowing its translation despite the overall reduction in protein synthesis.16 Although ER stress may activate SREBP and was implicated in AngII-induced cardiac pathology,17–19 whether AngII induces ER stress in MCs and the kidney is unknown.
We thus studied the role of SREBP in AngII profibrotic effects in MCs. Our data identify ER stress/Akt-mediated SREBP-1 activation by AngII as a novel mediator of TGF-β1 and matrix upregulation. SREBP inhibition in vivo prevents AngII-induced glomerular TGF-β and matrix synthesis, providing a promising potential therapeutic target for the treatment of CKD.
Results
AngII Activates SREBP-1
To determine whether AngII activates SREBP-1 in MC, we detected appearance of the mature 68-kD form by immunoblotting. This was seen within 1 hour in both nuclear (Figure 1A) and total cell (Supplemental Figure 1A) lysates. Subsequent studies assessed SREBP-1 in total cell lysate, with AngII for 3 hours, since activation was most consistent at this time. While increasing concentrations of AngII further increased SREBP-1 activation (Supplemental Figure 1B), all further studies used 100 nM. We next confirmed functionality of SREBP-1 activation by AngII using a plasmid in which green fluorescent protein (GFP) expression is under control of the SRE (Figure 1B),20 with corresponding immunofluorescence images in Supplemental Figure 1C. The well established downstream target fatty acid synthase (Figure 1C) and lipid synthesis, assessed by Oil Red O staining (Figure 1D, Supplemental Figure 1D), were also increased by AngII.
Figure 1.

AngII activates SREBP-1. (A) MCs were treated with AngII (100 nM) for the indicated times, and SREBP-1 activation was assessed by immunoblotting of nuclear extracts for the cleaved, mature form (mSREBP-1) (†P<0.01 control (con) versus others, n=5). (B) MCs were transfected with the SREBP responsive plasmid SRE-GFP and treated with AngII for 1 or 3 hours. GFP production, driven by SREBP activation, was quantified by fluorescence measurement (*P<0.05 con versus others, n=8). The immunoblot shows cell lysates probed for GFP under the same conditions. (C) Upregulation of the SREBP-1 target gene FAS by AngII was assessed after treatment for 6 hours (‡P<0.01 AngII versus con, n=3). (D) Intracellular lipid accumulation was assessed by Oil Red O (ORO) staining after AngII for 24–48 hours (†P<0.01 versus con, n=3).
AT1R/PI3K/Akt Signaling, but Not EGFR, Are Required for SREBP-1 Activation by AngII
We first established that AngII-induced SREBP-1 activation was mediated by the AT1R (Figure 2A). PI3K/Akt activation is downstream of the AT1R10 and enable SREBP-1 activation in other settings.4,5 Figures 2, B and C show that PI3K and Akt inhibition prevented AngII-induced SREBP-1 activation. In some cases Akt may signal through the mammalian target of rapamycin complex-1 to activate SREBP-1.21 Its inhibitor rapamycin, however, did not prevent AngII-induced SREBP-1 activation (Figure 2D). This suggests that Akt may directly regulate SREBP-1, as shown in response to insulin in hepatocytes,5 or may function through other downstream regulators such as GSK-3β.22,23
Figure 2.

AT1R/PI3K/Akt signaling, but not EGFR, is required for SREBP-1 activation. MCs were treated with various inhibitors before AngII, and mature SREBP-1 was assessed by immunoblotting. (A) The AT1R inhibitor losartan (10 µM, 30 minutes) prevented SREBP-1 activation (*P<0.05 versus con, #P<0.05 versus Ang, n=6). (B) Two structurally distinct PI3K inhibitors—LY294002 (20 µM, 30 minutes) or wortmannin (100 nM, 60 minutes)—blocked SREBP-1 activation (†P<0.01 versus con; #P<0.01 versus Ang; ∧P<0.05 versus Ang, n=5), as did the Akt inhibitor VIII (10 µM, 1 hour) shown in C (*P<0.01 versus con, #P<0.05 versus Ang, n=6). (D) SREBP-1 activation was unaffected by the mTOR inhibitor rapamycin (20 ng/ml, 1 hour). (E and F) Two EGFR inhibitors—AG1478 (1 µM, 30 minutes) and PD168393 (0.5 µM, 30 minutes)—did not block SREBP-1 activation. Immunoblots show effective inhibition of EGFR activation, as assessed by its autophosphorylation on Y1068, and minimal inhibition of Akt activation, as assessed by its phosphorylation on S473. Quantitative changes are shown in the accompanying bar graphs (G and H). For SREBP-1, *P<0.01 versus con (n=6); for pAkt, *P<0.01 versus con only (n=8).
AngII transactivation of the EGFR mediates TGF-β upregulation in MC.11,12 Surprisingly, two different EGFR inhibitors did not affect SREBP-1 activation (Figure 2, E–G). Akt activation was also not significantly decreased (Figure 2, E, F, and H). Thus, our data suggest that SREBP-1 activation by AngII depends on PI3K/Akt signaling through the AT1R but might be independent of EGFR transactivation.
AngII-Induced SREBP-1 Activation Requires SCAP and Serine Proteases
Cholesterol-regulated activation of SREBP-1 involves SCAP-mediated translocation to the Golgi for cleavage to the mature transcription factor by proteases S1P/S2P.2 However, SREBP cleavage may also be mediated independently by other enzymes.24–26 We first tested the SCAP inhibitor fatostatin. This prevented AngII-induced SREBP-1 activation (Figure 3A), as did the S1P inhibitor AEBSF (Figure 3B). As expected, fatostatin also prevented SREBP transcriptional activity (Supplemental Figure 2). In vivo, AngII-induced glomerular SREBP-1 induction, with nuclear localization suggesting its activation, was attenuated by fatostatin (Figure 3C). This was confirmed by immunoblotting of renal cortical lysate (Figure 3D). Localization of SREBP-1 to MC was further shown by coimmunofluorescence for SREBP-1 and α-smooth muscle actin (Supplemental Figure 3).
Figure 3.

SREBP-1 activation requires SCAP and serine proteases. (A) The SCAP inhibitor fatostatin (20 µM, 4 hours) prevented SREBP-1 activation by AngII (*P<0.001 versus con, #P<0.001 versus Ang, n=6). (B) SREBP-1 activation was similarly inhibited by the S1P protease inhibitor AEBSF (500 µM, 1 hour) (*P<0.001 versus con, #P<0.001 versus Ang, n=5). (C) Cortical sections from AngII-infused mice were stained by IHC for SREBP-1 (at 40× magnification). These show increased glomerular SREBP-1 expression. Nuclear localization supports SREBP-1 activation. Quantification is shown in the accompanying graph (∧P<0.001 versus Ang, *P<0.001 versus con, #P<0.001 versus Ang). (D) Immunoblotting of protein from kidney cortex confirms that AngII increases SREBP-1 activation, as assessed by appearance of its mature form, and that this is inhibited by fatostatin (∧P<0.001 versus Ang, *P<0.01 versus con, #P<0.05 versus Ang).
ER Stress Mediates SREBP-1 Activation by AngII
AngII induces ER stress in cardiomyocytes and heart disease models,18,19,27 but whether this occurs in MCs is unknown and was first assessed. AngII increased GRP78 expression (3 hours) and eIF2α phosphorylation (30 minutes), two markers of ER stress (Figure 4A), both of which required the AT1R (Figure 4B). EGFR inhibitors did not block ER stress, further supporting independence of SREBP-1 activation from the EGFR (Supplemental Figure 4). Induction of ER stress by AngII was also seen in vivo in glomeruli (Figures 4, C and D).
Figure 4.


ER stress mediates SREBP activation by AngII. (A) ER stress is induced by AngII, as assessed by upregulation of GRP78 and phosphorylation of eIF2α. (B) This is mediated by the AT1R, as both GRP78 induction (AngII, 3 hours) and eIF2α phosphorylation (AngII, 30 minutes) were prevented by losartan (10 µM, 30 minutes) (for GRP78, *P<0.01 versus con, #P<0.01 versus Ang, n=5; for pEIF2α, *P<0.01 versus con, #P<0.01 versus Ang, n=3). (C and D) AngII infusion induced glomerular GRP78 upregulation, as assessed by IHC (*P<0.05, 60× magnification) and by immunoblotting of total cortical lysate (*P<0.05). (E) ER stress induction with tunicamycin (Tm, 500 ng/ml, 1 hour) or thapsigargin (Tg, 200 nM, 1 hour) induced SREBP-1 activation in MCs (*P<0.001 versus control, n=3). (F and G) SREBP-1 activation by AngII was blocked by two ER stress inhibitors: 4-PBA (5 mM, 2 hours) and salubrinal (30 µM, 1 hour). (F) *P<0.001 versus con; #P<0.01 versus Ang, n=6. (G) *P<0.01 versus con, #P<0.01 versus Ang, n=4.
As shown in other models,17 ER stress itself led to SREBP-1 activation (Figure 4E). Furthermore, ER stress inhibition with the chemical chaperone 4-PBA or eIF2α phosphatase inhibitor salubrinal prevented AngII-induced SREBP-1 activation (Figures 4, F and G) and its transcriptional activity (Supplemental Figure 5). We then determined whether there was crosstalk between PI3K/Akt and ER stress, both required for AngII-induced SREBP-1 activation. ER stress induced Akt activation, and this was PI3K-dependent (Figures 5, A and B, Supplemental Figure 6). Furthermore, AngII-induced Akt activation was attenuated by several ER stress inhibitors (Figure 5C). Taken together, these data suggest that AngII-induced ER stress mediates SREBP-1 activation at least partially through Akt.
Figure 5.

PI3K/Akt activation upstream of SREBP-1 activation is also regulated by ER stress. (A) ER stress induction with thapsigargin (Tg, 200 nM, 30 minutes) or tunicamycin (Tm, 500 ng/ml, 30 minutes) activated Akt, as assessed by its phosphorylation on S473 (*P<0.01 versus con, n=6). (B) ER stress–induced Akt activation was blocked by PI3K inhibition with LY294002 (20 µM, 30 minutes) or wortmannin (100 nM, 60 minutes). (C) AngII (30 minutes)–induced Akt activation was attenuated by the ER stress inhibitors 4-PBA (5 mM, 2 hours), TUDCA (300 µg/ml, 1 hour), and salubrinal (30 µM, 1 hour) (n=5; *P<0.01 versus con, #P<0.05 versus Ang).
SREBP-1 Mediates AngII Upregulation of TGF-β1 and Fibronectin Synthesis
We next asked whether SREBP-1 mediates AngII upregulation of the well established target TGF-β1.11,12 Both SCAP and S1P inhibition prevented activation of a TGF-β1 promoter-luciferase reporter by AngII (Figure 6A, Supplemental Figure 7A), as did downregulation of SREBP-1 with siRNA (Figure 6B). Supplemental Figure 7B shows efficacy of SREBP-1 siRNA. TGF-β1 transcript upregulation and its secretion into the medium also required SREBP activation (Figure 6, C–E). Furthermore, overexpression of dominant negative SREBP1a-Y335A or SREBP1c-Y321A prevented promoter activation (Figure 6F). Finally, deletion of the SREBP-1 binding site in the TGF-β1 promoter reporter prevented its activation by AngII (Figure 6G).
Figure 6.

SREBP-1 mediates AngII induction of TGF-β1 and downstream fibronectin upregulation. MCs were treated with AngII for 24 hours. (A) AngII-induced activation of a transfected TGF-β1 promoter-luciferase construct was inhibited by fatostatin (20 µM, 4 hours) (∧P<0.001 versus Ang, *P<0.001 versus con, #P<0.001 versus Ang, n=9). (B) Transfection of MCs with SREBP-1 sRNA prevented AngII-induced activation of the TGF-β1 promoter-luciferase construct compared with control siRNA (*P<0.05 versus control, n=3). (C) Fatostatin inhibited AngII-induced upregulation of the TGF-β1 transcript (*P<0.01 versus con, #P<0.01 versus Ang, n=6). (D) SREBP-1 siRNA also prevented TGF-β1 transcript upregulation by AngII (*P<0.05 versus con, n=3). (E) Both fatostatin and the S1P inhibitor AEBSF (500 µM, 1 hour) prevented AngII-induced TGF-β1 secretion into the medium as assessed by ELISA (*P<0.001 versus con, #P<0.001 versus Ang, n=4). (F) Transfection of MCs with dominant negative SREBP-1a (dnSREBP1a) or -1c prevented AngII-induced TGF-β1 promoter-luciferase activation. Con and AngII groups were transfected with the empty vector pcDNA (*P<0.001 versus con, #P<0.001 versus Ang, n=9). (G) Deletion of the SREBP-1 binding site in the TGF-β1 promoter-luciferase construct prevented its activation by AngII (‡P<0.001 AngII versus con, n=6). (H) Fatostatin inhibited AngII-induced upregulation of fibronectin (*P<0.001 versus con, #P<0.001 versus Ang, n=5). WT, wild type.
It is well known that AngII activates Smad3, the canonical downstream mediator of TGF-β1 signaling. Although early activation (within minutes) is TGF-β independent, requiring Erk and p38, later activation is TGF-β dependent.28,29 Because we showed that SREBP-1 mediates TGF-β1 upregulation by AngII at 24 hours, it should also mediate downstream signaling. To verify this, we assessed Smad3 transcriptional activity using the CAGA12-luciferase reporter after 24 hours of AngII. Supplemental Figure 8 confirms its inhibition by fatostatin. Figure 6H shows that upregulation of fibronectin, an important matrix protein component of sclerotic glomeruli induced by TGF-β, was also prevented by fatostatin.
ER Stress Is Required for TGF-β Upregulation by AngII
Figure 7A shows that ER stress inducers alone cannot activate the TGF-β promoter. Their tendency to reduce baseline promoter activity may be due to translational inhibition by ER stress.30 Indeed, cycloheximide (5 µg/ml) attenuated promoter activity to a similar degree (not shown). Blocking ER stress with various agents, however, inhibited AngII-induced promoter activation, TGF-β secretion into the medium, and fibronectin upregulation (Figures 7, B–D). These data support an important role for ER stress–induced SREBP-1 activation in matrix upregulation by AngII but demonstrate that ER stress alone is insufficient for this response. Because ER stress does, however, activate SREBP-1, we postulated that AngII signaling provides an important second signal that enables TGF-β upregulation.
Figure 7.

ER stress alone does not induce TGF-β upregulation but mediates TGF-β and fibronectin upregulation by AngII. (A) MCs were transfected with a TGF-β1 promoter-luciferase construct. AngII, but not ER stress inducers tunicamycin (Tm, 500 ng/ml) or thapsigargin (Tg, 200 nM), induced promoter activation after 24 hours (*P<0.01 versus con, #P<0.01 versus Ang, n=12). (B) ER stress inhibition with TUDCA (300 µg/ml, 1 hour) or salubrinal (30 µM, 1 hour) prevented TGF-β1 promoter activation by AngII (*P<0.01 versus con, #P<0.01 versus Ang, n=9). (C) Increased TGF-β1 secretion into the medium in response to AngII (24 hours) was prevented by the ER stress inhibitors TUDCA and salubrinal (*P<0.001 versus con, #P<0.01 versus Ang; &P<0.001 versus Ang, n=4). (D) The ER stress inhibitors salubrinal, 4-PBA (5 mM, 2 hours), and TUDCA prevented AngII (24 hours)-induced fibronectin upregulation (*P<0.001 versus con, #P<0.01 versus Ang; &P<0.001 versus Ang, n=3).
EGFR, but Not ER Stress, Regulates Activation of Sp1
As a relatively weak transcription factor, SREBP-1 functions in coordination with cofactors, most commonly Sp1.31,32 A potential SREBP-binding sequence lies close to an Sp1 site within −100 bp of the start codon in the human, mouse, and rat TGF-β promoters (J04431, M57902, NM021578). Using its inhibitor mithramycin, we first established that Sp1 was required for AngII-induced TGF-β promoter upregulation (Figure 8A). Furthermore, AngII, but not ER stress, induced activation of an Sp1-responsive luciferase plasmid (Figure 8B). The attenuation of baseline Sp1 transcriptional activity by ER stress may also be related to translational inhibition because cycloheximide similarly decreased Sp1-luciferase activity (not shown). Finally, AngII-induced Sp1 activation was prevented by EGFR inhibitors, but not ER stress blockers (Figure 8, C and D), suggesting that although EGFR is not required for SREBP-1 activation, it provides an important second signal for TGF-β upregulation through its effects on Sp1. Indeed, deletion of the Sp1 binding site close to the SREBP-1 site in the TGF-β1 promoter prevented the AngII response (Figure 8E).
Figure 8.

EGFR, but not ER stress, regulates activation of the coregulatory transcription factor Sp1 required for TGF-β upregulation. (A) AngII-induced activation of a transfected TGF-β1 promoter-luciferase construct was inhibited by the Sp1 inhibitor mithramycin (100 nM, 1 hour) (*P<0.05 Ang versus con; ∧P<0.01 versus Ang, #P<0.001 versus Ang, n=12). (B) AngII, but not the ER stress inducers tunicamycin (Tm, 500 ng/ml) or thapsigargin (Tg, 200 nM), induced activation of an Sp1-responsive luciferase construct after 24 hours (*P<0.001 versus con, #P<0.001 versus Ang, n=9). (C) ER stress inhibitors 4-PBA (5 mM, 2 hours) and salubrinal (30 µM, 1 hour) did not prevent AngII-induced Sp1 luciferase activation (*P<0.01 versus con, n=6). (D) Two distinct EGFR inhibitors—AG1478 (AG, 1 µM, 30 minutes) and PD168393 (PD, 0.5 µM, 30 minutes)—prevented AngII-induced Sp1 luciferase activation (*P<0.01 versus con, #P<0.01 versus Ang, n=6). (E) Deletion of the Sp1 binding site close to the SREBP-1 binding site in the TGF-β1 promoter-luciferase construct prevented its activation by AngII (*P<0.001 versus con, n=6). WT, wild type.
TGF-β1 Promoter Binding by SREBP-1 Is Regulated by ER Stress, but Not the EGFR
We confirmed that AngII induces SREBP-1 binding to the TGF-β promoter by chromatin immunoprecipitation, using primers encompassing the region −230–0. This was blocked by fatostatin (Figure 9A), with no binding detected in IgG immunoprecipitates. ER stress induction effected a similar degree of SREBP-1 binding (Figure 9B). Conversely, ER stress inhibition blocked AngII-induced SREBP-1 binding, in agreement with our data showing that ER stress is required for SREBP-1 activation (Figure 9C). Binding was unaffected by EGFR inhibition (Figure 9D). In keeping with a coactivator role for Sp1, AngII, but not ER stress, also induced its binding to the TGF-β1 promoter (Figure 9E). Sp1 binding was prevented by EGFR inhibition (Figure 9F). In aggregate, these data support an important role for ER stress–mediated SREBP-1 activation in TGF-β upregulation by AngII, in cooperation with EGFR-mediated Sp1 activation.
Figure 9.

SREBP-1 binding to the TGF-β1 promoter is regulated by ER stress, but not the EGFR. Chromatin immunoprecipitation (ChIP) was performed as described in the Concise Methods to detect association of SREBP-1 or Sp1 with the TGF-β1 promoter after treatment of MCs with AngII for 24 hours. (A) AngII induced SREBP-1 binding to the TGF-β1 promoter. This was inhibited by pretreatment with fatostatin (20 µM, 4 hours) (*P<0.001 versus con, #P<0.05 versus Ang, n=6). (B) ER stress induction with tunicamycin (Tm, 500 ng/ml) or thapsigargin (Tg, 200 nM) similarly induced SREBP-1 binding to the TGF-β1 promoter. (C) AngII-induced SREBP-1 binding to the TGF-β1 promoter was inhibited by the ER stress blockers salubrinal (30 µM, 1 hour) and TUDCA (300 µg/ml, 1 hour) (*P<0.01 versus con, #P<0.01 versus Ang, n=3). (D) AngII-induced SREBP-1 binding to the TGF-β1 promoter was unaffected by the EGFR inhibitors AG1478 (1 µM, 30 minutes) and PD168393 (0.5 µM, 30 minutes). (E) ER stress induction with tunicamycin (Tm, 500 ng/ml) or thapsigargin (Tg, 200 nM) did not affect Sp1 binding to the TGF-β1 promoter (*P<0.01 versus con, #P<0.01 versus Ang, n=3). (F) Two distinct EGFR inhibitors—AG1478 (AG, 1 µM, 30 minutes) and PD168393 (PD, 0.5 µM, 30 minutes)—prevented AngII-induced Sp1 binding to the TGF-β1 promoter (*P<0.01 versus con, #P<0.01 versus Ang, n=3).
Fatostatin Attenuates AngII-Induced Renal Fibrosis
We next assessed the importance of SREBP-1 activation to glomerular fibrosis induced by AngII in vivo. Mice infused with AngII develop glomerular fibrosis characterized by TGF-β upregulation and matrix deposition.33 These mice developed significant hypertension that was not attenuated by fatostatin (systolic BP: control, 129.8±8.3 mmHg; fatostatin, 129.0±1.7 mmHg; AngII, 208.8±15.5 mmHg, AngII/fatostatin, 210.2±24.8 mmHg). Proteinuria was attenuated by fatostatin, although there was no effect on serum creatinine (Supplemental Figures 9, A and B).
Fatostatin inhibited AngII-induced TGF-β upregulation, which colocalized to MCs (Figure 10A, Supplemental Figure 10). TGF-β1 transcript induction was similarly prevented by fatostatin (Supplemental Figure 11A), as was TGF-β signaling as assessed by Smad3 activation (Figure 10, A and B). The lack of complete inhibition to basal levels may represent a small contribution of TGF-β–independent activation of Smad3 in vivo.29
Figure 10.


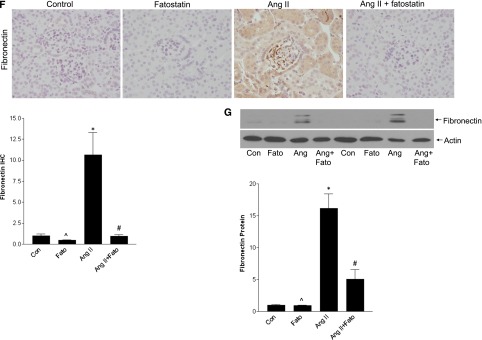
AngII-induced TGF-β and matrix upregulation are inhibited by fatostatin. (A) IHC of cortical sections showed a significant increase in TGF-β1 expression, quantified in the graph (*P<0.01 versus con, ∧P<0.01 versus Ang, #P<0.01 versus Ang). Activation of Smad3, a canonical downstream mediator of TGF-β1, was also seen, as assessed by IHC for its C-terminal S423/S425 phosphorylated form (pSmad3) (*P<0.001 versus con, ∧P<0.001 versus Ang, #P<0.001 versus Ang). (B) Smad3 activation, as assessed by its phosphorylation, was also confirmed by immunoblotting (*P<0.05 versus con, ∧P<0.05 versus Ang, #P<0.05 versus Ang). (C) AngII induced matrix upregulation as seen by increased periodic acid-Schiff staining. Increased collagen deposition was assessed by both Picrosirius red, visualized under polarized light, as well as by IHC for collagen I. These were quantified in (D) (*P<0.001 versus con, ∧P<0.001 versus Ang, #P<0.001 versus Ang). (E) Inhibition of AngII-induced collagen I upregulation by fatostatin was confirmed by immunoblotting (*P<0.01 versus con, ∧P<0.05 versus Ang, #P<0.05 versus Ang). (F) Increased fibronectin upregulation by AngII, assessed by IHC, was prevented by fatostatin (*P<0.05 versus con, ∧P<0.05 versus Ang, #P<0.05 versus Ang), and this was confirmed by immunoblotting as seen in (G) (*P<0.001 versus con, ∧P<0.001 versus Ang, #P<<0.01 versus Ang). All images are taken at 40× magnification.
TGF-β is a major profibrotic regulator. Figure 10C shows that AngII-induced matrix accumulation, identified by periodic acid-Schiff staining, was also inhibited by fatostatin. Treatment similarly prevented collagen and fibronectin upregulation, determined by immunohistochemistry (IHC), immunoblotting (Figure 10, C–G), and real-time PCR (Supplemental Figure 11, B and C). Supplemental Figure 12 shows that fatostatin also attenuated tubulointerstitial fibrosis. Thus, fatostatin inhibits AngII-induced SREBP-1 activation in vivo, and this is associated with inhibition of TGF-β upregulation and activity and downstream matrix accumulation.
Discussion
Our studies present several novel observations in MCs: (1) AngII induces SREBP-1 activation and mediates matrix synthesis through direct transcriptional upregulation of TGF-β1; (2) AngII induces ER stress, and this is a key mediator of SREBP-1 activation; (3) independent activation of both SREBP-1 and EGFR signaling is required for the profibrotic response; (4) SREBP activation in vivo is an important contributor to glomerular fibrosis. These studies provide a basis for further exploration of the therapeutic potential of targeting SREBP-1 or ER stress in CKD.
Renal overexpression of active SREBP-1a or 1c leads to glomerulosclerosis, with elevated TGF-β, fibronectin, and collagen expression.7,8 Although AngII infusion was shown to increase mature SREBP-1 in whole kidney, with increased lipid and TGF-β accumulation in tubular cells, glomeruli were not assessed.9 Our data showing AngII-induced glomerular SREBP-1 activation now extend this and support a potential role for SREBP-1 in glomerular sclerosis. Indeed, in a second model of hypertension characterized by elevated intrarenal AngII despite suppressed systemic levels,34 the Dahl salt-sensitive rat, both glomerular SREBP-1 and GRP78 were increased in rats on a high-salt diet (Supplemental Figure 13A). Systolic BPs were 148.2±7.6 mmHg (low salt) and 183.3±8.4 mmHg (high salt) (P<0.05). However, in the deoxycorticosterone acetate (DOCA)/salt model of systemic hypertension in which glomerular and systemic AngII levels are low,35,36 neither SREBP-1 nor GRP78 were elevated (Supplemental Figure 13, B and C). Systolic BPs were 131.6±2.5 mmHg (control) and 159.5±3.0 mmHg (DOCA/salt) (P<0.0001). These findings support an important role for SREBP-1 and ER stress signaling as mediators of renal pathology associated with elevated intrarenal AngII.
SREBP-1 is best known for its role in lipid homeostasis. A direct and lipid-independent effect on regulating matrix synthesis is less well appreciated. We were the first to demonstrate that SREBP-1 directly regulates TGF-β transcript upregulation by glucose,4 and now show this also occurs in response to AngII both in vitro and in vivo. In addition to its effects on TGF-β synthesis, SREBP-1 may also affect matrix accumulation in alternate ways, such as its regulation of collagen 6a1 transcription in 3T3 cells.37 Plasminogen activator inhibitor-1, which inhibits matrix breakdown, was also identified as an SREBP-1c transcriptional target in adipocytes.38 These data suggest that SREBP-1 is an important regulator of organ fibrosis.
The EGFR is a well described mediator of AngII-induced TGF-β upregulation.11,39 We previously showed that SREBP-1 activation by glucose requires the EGFR.4 Surprisingly, this is not the case with AngII, despite a requirement for both the EGFR and SREBP-1 in regulating TGF-β synthesis. This suggested the presence of two complementary signals for TGF-β induction. Indeed, SREBPs are known weak transcription factors requiring cooperation with other coactivators.31,32 Our studies now show that Sp1, downstream of the EGFR, is required for TGF-β upregulation. These data are consistent with functional input from both SREBP-1 and EGFR/Sp1 to induce TGF-β synthesis.
AngII induces ER stress in heart and cardiomyocytes.18,19,27 Our data now identify that this also occurs in MCs and glomeruli and is an important mediator of SREBP-1 activation. Several different mechanisms by which ER stress activates SREBP have been proposed, depending on cell and stimulus:17 (1) Caspase activation may lead to SREBP cleavage at a site different from S1P. However, in conditions that cause ER stress without apoptosis, SREBP is cleaved through S1P/S2P as we found for AngII; (2) ER stress may decrease Insig through eIF2α-mediated translational inhibition, thereby releasing SCAP/SREBP for activation; and (3) GRP78 may retain SCAP/SREBP-1 in the ER through direct interaction, with complex release upon ER stress.17 We now describe an additional mechanism, showing that AngII-induced ER stress regulates PI3K/Akt activation. This may enable SREBP activation at different levels, including facilitating its ER-to-Golgi translocation and protecting nuclear SREBP from degradation.5,22,23 How ER stress leads to Akt activation, however, remains to be elucidated.
Recent studies suggested that ER stress may lead to the production of extracellular matrix and organ fibrosis in some settings.40,41 Interestingly, alleviation of ER stress with 4-PBA attenuated AngII-induced cardiac fibrosis and TGF-β upregulation.19 Our data support an important role for ER stress in AngII-induced matrix synthesis through activation of SREBP-1. ER stress alone, however, was not profibrotic in our cells despite its induction of SREBP-1 binding to the TGF-β promoter. This is in contrast to increased fibronectin synthesis by ER stress in renal tubular cells42 but agrees with the lack of fibrotic response in glial and alveolar cells and in vivo in lungs.43,44 With a second insult (bleomycin), however, mice previously exposed to ER stress developed more severe lung fibrosis. This was postulated to occur through increased apoptotic cells stimulating matrix production in neighboring cells.44,45 Our data, however, show that AngII activates two parallel but necessary signals for TGF-β promoter activation, namely ER stress-induced SREBP-1 activation and EGFR-mediated activation of the cotranscription factor Sp1.
Taken together, our studies have identified ER stress and SREBP-1 activation as novel mediators of AngII-induced TGF-β1 upregulation and matrix synthesis in MCs. SREBP inhibition prevented glomerular fibrosis in vivo. Future studies will determine whether alleviation of ER stress can similarly inhibit AngII-induced glomerular fibrosis, and whether there is an effect on renal function using a more sensitive means than creatinine for assessment. Given the important role of AngII in the development of renal fibrosis, these also represent promising potential therapeutic targets that should be evaluated for their efficacy in the prevention of diabetic and nondiabetic glomerular fibrosis.
Concise Methods
Cell Culture
Primary MCs were obtained from glomeruli of Sprague-Dawley rats by differential sieving and cultured in DMEM with 20% FCS (Invitrogen), streptomycin (100 µg/ml), and penicillin (100 units/ml). Experiments used cells between passages 6 and 15. MCs were serum deprived for 24 hours before treatment with AngII (100 nM, Sigma-Aldrich). Inhibitors were added before AngII as follows: AEBSF 500 µM, 1 hour (Calbiochem); fatostatin 20µM, 4 hours (Chembridge); AG1478 1 µM, 30 minutes (Sigma-Aldrich); PD168393 0.5µM, 30 minutes (Calbiochem); LY294002 20 µM, 30 minutes (Sigma-Aldrich); wortmannin 100 nM, 1 hour (Sigma-Aldrich); Akt inhibitor VIII 10 µM, 1 hour (EMD Millipore); rapamycin 20 ng/ml, 1 hour (Sigma-Aldrich); 4-PBA 5 mM, 2 hours (Sigma-Aldrich); tauroursodeoxycholic acid (TUDCA) 300 µg/ml, 1 hour (Sigma-Aldrich); salubrinal 30 µM, 1 hour (Selleckchem); and mithramycin 100 nM, 1 hour (AG Scientific). ER stress inducers were tunicamycin 500 ng/ml and thapsigargin 200 nM (both Sigma-Aldrich).
Protein Analysis
Cells were lysed and protein extracted as published, with the addition of ALLN to the lysis buffer at 25 µg/ml (Calbiochem).46 Lysates were centrifuged at 4°C, 14,000 rpm for 10 minutes. Supernatant (50 µg) was separated on SDS-PAGE, and Western blotting was performed. Antibodies used were SREBP-1 (mouse; Santa Cruz Biotechnology), SREBP-2 (mouse; BD Biosciences Pharmingen), GFP (rabbit; Cell Signaling Technology), FAS (rabbit monoclonal, Cell Signaling), pEGFR Y1068 (mouse; Cell Signaling Technology), pAkt S473 (rabbit; Cell Signaling Technology), peIF2α S51 (rabbit; Cell Signaling Technology), GRP78 (mouse; BD Transduction Laboratories), and fibronectin (mouse; BD Transduction Laboratories). Equality of loading was assessed by immunoblotting for actin (mouse; Sigma-Aldrich) and for nuclear lysates for CREB (rabbit; Cell Signaling Technology).
GFP Quantification
MCs plated to 85% confluence were transfected with 1 µg of an SRE-GFP plasmid (kindly provided by Dr. R. Austin20) in six-well plates using Effectene (Qiagen). MCs were serum-deprived overnight 24 hours after transfection, then exposed to AngII. GFP was quantified as in Bradburne et al.47: Cells were washed in cold PBS, lysed in 0.2 N HCl, and centrifuged to remove cell debris. GFP fluorescence in the supernatant was measured in a fluorometer (Gemini EM, Molecular Device) at excitation of λ 475 and emission of λ 510. Readings were normalized to protein concentration.
Oil Red O Staining
After treatment, MCs were fixed in formaldehyde and incubated in Oil Red O (Sigma-Aldrich) for 1 hour. Following a wash with water, lipids were recovered by incubation with 60% isopropanol and quantified at 510 nm. Readings were normalized to protein concentration.
Luciferase Assay
MCs plated to 85% confluence were transfected with 0.5 µg of a TGF-β1 promoter-luciferase construct (kindly provided by Dr. N. Kato), CAGA12-luc with 12 copies of the Smad3-binding element (kindly provided by Dr. M. Bilandzic), or 3xSp1-luciferase construct (kindly provided by Dr. Y.P. Di48) and 0.05 µg pCMV-β-galactosidase (β-gal) (Clontech) using Effectene (Qiagen). The TGF-β1 promoter-luciferase plasmid was also used as a template to construct new plasmids with deletion of the SREBP-1 or the Sp1 binding site using overlapping primer pairs. Primer sequences will be provided on request. Mutant constructs were sequenced for confirmation.
Where indicated, cells were also transfected with empty vector pcDNA or dominant negative SREBP-1a (Y335A) or -1c (Y321A), kindly provided by Dr. A. Schulze.49 MCs were serum-deprived overnight 24 hours after transfection, then exposed to AngII for 24 hours. Lysis was achieved with reporter lysis buffer (Promega, Madison, WI) using one freeze-thaw cycle, and luciferase and β-gal activities were measured on clarified lysate using specific kits (Promega) with a Berthold luminometer and a plate reader (420 nm), respectively. β-gal activity was used to adjust for transfection efficiency.
Quantitative Real-Time PCR
Total RNA was extracted using Trizol according to manufacturer’s instructions (Life Technologies). Reverse transcription was performed using standard methods and cDNA analyzed by real-time PCR using a SYBR Green PCR Master Mix kit (Applied Biosystems). Amplification using specific primers for TGF-β1, SREBP-1a and 1c, fibronectin and collagen Iα1, with 18S as an internal control, was measured continuously using an ABI 7500 Sequence Detector (Applied Biosystems). Fold changes over the control values were calculated using the ΔΔCt method.
TGF-β ELISA
To quantify TGF-β secretion, media were collected after treatment with AngII for 24 hours. After heat activation of the samples, total TGF-β1 was determined according to kit instructions (R&D Systems).
Small Interfering RNA
On-target plus SMART pool small interfering RNA (siRNA) for SREBP-1 and control nontargeting siRNA were obtained from Dharmacon. MCs were transfected with 100nM siRNA using Lipofectamine RNAiMAX (Life Technologies) at 60% confluence. Protein expression was used to assess efficacy of downregulation by RNAi.
Chromatin Immunoprecipitation
After treatment, MCs were cross-linked with 1% formaldehyde, then washed and scraped into cold PBS with protease inhibitors. After centrifugation, the cell pellet was resuspended in buffer (20 mM HEPES [pH, 7.9], 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, protease inhibitors), incubated on ice for 20 minutes and centrifuged. The pellet (nucleus) was resuspended in breaking buffer (50 mM Tris-HCl [pH, 8.0], 1 mM EDTA, 150 mM NaCl, 1% SDS, 2% Triton X-100, protease inhibitors), sonicated 5×10s, and Triton buffer added (50 mM Tris-HCl [pH, 8.0], 1 mM EDTA, 150 mM NaCl, 0.1% Triton X-100). Samples were precleared with blocked protein G Sepharose beads, an aliquot reserved as the input, and the remainder divided to immunoprecipitate with control rabbit IgG (Jackson ImmunoResearch), SREBP-1 (sc-8984×; Santa Cruz Biotechnology), or Sp1 (Abcam, Inc.) antibodies followed by incubation with protein G beads. Samples were washed three times in Triton buffer and SDS buffer added (62.5 mM Tris HCl [pH, 6.8], 200 mM NaCl, 2% SDS, 10 mM DTT, 2 µl of proteinase K [40 mg/ml]), then vortexed and incubated at 65°C overnight to reverse crosslinking. DNA was isolated using phenol/chloroform extraction and resuspended in dH2O. PCR was performed using the rat TGF-β1 promoter primers 5′-ATCCCGGTGGCATACTGAG (F) and 5′-CACGGAACTTCGGAGAGC (R), at 60°C annealing temperature for 35 cycles.
Animal Studies
All animal studies were carried out in accordance with McMaster University and Canadian Council on Animal Care guidelines. Male 129Sv/Ev mice (Taconic), 7–8 weeks old, underwent a left nephrectomy followed by infusion with AngII (1000ng/kg/min (Sigma-Aldrich) or saline by osmotic minipump (Alzet). Fatostatin was given to vehicle- or AngII-infused mice at 30 mg/kg per day intraperitoneally as described previously.50 Control groups received vehicle alone (5% DMSO in PBS). There were seven mice in each AngII group, five in the control group, and three in the fatostatin alone group. Tail cuff BP was measured using tail cuff plethysmography (CODA 2 system; Kent Scientific). Mice were euthanized after 8 weeks. Before euthanasia, urine was collected in a metabolic cage, and albumin and creatinine were measured using commercially available kits (Exocell for albumin, Crystal Chem for creatinine) to yield an albumin-to-creatinine ratio. Serum creatinine was also measured as per kit instructions (Crystal Chem).
Male Dahl salt-sensitive rats (Charles River Laboratories) were maintained on a regular chow diet until 12 weeks of age, when diets were changed to normal salt (AIN-76A; 0.4% NaCl, n=4) or high salt (AIN-76A; 8% NaCl, n=4; Research Diets) available ad libitum. After 4 weeks, BPs were measured as described earlier, after which kidneys were perfused with Hank's balanced salt solution and collected for IHC.
For the DOCA-salt model, 10-week-old male C57BL/6 mice (Charles River Laboratories) underwent a right uninephrectomy. After 1 week of recovery, a 50 mg 21-day release DOCA pellet (Innovative Research of America) was implanted subcutaneously. Mice were then given 1% NaCl in their drinking water (n=6). These animals were compared with a uninephrectomized group that did not receive a DOCA pellet and were maintained on normal drinking water (n=5). After 3 weeks, BPs were measured as above and kidneys perfused with Hank's balanced salt solution and collected for IHC.
IHC was performed on deparaffinized 4-µm paraffin sections after heat-induced epitope retrieval, with the exception of fibronectin IHC, for which antigen retrieval was not used. Picrosirius red staining was performed according to the manufacturer’s protocol (Polysciences Inc.) and viewed under polarized light. Positive staining for all imaging including Picrosirius red was quantified using ImagePro 6.2 from five different fields at a magnification of 20×, and expressed as a ratio to total image area. These were normalized to control samples to yield fold-change values. Immunofluorescence was conducted on cortical cryosections (6 µm) as previously described.51 Primary antisera used were FITC-conjugated α-smooth muscle actin (Abcam, Inc.), rabbit SREBP-1 (Abcam, Inc.), and rabbit TGF-β1 (R&D Systems). An AF568-conjugated secondary antibody (Invitrogen) was used. For protein and RNA assessment, snap-frozen kidney cortex was homogenized in lysis buffer or Trizol respectively, and samples analyzed as described above.
Statistical Analyses
Statistical analyses were performed with SPSS software, version 21 for Windows (IBM, Chicago, IL), using one-way ANOVA and Tukey honest significant difference for post hoc analysis. A t test was used for experiments with only two groups. A P value <0.05 (two-tailed) was considered to represent a statistically significant difference. Data are presented as the mean±SEM, and number of repetitions are denoted as “n=”.
Disclosures
None.
Supplementary Material
Acknowledgments
J.K. gratefully acknowledges the support of the Canadian Diabetes Association and St. Joseph’s Healthcare for their support of nephrology research. We thank Dr. N. Kato (University of Tokyo, Japan) for providing the TGF-β1 promoter-luciferase construct, Dr. M. Bilandzic (Prince Henry’s Institute, Australia) for providing CAGA12-luc, Dr. Y.P. Di (University of Pittsburgh, PA) for providing the 3xSp1-Luc construct, Dr. R. Austin (McMaster University, Hamilton, ON, Canada) for providing the pSRE-GFP plasmid and Dr. A. Schulze (London Research Institute, UK) for providing the SREBP-1a Y335A and SREBP-1c Y321A constructs.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013121332/-/DCSupplemental.
References
- 1.Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F: SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 86: 839–848, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Bengoechea-Alonso MT, Ericsson J: SREBP in signal transduction: Cholesterol metabolism and beyond. Curr Opin Cell Biol 19: 215–222, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY: Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol 22: 76–81, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Uttarwar L, Gao B, Ingram AJ, Krepinsky JC: SREBP-1 activation by glucose mediates TGF-β upregulation in mesangial cells. Am J Physiol Renal Physiol 302: F329–F341, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Mansbach CM, 2nd, Siddiqi SA, Park EA, Raghow R, Elam MB: Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J Biol Chem 284: 7518–7532, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou RH, Yao M, Lee TS, Zhu Y, Martins-Green M, Shyy JY: Vascular endothelial growth factor activation of sterol regulatory element binding protein: A potential role in angiogenesis. Circ Res 95: 471–478, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Sun L, Halaihel N, Zhang W, Shimomura I, Horton JD, Rogers T, Levi M: Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem 277: 18919–18927, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Ishigaki N, Yamamoto T, Shimizu Y, Kobayashi K, Yatoh S, Sone H, Takahashi A, Suzuki H, Yamagata K, Yamada N, Shimano H: Involvement of glomerular SREBP-1c in diabetic nephropathy. Biochem Biophys Res Commun 364: 502–508, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Saito K, Ishizaka N, Hara M, Matsuzaki G, Sata M, Mori I, Ohno M, Nagai R: Lipid accumulation and transforming growth factor-beta upregulation in the kidneys of rats administered angiotensin II. Hypertension 46: 1180–1185, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Mehta PK, Griendling KK: Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Perlman A, Lawsin LM, Kolachana P, Saji M, Moore J, Jr, Ringel MD: Angiotensin II regulation of TGF-beta in murine mesangial cells involves both PI3 kinase and MAP kinase. Ann Clin Lab Sci 34: 277–286, 2004 [PubMed] [Google Scholar]
- 12.Uchiyama-Tanaka Y, Matsubara H, Nozawa Y, Murasawa S, Mori Y, Kosaki A, Maruyama K, Masaki H, Shibasaki Y, Fujiyama S, Nose A, Iba O, Hasagawa T, Tateishi E, Higashiyama S, Iwasaka T: Angiotensin II signaling and HB-EGF shedding via metalloproteinase in glomerular mesangial cells. Kidney Int 60: 2153–2163, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Dickhout JG, Krepinsky JC: Endoplasmic reticulum stress and renal disease. Antioxid Redox Signal 11: 2341–2352, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Kitamura M: Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol 295: F323–F334, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Tabas I, Ron D: Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13: 184–190, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damiano F, Alemanno S, Gnoni GV, Siculella L: Translational control of the sterol-regulatory transcription factor SREBP-1 mRNA in response to serum starvation or ER stress is mediated by an internal ribosome entry site. Biochem J 429: 603–612, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Colgan SM, Hashimi AA, Austin RC: Endoplasmic reticulum stress and lipid dysregulation. Expert Rev Mol Med 13: e4, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, Cai L: Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med 13[8A]: 1499–1512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K: Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol 32: 1652–1661, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colgan SM, Tang D, Werstuck GH, Austin RC: Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell Biol 39: 1843–1851, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A: SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8: 224–236, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J: Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1: 379–391, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Bengoechea-Alonso MT, Ericsson J: A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem 284: 5885–5895, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Higgins ME, Ioannou YA: Apoptosis-induced release of mature sterol regulatory element-binding proteins activates sterol-responsive genes. J Lipid Res 42: 1939–1946, 2001 [PubMed] [Google Scholar]
- 25.Pastorino JG, Shulga N: Tumor necrosis factor-alpha can provoke cleavage and activation of sterol regulatory element-binding protein in ethanol-exposed cells via a caspase-dependent pathway that is cholesterol insensitive. J Biol Chem 283: 25638–25649, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL: Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J 15: 1012–1020, 1996 [PMC free article] [PubMed] [Google Scholar]
- 27.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M: Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 110: 705–712, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP, Lan HY: Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res 98: 1032–1039, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang F, Chung AC, Huang XR, Lan HY: Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54: 877–884, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Inagi R: Endoplasmic reticulum stress as a progression factor for kidney injury. Curr Opin Pharmacol 10: 156–165, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M: Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4: e1000133, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seo YK, Chong HK, Infante AM, Im SS, Xie X, Osborne TF: Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc Natl Acad Sci U S A 106: 13765–13769, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G, Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L, Birbaumer L, Rosenberg P, Winn MP: TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol 22: 526–535, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobori H, Nishiyama A, Abe Y, Navar LG: Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG: Urinary angiotensinogen as an indicator of intrarenal Angiotensin status in hypertension. Hypertension 41: 42–49, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinto YM, Paul M, Ganten D: Lessons from rat models of hypertension: From Goldblatt to genetic engineering. Cardiovasc Res 39: 77–88, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Ferrari A, Maretto S, Girotto D, Volpin D, Bressan GM: SREBP contributes to induction of collagen VI transcription by serum starvation. Biochem Biophys Res Commun 313: 600–605, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Le Lay S, Lefrère I, Trautwein C, Dugail I, Krief S: Insulin and sterol-regulatory element-binding protein-1c (SREBP-1C) regulation of gene expression in 3T3-L1 adipocytes. Identification of CCAAT/enhancer-binding protein beta as an SREBP-1C target. J Biol Chem 277: 35625–35634, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F: Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: A new therapeutic approach. Nat Med 11: 867–874, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Tanjore H, Lawson WE, Blackwell TS: Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta 1832: 940–947, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenna S, Trojanowska M: The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol 24: 663–668, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, Legendre C, Beaune P, Thervet E, Anglicheau D: Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am J Transplant 8: 2283–2296, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Natori T, Nagai K: Endoplasmic reticulum stress upregulates the chondroitin sulfate level which thus prevents neurite extension in C6 glioma cells and primary cultured astrocytes. Cell Mol Neurobiol 28: 857–866, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS: Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A 108: 10562–10567, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I, Uemoto S: CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 294: G498–G505, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Krepinsky JC, Ingram AJ, Tang D, Wu D, Liu L, Scholey JW: Nitric oxide inhibits stretch-induced MAPK activation in mesangial cells through RhoA inactivation. J Am Soc Nephrol 14: 2790–2800, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Bradburne C, Robertson K, Thach D: Assessment of methods and analysis of outcomes for comprehensive optimization of nucleofection. Genet Vaccines Ther 7: 6, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di YP, Zhao J, Harper R: Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J Biol Chem 287: 27948–27958, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A: PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24: 6465–6481, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ, Uesugi M: A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 16: 882–892, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Wu D, Peng F, Zhang B, Ingram AJ, Gao B, Krepinsky JC: Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signalling in mesangial cells. Diabetologia 50: 2008–2018, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.