

Abstract
Recent studies have highlighted the renoprotective effect of sirtuin1 (SIRT1), a deacetylase that contributes to cellular regulation. However, the pathophysiologic role of SIRT1 in podocytes remains unclear. Here, we investigated the function of SIRT1 in podocytes. We first established podocyte-specific Sirt1 knockout (SIRT1pod−/−) mice. We then induced glomerular disease by nephrotoxic serum injection. The increase in urinary albumin excretion and BUN and the severity of glomerular injury were all significantly greater in SIRT1pod−/− mice than in wild-type mice. Western blot analysis and immunofluorescence showed a significant decrease in podocyte-specific proteins in SIRT1pod−/− mice, and electron microscopy showed marked exacerbation of podocyte injury, including actin cytoskeleton derangement in SIRT1pod−/− mice compared with wild-type mice. Protamine sulfate-induced podocyte injury was also exacerbated by podocyte-specific SIRT1 deficiency. In vitro, actin cytoskeleton derangement in H2O2-treated podocytes became prominent when the cells were pretreated with SIRT1 inhibitors. Conversely, this H2O2-induced derangement was ameliorated by SIRT1 activation. Furthermore, SIRT1 activation deacetylated the actin-binding and -polymerizing protein cortactin in the nucleus and facilitated deacetylated cortactin localization in the cytoplasm. Cortactin knockdown or inhibition of the nuclear export of cortactin induced actin cytoskeleton derangement and dissociation of cortactin from F-actin, suggesting the necessity of cytoplasmic cortactin for maintenance of the actin cytoskeleton. Taken together, these findings indicate that SIRT1 protects podocytes and prevents glomerular injury by deacetylating cortactin and thereby, maintaining actin cytoskeleton integrity.
Keywords: podocyte, SIRT1, cytoskeleton, cortactin
Podocytes, which are characterized as visceral glomerular epithelial cells, are highly differentiated cells that play an essential role in maintenance of the glomerular tuft and filtration barrier. Foot processes (FPs) of adjacent podocytes regularly interdigitate to form narrow filtration slits that are bridged by the slit diaphragm, a zipper-like structured modified adherens junction.1 Podocyte injury is a common feature in glomerular diseases.1–6 Among several pathophysiologic mechanisms of podocyte injury, derangement of actin cytoskeleton is a critical pathologic manifestation, because the actin cytoskeleton plays an essential role in maintaining the podocyte structure, including FPs. Actin fibers in podocytes function in not only the maintenance of cellular structure but also, interactions with various molecules, including structural molecules in the slit diaphragm.1,7,8 These findings strongly suggest that the maintenance of actin cytoskeleton is essential to glomerular function and podocyte homeostasis.
Sirtuin1 (SIRT1; silent mating type information regulation 2 homolog) is an NAD+-dependent deacetylase that regulates a variety of physiologic phenomena.9–15 Most of the effects of SIRT1 depend on its deacetylation activity.9,10 Recent studies have shown that SIRT1 is associated with a number of pathologic conditions.16–21 With regard to the kidney, SIRT1 is reported to have renoprotective functions.22,23 As examples, SIRT1 regulates glucose or lipid metabolism, BP, and oxidative stress, which are all closely associated with kidney disease.24–28 In addition, SIRT1 protects the proximal tubular,29–31 medullary,32 and mesangial cells.33,34 Several studies have also suggested the possibility of a protective effect of SIRT1 on podocytes35–37; however, the molecular mechanism of the function of SIRT1 expressed in podocytes remains unclear.
Here, we hypothesized that the protective effect of SIRT1 on podocytes results from its maintenance of actin cytoskeleton, which is a critical factor for podocyte homeostasis. To address this question, we established podocyte-specific Sirt1 knockout (SIRT1pod−/−) mice and investigated the effect of SIRT1 deletion in podocytes.
Results
Podocyte Injury Is Exacerbated by Deficiency of Podocyte SIRT1 Expression in GN Induced by Nephrotoxic Serum
In this study, we used SIRT1pod−/− mice, established by crossing Sirt1flox/flox mice with podocin-Cre mice, to investigate the function of SIRT1 in podocytes. SIRT1 deletion in SIRT1pod−/− mice was confirmed by Western blot (WB) analysis using isolated glomeruli (Figure 1A). SIRT1pod−/− mice did not show any physiologic changes (Table 1), and neither histologic change nor biochemical alteration was observed (Supplemental Figure 1, A–C). To reveal the vulnerability of podocytes in SIRT1pod−/− mice, we used a nephrotoxic serum (NTS) -induced GN model as a direct podocyte injury model in mice. Our NTS, which contains sheep antiglomerular IgG, could induce podocyte damage with albuminuria by single injection (heterologous reaction to sheep IgG) in mice (Concise Methods). Seven days after disease induction, the ratio of damaged glomeruli was significantly higher in SIRT1pod−/− mice and associated with increases in tubular cast formation and glomerular macrophage infiltration (Figure 1, B–D, Supplemental Figure 2). Although the number of glomeruli with tuft necrosis and/or crescent formation was increased in SIRT1pod−/− mice (Figure 1C), the proportion of tuft necrosis and/or crescent formation within each affected glomerulus was the same in both groups (Supplemental Figure 3), showing that the disease phenotype was exaggerated in quantity but equivalent in quality. We confirmed that the reactivity of NTS (the binding level of sheep IgG in NTS) to glomeruli of the two groups was similar (Figure 1E). Moreover, the deposition of mouse IgG on glomeruli was similar between wild-type and SIRT1pod−/− mice (Supplemental Figure 4), suggesting that the immune response against the NTS was equivalent. Both urinary albumin/creatinine ratio (U-alb/cre; 28.35.5 in wild-type mice versus 93.5±18.5 in SIRT1pod−/− mice) and BUN (21.6±0.9 in wild-type mice versus 26.1±1.0 mg/dl in SIRT1pod−/− mice) were also significantly exacerbated in SIRT1pod−/− mice compared with wild-type mice (Figure 1, F and G). Furthermore, on WB analysis using isolated glomeruli, the expression of nephrin, synaptopodin, and Wilms’ tumor 1 protein (WT-1) after the induction of GN was significantly decreased in SIRT1pod−/− mice compared with wild-type mice (Figure 2A). Immunofluorescence analyses revealed that synaptopodin or WT-1 expression level in SIRT1pod−/− and wild-type mice was similar before disease induction (Supplemental Figure 1D) and significantly decreased after disease induction. Importantly, this decrease was also aggravated in SIRT1pod−/− mice (Figure 2B). These results are compatible with the exacerbation of histologic and biochemical data in NTS-injected SIRT1pod−/− mice.
Figure 1.

Glomerular injury induced by NTS was exacerbated in SIRTpod−/− mice. (A) WB analysis using isolated glomeruli from wild-type or SIRT1pod−/− mice. A reduction in SIRT1 expression in SIRT1pod−/− mice was confirmed. (B) Histologic analysis by periodic acid–Schiff staining. At 7 days after glomerular disease induction with NTS injection, glomerular damage was more severe in SIRT1pod−/− mice than in wild-type mice. Scale bars, 100 μm. (C) Ratio of injured glomeruli in wild-type or SIRT1pod−/− mice (n=9 for each). The ratio of damaged glomeruli in SIRT1pod−/− mice with the GN was significantly higher than that in wild-type mice. ***P<0.001 versus wild-type mice. (D)The relative area of tubulointerstitial cast in wild-type or SIRT1pod−/− mice (n=4 for each). Tubular cast formation in SIRT1pod−/− mice with the GN was significantly higher than that in wild-type mice. **P<0.01 versus wild-type mice. (E) Detection of the reactivity of NTS to the glomeruli of experimental mice. Immunofluorescence analysis showed a similar level of binding of sheep IgG in NTS to glomeruli in wild-type and SIRT1pod−/− mice, indicating that disease induction in both groups was identical. Scale bar, 100 μm. (F) BUN or (G) U-alb/cre in experimental mice; 7 days after NTS injection, blood and urine were collected from wild-type (n=14) and SIRT1pod−/− (n=16) mice. Both BUN and U-alb/cre were significantly higher in SIRT1pod−/− mice than in wild-type mice under disease conditions. **P<0.01.
Table 1.
Physiologic data of the experimental mice
| SIRT1+/+ | SIRT1pod−/− | P Value | |
|---|---|---|---|
| n | 24 | 22 | — |
| Age (wk) | 13.96±0.12 | 14.09±0.09 | 0.41 |
| Body weight (g) | 26.8±0.36 | 26.6±0.62 | 0.28 |
| SBP (mmHg) | 97.2±2.9 | 99.3±3.5 | 0.66 |
| DBP (mmHg) | 50.2±5.5 | 54.83±4.7 | 0.53 |
| Pulse rate (bpm) | 600.6±45.8 | 608.4±53.4 | 0.91 |
Data are means±SEMs of wild-type mice and SIRT1pod−/− mice before disease induction. DBP, diastolic BP; SBP, systolic BP.
Figure 2.

Podocyte injury induced by NTS was exacerbated in SIRTpod−/− mice. (A) Podocyte injury level in isolated glomeruli of wild-type or SIRT1pod−/− mice at 7 days after NTS treatment. WB analysis followed by densitometry was performed to assess podocyte injury level, which was estimated by the expression of podocyte-specific proteins, such as nephrin, synaptopodin (Synpo), and WT-1. For densitometry, α-tubulin was used as an internal control. Actin expression was also assessed to confirm that the expression of actin was not changed in SIRT1-deficient podocytes. Expression of these proteins was significantly reduced in SIRT1pod−/− mice compared with wild-type mice. *P<0.05 versus wild-type mice. (B) Representative immunohistochemical pictures of Synpo or WT-1 in experimental mice. The results of quantitative analysis of fluorescence intensity are shown below. Decreased podocyte-specific protein expression was significantly more severe in the SIRT1pod−/− mice compared with wild-type mice under disease condition. Scale bars, 20 μm. ***P<0.001 versus wild-type mice.
FP Effacement and Actin Cytoskeleton Derangement Are Exacerbated by Podocyte-Specific Sirt1 Knockout
To elucidate the mechanism by which SIRT1 deficiency deranges podocyte homeostasis, we evaluated structural alterations in podocytes in NTS-injected SIRT1pod−/− mice by electron microscopy. At 7 days after NTS treatment, FP effacement was more severe in SIRT1pod−/− than wild-type mice, and the accumulation of F-actin, which indicates actin cytoskeleton derangement, was higher (Figure 3, A and B). These findings are consistent with the marked increase in albuminuria in glomerular disease-induced SIRT1pod−/− mice (Figure 1G) and suggest that the major features of podocyte vulnerability after Sirt1 knockout were disruption of the actin cytoskeleton and slit diaphragm.
Figure 3.

FP effacement and actin cytoskeleton damage were exacerbated in SIRTpod−/− mice with GN induced by NTS. (A) Electron microscopic images of glomeruli of (a, c, e, and g) wild-type and (b, d, f, and h) SIRT1pod−/− mice at 7 days after NTS treatment. (b and d) In SIRT1pod−/− mice, FP effacement was more severe compared with (a and c) that in wild-type mice. g and h are enlargements of the parts indicated by arrows in e and f. Accumulation of F-actin (arrowheads) was also increased by SIRT1 deficiency in podocytes. Scale bars, 2 μm in a, b, and e–h; 1 μm in c and d. (B) Quantitative analysis of FP effacement in the mice at 7 days after NTS treatment. ***P<0.001. (C) Electron microscopic images of glomeruli of wild-type and SIRT1pod−/− mice at 2 days after NTS treatment. FP effacement observed at an early phase of NTS-induced GN was aggravated by SIRT1 deficiency in podocytes. Accumulation of F-actin (arrowheads) was also increased in SIRT1pod−/− mice compared with wild-type mice. Scale bar, 1 µm. (D) Quantitative analysis of FP effacement in the mice at 2 days after NTS treatment. ***P<0.001 versus wild-type mice.
At an early stage (day 2) after NTS injection (or heterologous phase), exacerbation of FP effacement and accumulation of F-actin were observed in SIRT1pod−/− mice (Figure 3, C and D) in association with the tendency of the increase in U-alb/cre (284.2±118.1 in wild-type mice versus 427.3±108.5in SIRT1pod−/− mice; P=0.39). Glomerular macrophage infiltration was not increased in either group, showing that the exacerbation of podocyte injury by SIRT1 deletion is probably caused by the direct effect of the antibody rather than an inflammatory process (Supplemental Figure 5). Moreover, FP effacement induced by protamine sulfate (PS) perfusion in SIRT1pod−/− mice was also significantly aggravated compared with in wild-type mice (Figure 4).
Figure 4.

FP effacement induced by perfusion of PS was exacerbated in SIRT1pod−/− mice. (A) Electron microscopic images of glomeruli in the mice with PS perfusion. FP effacement induced by perfusion of PS was significantly exacerbated in SIRT1pod−/− mice compared with wild-type mice. Scale bars, 2 µm. (B) The result of quantitative analysis is shown. ***P<0.001 versus wild-type mice.
SIRT1 Activity Is Necessary for Maintenance of Actin Cytoskeleton in Podocytes
To assess the effect of SIRT1 on actin cytoskeleton dynamics, we investigated structural changes in cultured murine podocytes treated with SIRT1 inhibitors. Having found evidence that oxidative stress, as measured by nitrotyrosine staining, is induced in podocytes of NTS-treated mice (Figure 5A), we used hydrogen peroxide (H2O2) to induce cellular damage of cultured podocytes. The cultured podocytes were pretreated with the SIRT1-specific inhibitor EX-527 or vehicle (ethanol) for 24 hours and incubated with or without H2O2 (300 μM) for 24 hours. The SIRT1 inhibitory effect of EX-527 was confirmed by WB analysis for acetyl-histone H3 (Figure 5B). Although mild derangement of actin cytoskeleton was seen in the podocytes treated with either H2O2 or EX-527, severe derangement was observed in SIRT1-inactivated podocytes treated with H2O2 (Figure 5, C–E). Similar results were observed in experiments using other SIRT1 inhibitors, such as cambinol or nicotinamide (NAM) (Figure 5C). We confirmed that the inhibition of SIRT1 activity did not influence the actin expression level (Figure 5B). We next confirmed the effect of SIRT1 on maintenance of actin cytoskeleton using resveratrol, an SIRT1 activator. Pretreatment with resveratrol prevented the derangement induced in cultured podocytes by a high concentration of H2O2 (700 μM) (Figure 5, F–H). Moreover, PS-induced actin cytoskeletal derangement was also exacerbated by EX-527 (Supplemental Figure 6). These loss- and gain-of-function analyses revealed that SIRT1 function is associated with maintenance of the actin cytoskeleton.
Figure 5.


SIRT1 activity was necessary for actin cytoskeleton maintenance in podocytes. (A) Oxidative stress detection by immunohistochemistry for nitrotyrosine in glomeruli of control or GN-induced mice. In NTS-injected mice, podocytes were positive for nitrotyrosine, indicating that oxidative stress was induced in GN induced by NTS. Scale bar, 50 μm. (B) WB analysis and densitometry for acetyl-histone H3 (Ac-HisH3) in cultured podocytes. EX-527, an SIRT1 inhibitor, significantly increased acetylation level of histone H3 in a dose-dependent manner in podocytes. (C) Detection of actin fiber by fluorescein–phalloidin staining in cultured podocytes. Lower panels are enlargements. Cultured podocytes were treated with SIRT1 inhibitors (EX-527, 100 µM; cambinol, 50 µM; or NAM, 10 mM) or vehicle (ethanol) for 24 hours and then treated with or without H2O2 (300 µM) for 24 hours. H2O2 was used to induce podocyte injury to mimic that in GN induced by NTS. Actin cytoskeleton derangement was markedly deteriorated in podocytes treated with both EX-527 and H2O2 compared with the cells treated with either EX-527 or H2O2. Similar results were confirmed using the other SIRT1 inhibitors cambinol or NAM. Scale bars, 50 μm. (D and E) Quantitative analysis of actin cytoskeleton derangement in podocytes treated with EX-527 (EX; 100 µM) and H2O2 (300 µM). (D) Mean score of actin cytoskeleton derangement and (E) ratio of the cells with severe derangement (corresponds to score=4) were measured. The derangement was significantly exacerbated by SIRT1 inhibition under oxidative stress conditions. (F) Fluorescein–phalloidin staining of cultured podocytes treated with SIRT1 activator under oxidative stress conditions. Lower panels are enlargements. Cultured podocytes were pretreated with SIRT1 activator resveratrol (200 µM) or vehicle (ethanol) for 3 hours and then cultured with high-dose H2O2 (high H2O2; 700 μM) for 1 hour. Resveratrol prevented actin cytoskeleton derangement induced by oxidative stress. Scale bars, 50 μm. (G and H) Quantitative analysis of actin cytoskeleton derangement in podocytes treated with resveratrol and H2O2. (G) Mean score of actin cytoskeleton derangement and (H) ratio of the cells with severe derangement were measured. The derangement was significantly attenuated by SIRT1 activation under oxidative stress conditions. RSV, resveratrol; vehi, vehicle (ethanol). *P<0.05 versus vehicle; **P<0.01 versus vehicle; ***P<0.001.
Podocyte Motility Is Reduced by SIRT1 Inhibition
Because the cellular motility of podocytes is dependent on their dynamic assembly of actin,38–41 we hypothesized that podocyte migration ability is attenuated when the regulation of actin cytoskeleton is disorganized by SIRT1 inhibition. To address this, we examined the change in cellular motility by scratch assay. Motility was markedly reduced by EX-527 treatment compared with vehicle (Figure 6A). Similar results were observed using other SIRT1 inhibitors (Figure 6, B and C), supporting that SIRT1 activity plays an essential role in actin cytoskeleton dynamics.
Figure 6.

SIRT1 activity contributed to podocyte motility. Scratch assay followed by counting of migrated podocytes treated with the SIRT1 inhibitions (A) EX-527, (B) cambinol, or (C) NAM. The cells were cultured with 100 μM EX-527, 50 μM cambinol, or 5 mM NAM for 3 days after scratch, and then, the number of migrated cells was counted. Inhibition of SIRT1 activity significantly retarded podocyte migration. Results were confirmed by three independent experiments. Vehi, vehicle (ethanol). Scale bars, 200 μm. *P<0.05 versus vehicle; **P<0.01 versus vehicle; ***P<0.001 versus vehicle.
SIRT1 Deacetylates Cortactin in Nuclei of Podocytes
We focused on cortactin, which is an actin-binding protein for polymerization and stabilization of F-actin,38,42 and it is controlled by SIRT1 deacetylation.42–44 We investigated the effect of SIRT1 on the acetylation state of cortactin and subsequent actin cytoskeleton maintenance and dynamics in podocytes. On WB analysis, the ratio of acetylated cortactin to total cortactin was increased in cultured podocytes treated with SIRT1 inhibitors in a dose-dependent manner (Figure 7, A and B). Conversely, this ratio was reduced in resveratrol-treated podocytes (Figure 7C). Moreover, these in vitro data were consistent with an in vivo study showing the increased acetylated cortactin level in isolated glomeruli from SIRT1pod−/− mice (Figure 7D). This interaction between SIRT1 and cortactin was confirmed in immunoprecipitation analysis in cultured podocytes (Figure 7E). Immunofluorescence analysis, which showed colocalization of SIRT1 with cortactin in nuclei, also supported the interaction between SIRT1 and cortactin (Figure 7F), suggesting that SIRT1 deacetylates cortactin in the nuclei of podocytes.
Figure 7.

SIRT1 regulated the cortactin acetylation level in vitro and in vivo. (A–C) WB analysis of acetylated cortactin (Ac-cort) and total cortactin in the whole-cell extract of cultured podocytes treated with the SIRT1 inhibitors (A) EX-527 or (B) cambinol or (C) the SIRT1 activator resveratrol (RSV). Right panels show quantitative analyses of the ratio of Ac-cort to cortactin. The ratio of Ac-cort was significantly increased by EX-527 or cambinol in a dose-dependent manner, whereas it was decreased by RSV. *P<0.05 versus vehicle; **P<0.01 versus vehicle. (D) WB analysis of Ac-cort and total cortactin in the isolated glomeruli of wild-type or SIRT1pod−/− mice. Quantitative analysis of the ratio of Ac-cort to cortactin is shown in the right panel. The ratio of Ac-cort was significantly increased in SIRT1pod−/− mice compared with wild-type mice. **P<0.01 versus wild-type mice. (E) Immunoprecipitation (IP) analysis for the detection of SIRT1 binding with cortactin. Whole-cell extract of cultured podocytes without stimulation was immunoprecipitated with anticortactin antibody or normal anti-rabbit IgG, and the precipitate was analyzed by WB analysis using anti-SIRT1 or anticortactin antibodies (upper panel). The interaction from the opposite direction was confirmed (lower panel). A protein sample without IP was also analyzed (input) as a control. The results showed that SIRT1 interacts with cortactin. (F) Immunofluorescence analysis for detection of SIRT1 and cortactin in cultured podocytes. Staining of SIRT1, cortactin, and nuclei (Hoechst 33258) and a merged image are shown. The merged image shows that SIRT1 was colocalized with cortactin in the nuclei, suggesting that SIRT1 colocalizes with cortactin in the nuclei. Scale bar, 100 μm.
SIRT1 Regulates Cortactin–Actin Interaction in Podocytes
Next, we investigated cortactin localization and its contribution to actin cytoskeleton maintenance and dynamics. Immunocytochemistry showed that total (acetylated and deacetylated) cortactin was localized in both the cytoplasm and nucleus and that the cytoplasmic cortactin was partly colocalized with actin fibers (Figure 8, A and B). When actin cytoskeleton derangement was induced by H2O2 in cells pretreated with EX-527, the fiber-like distribution of cytoplasmic cortactin was simultaneously disrupted, and cortactin was dissociated from actin fiber (Figure 8, A and B). Similar results were confirmed using knockdown of Sirt1 in podocytes (Figure 8, C–F). In contrast, resveratrol prevented actin cytoskeleton derangement induced by high concentrations of H2O2 through amelioration of altered distribution of cortactin and dissociation of cortactin from actin fiber (Figure 8G). Along with the cortactin localization, we assessed the alteration of actin cytoskeleton by cortactin knockdown by siRNA transfection. In podocytes, the reduction of cortactin induced actin cytoskeleton derangement without any stimulation, suggesting that cortactin has a crucial role in the maintenance of actin cytoskeleton (Figure 9).
Figure 8.


SIRT1 was necessary for cortactin binding to actin fiber and maintenance of actin cytoskeleton under oxidative stress. (A) Detection of cortactin by immunofluorescence in cultured podocytes treated with SIRT1 inhibitor under oxidative stress. Staining of cortactin, actin fibers (phalloidin), and nuclei (Hoechst 33258) and their merged images are shown. Cultured podocytes were treated with vehicle or EX-527 (100 μM) for 24 hours and subsequently incubated for 24 hours with or without H2O2 (300 μM). In vehicle-treated cells, cortactin was observed in both cytoplasm and nucleus, and cytoplasmic cortactin was partly colocalized with actin fibers. Cells treated with either H2O2 or EX-527 showed moderate actin cytoskeleton derangement. Treatment with both EX-527 and H2O2, namely inhibition of SIRT1 activity under oxidative stress conditions, produced severe derangement of actin cytoskeleton and the apparent absence of fiber-like distribution of cytoplasmic cortactin; in contrast, no changes in nuclear cortactin were observed. Scale bar, 100 μm. (B) Enlargements of the merged images shown in A. Actin cytoskeleton derangement associated with the disruption of fiber-like distribution of cytoplasmic cortactin is clearly observed in SIRT1-inhibited podocytes under oxidative stress conditions. Scale bar, 100 μm. (C) Quantitative analysis of mRNA of Sirt1 in cultured podocytes transfected with Sirt1 small interfering RNA (siRNA) (10 nM). Real-time PCR showed that Sirt1 siRNA induced a significant decrease of Sirt1 expression (16%) compared with negative control siRNA (10 nM). ***P<0.001 versus control siRNA. (D) Immunofluorescence images of cultured podocytes transfected with Sirt1 siRNA under oxidative stress. Staining of cortactin, actin fibers (phalloidin), and nuclei (Hoechst 33258) and their merged images are shown. Cultured podocytes were transfected with Sirt1 siRNA (10 nM) or negative control siRNA (10 nM) and subsequently incubated for 24 hours with or without H2O2 (300 μM). Sirt1 knockdown induced vulnerability to oxidative stress. Scale bar, 100 μm. (E and F) Quantitative analysis of actin cytoskeleton derangement in the cells transfected with Sirt1 siRNA under oxidative stress conditions. (E) Mean score of actin cytoskeleton derangement and (F) ratio of the cells with severe derangement were measured. The derangement was significantly exacerbated by Sirt1 knockdown under oxidative stress. **P<0.01 versus control siRNA. (G) Immunofluorescence images of cultured podocytes treated with resveratrol under oxidative stress. Staining of cortactin, actin fibers, and nuclei and their merged images are shown. Cultured podocytes were pretreated with SIRT1 activator resveratrol (RSV; 200 μM) or vehicle for 3 hours and then cultured with high-dose H2O2 (high H2O2; 700 μM) for 1 hour. RSV prevented derangement of cortactin distribution and dissociation of cortactin from actin fiber, attenuating actin cytoskeleton derangement induced by oxidative stress. Vehi, vehicle (ethanol). Scale bar, 100 μm.
Figure 9.
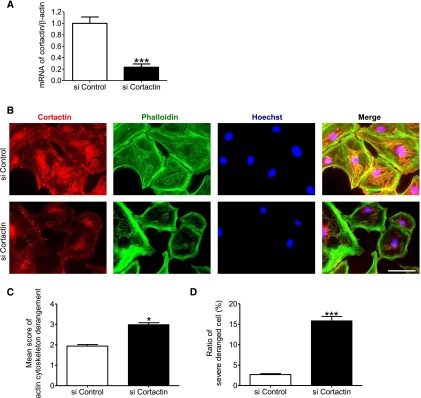
Cortactin was essential for maintenance of actin cytoskeleton. (A) Quantitative analysis of mRNA of cortactin in cultured podocytes transfected with cortactin small interfering RNA (siRNA) (20 nM). Real-time PCR showed that the cortactin siRNA significantly decreased cortactin expression (23%). (B) Immunofluorescence analysis of cultured podocytes transfected with negative control siRNA or cortactin siRNA. Staining of cortactin, actin fibers (phalloidin), and nuclei (Hoechst 33258), and their merged images are shown. Cultured podocytes were transfected with negative control siRNA (20 nM) or cortactin siRNA (20 nM). Cortactin knockdown caused reduction of cortactin expression and marked actin cytoskeleton derangement in podocytes. Scale bar, 100 μm. (C and D) Quantitative analysis of actin cytoskeleton derangement in the cells transfected with cortactin siRNA. (C) Mean score of actin cytoskeleton derangement and (D) ratio of the cells with severe derangement were measured. The derangement was significantly exacerbated by cortactin knockdown. *P<0.05 versus control siRNA; ***P<0.001 versus control siRNA.
SIRT1 Deacetylated Cortactin in the Nuclei of Podocytes and Changed Localization of Cortactin
Finally, we assessed the change of localization of cortactin by deacetylation. In contrast to total cortactin, acetylated cortactin was detectable only in the nucleus and remained colocalized with SIRT1, regardless of the state of actin cytoskeleton (Figure 10, A and B). We confirmed the nuclear localization of acetylated cortactin in podocytes in vivo (Figure 10C). Taking these findings together, we speculated that the deacetylation of cortactin by SIRT1 regulates its localization and subsequent cortactin–actin interaction in cytoplasm. We then examined the effect of SIRT1 on the acetylation level of cortactin in the cytoplasm and nucleus of cultured podocytes or isolated glomeruli. In nuclear extract, acetylated cortactin was increased by SIRT1 inhibition, whereas it was undetectable in cytoplasmic extract (Figure 10, D and E). The nuclear total cortactin was increased by SIRT1 inhibition, whereas cytoplasmic total cortactin was conversely decreased (Figure 10, D and E). Similar changes were observed in isolated glomeruli from wild-type and SIRT1pod−/− mice (Figure 10, F and G). In contrast, SIRT1 activator increased cytoplasmic total cortactin and conversely, decreased total and acetylated cortactin in the nucleus (Figure 10, H and I). These results suggest that the deacetylation activity of SIRT1 regulates the cortactin localization and function.
Figure 10.
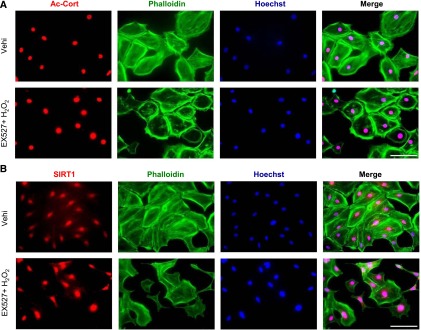



SIRT1 deacetylated cortactin in the nuclei of podocytes and promoted cytoplasmic localization of cortactin. (A and B) Detection of (A) acetylated cortactin (Ac-cort) and (B) SIRT1 by immunofluorescence in cultured podocytes treated with SIRT1 inhibitor under oxidative stress. Cultured podocytes were analyzed for staining of (A) Ac-cort or (B) SIRT1 with actin fiber (phalloidin) and nuclei (Hoechst 33258). Ac-cort was observed only in nuclei, regardless of the state of actin cytoskeleton in association with colocalization of SIRT1. Scale bars, 100 μm. (C) Immunostaining of Ac-cort in the glomerulus of the experimental mice. Lower images are enlargements focusing on podocytes (arrowheads). In the podocytes, Ac-cort was detected only in nuclei in each mouse, which was compatible with the results in vitro shown in A. Scale bars, 20 μm. (D and E) Detection of cortactin by WB analysis using the (D) cytoplasmic or (E) nuclear extract of cultured podocytes treated with SIRT1 inhibitor. Cells treated with EX-527 (SIRT1 inhibitor; 100 μM) were analyzed for Ac-cort or total cortactin, and their expression levels were quantitatively analyzed. Total cortactin level in cytoplasm was reduced by SIRT1 inhibition, whereas nuclear cortactin level was conversely increased in association with nuclear accumulation of Ac-cort. *P<0.05 versus vehicle; **P<0.01 versus vehicle. (F and G) Detection of cortactin by WB analysis using (F) cytoplasmic or (G) nuclear extract of isolated glomeruli from wild-type mice or SIRT1pod−/− mice. Compared with wild-type mice, total cortactin level in cytoplasm was lower in SIRT1pod−/− mice, whereas nuclear cortactin level as well as Ac-cort were conversely higher, which was compatible with in vitro data shown in D and E. *P<0.05 versus wild-type mice; **P<0.01 versus wild-type mice; ***P<0.001 versus wild-type mice. (H and I) Detection of cortactin by Western blot analysis using (H) cytoplasmic or (I) nuclear extract of cultured podocytes treated with SIRT1 activator. Cells treated with resveratrol (RSV; 200 μM) were analyzed for Ac-cort or total cortactin, and their expression levels were quantitatively analyzed. Total cortactin level in cytoplasm was increased by SIRT1 activator, whereas nuclear cortactin level as well as Ac-cort were conversely decreased. *P<0.05 versus vehicle. α-Tubulin and histone H1 were used as internal controls of cytoplasmic and nuclear extracts, respectively, in D–I. (J) Immunofluorescence images of cultured podocytes treated with leptomycin B. Staining of cortactin (using mouse anticortactin antibody; red), actin fibers (phalloidin), and nuclei (Hoechst 33258) and their merged images are shown. Cultured podocytes were treated with nuclear export inhibitor leptomycin B (20 nM) for 12 hours. In leptomycin B-treated cells, cortactin was accumulated in nuclei, and actin cytoskeleton derangement was induced. Cytoplasmic cortactin was necessary for actin cytoskeleton maintenance. Scale bar, 100 μm. (K) Immunofluorescence of cultured podocytes treated with leptomycin B. Staining of cortactin (using rabbit anticortactin antibody; green) and nuclei (Hoechst 33258) and their merged images are shown. The similar change of cortactin localization was confirmed. Scale bar, 100 μm. (L and M) Quantitative analysis of actin cytoskeleton derangement in podocytes treated with leptomycin B. (L) Mean score of actin cytoskeleton derangement and (M) ratio of cells with severe derangement were measured. The derangement was significantly exacerbated by leptomycin B. Vehi, vehicle (ethanol). *P<0.05 versus vehicle; **P<0.01 versus vehicle.
To verify the necessity of cytoplasmic cortactin for maintenance of actin cytoskeleton, we used nuclear export inhibitor leptomycin B and assessed the link between localization of cortactin and actin cytoskeleton. As we expected, in the podocytes treated with leptomycin B, the nuclear accumulation of cortactin was induced, which was associated with severe actin cytoskeletal derangement (Figure 10, J–M, Supplemental Figure 7). These findings strongly support our hypothesis that cytoplasmic cortactin is required for maintenance of actin cytoskeleton (Figure 11).
Figure 11.

SIRT1 maintains podocyte homeostasis by deacetylation of cortactin and subsequent localization change of cortactin. (A and C, left) In the normal state, SIRT1 deacetylates and activates cortactin, and the deacetylated cortactin functions for the maintenance of actin cytoskeleton in the cytoplasm, leading in turn, to the maintenance of podocyte homeostasis. As a result, the glomerulus, including the slit diaphragm, is maintained. (B and C, right) Conversely, suppression of SIRT1 induces an increase in acetylated cortactin in the nucleus and decrease in active cortactin in the cytoplasm, which leads to the derangement of actin cytoskeleton, especially when cytotoxic stimulation is given. Consequent podocyte dysfunction or podocyte vulnerability is induced, leading to slit diaphragm injury followed by glomerular injury.
Discussion
In this study, we established SIRT1pod−/− mice. Podocyte-specific Sirt1 knockout mice showed no biochemical or histologic abnormalities in kidney. However, pathologic alterations, such as FP effacement and actin cytoskeleton derangement, were significantly exacerbated in (1) the podocytes of SIRT1pod−/− mice at both heterologous (day 2) and autologous (day 7) phases of GN induced by NTS and (2) the cultured podocytes with modulated SIRT1 activity under oxidative stress conditions, which is a major mediator of GN caused by NTS. Importantly, we also found that SIRT1 deacetylated cortactin, an actin-binding molecule, and thereby, enhanced cortactin activity, with subsequent polymerization and stabilization of branched actin network. Our study reveals that SIRT1 has the novel function in podocytes of maintaining actin cytoskeleton and subsequently, the structure of slit diaphragm through the deacetylation of cortactin. Furthermore, it indicates the possibility that SIRT1 is essential to the protection of podocyte function as well as structure against pathogenic microenvironments, such as oxidative stress. Our results, therefore, clarify the novel beneficial effect of SIRT1 on podocyte homeostasis.
The cytoskeletal element of FPs is composed of actin fibers only, indicating that actin cytoskeleton is essential to maintenance of the structure and function of the slit diaphragm.45 Mutations of actin-binding proteins, such as α–actinin-4 or CD2AP, lead to podocyte injury because of disorganization of the actin cytoskeleton and disruption of its filtration barrier.41,46,47 We performed a scratch assay and showed that SIRT1 was necessary for podocyte motility. Although the question of whether motile podocytes induce proteinuria and renal damage is still controversial,8,41 it is widely accepted that the reorganization of actin cytoskeleton is necessary for podocyte migration.40,41,48 Thus, the results of our experiments show that SIRT1 is required for the appropriate reorganization of actin cytoskeleton in podocytes.
SIRT1 belongs to the HDAC family, which acts to deacetylate proteins and histones and controls epigenetic changes and protein functions.9,49–52 SIRT1 contributes to the regulation of pathogenic intracellular signaling in podocytes. Not only calorie restriction but also, some cytotoxic conditions, such as oxidative stress, are reported to increase SIRT1 expression,22 which is probably one of the protective reactions against cellular damage. We, indeed, observed that SIRT1 expression was increased in GN induced by NTS in vivo and H2O2-treated podocytes in vitro (Supplemental Figure 8). Chuang et al.35 showed that SIRT1 overexpression in cultured murine podocytes prevents glycative stress-induced apoptosis under diabetic conditions, whereas Yuan et al.36 showed that SIRT1 prevents aldosterone-induced apoptosis of podocytes. Both studies highlight the link between the podocyte-protective effect of SIRT1 and podocyte apoptosis. Proximal tubule-specific SIRT1 knockout mice showed aggravation of the glomerular changes, such as podocyte function.37 We directly verified the function of SIRT1 in podocytes using novel genetically engineered podocyte-specific knockout mice and elucidated the underlying mechanisms for this phenomenon in vivo using pharmacologic methods in cultured podocytes in vitro. Our results revealed a novel function of SIRT1 in podocytes: SIRT1, particularly its deacetylase activity against cortactin, protects podocyte homeostasis through the maintenance of actin cytoskeleton. Our experiments, therefore, reveal the novel function of SIRT1 expressed in podocytes.
Cortactin is an actin filament-binding substrate of Src tyrosine kinase,53,54 which regulates the assembly, polymerization, and stabilization of the branched actin network.44,55–57 Recent works showed that deacetylation is also important in the association of cortactin with actin filaments in cancer cells.43,44 Although the colocalization of cortactin with actin filaments in podocytes has been reported,58,59 the function and regulation of cortactin have remained unclear. Our study revealed that the suppression of SIRT1 expression under pathogenic conditions, such as oxidative stress-related podocyte injury, induced the derangement of actin cytoskeleton through the acetylation of cortactin, indicating a critical link between SIRT1 and cortactin in podocyte homeostasis.
Another novel finding of our study was that SIRT1 is deeply associated with the change of localization of cortactin in podocytes. In this study, we showed that the expression and cytoplasmic localization of cortactin were critical for actin cytoskeleton maintenance through observation of cortactin siRNA-transfected podocytes and leptomycin B-treated cells. Moreover, we also assessed changes in the amount of total or acetylated cortactin in the cytoplasm and nuclei of glomeruli of SIRTpod−/− mice and cultured podocytes with modulated SIRT1 activity. Acetylated cortactin was mainly detected in the nuclei, and its level was inversely correlated with SIRT1 activity. Considered together with the finding that SIRT1 is coprecipitated with cortactin, SIRT1 is necessary for the cytoplasmic localization of cortactin. The regulatory role of post-translational acetylation on the subcellular localization of proteins is well known.60 In particular, nuclear protein LKB1 is exported to the cytoplasm after deacetylation by SIRT1.61 The tandem repeat domain of cortactin is considered to contain lysine residues targeted by SIRT1,38,42,43 and the deletion of this domain led to nuclear accumulation of cortactin in association with inhibition of cortactin–actin binding in cultured hippocampal neurons.62 On the basis of these findings, our results strongly suggest that SIRT1 regulates cortactin localization by deacetylation as one of multiple functions of SIRT1.
In conclusion, we found that SIRT1 deacetylates cortactin in the nucleus and that the deacetylated cortactin in cytoplasm predominantly interacts with actin fibers to enhance maintenance of actin cytoskeleton in podocytes. In contrast, deletion or inhibition of SIRT1 enhances the accumulation of acetylated cortactin in the nucleus and accelerates actin cytoskeleton derangement. The SIRT1–cortactin–actin axis regulates podocyte homeostasis at least in part and protects the cells against pathogenic conditions, suggesting that the optimization of SIRT1 activity may be important for podocyte biology.
Concise Methods
Reagents and Antibodies
Three SIRT1 inhibitors were used: EX-527 (Tocris Bioscience, Bristol, UK), cambinol (Santa Cruz Biotechnology, Santa Cruz, CA), and NAM (Sigma-Aldrich, St. Louis, MO). SIRT1 activator resveratrol was purchased from Tokyo Chemical Industry (Tokyo, Japan). H2O2 was purchased from Wako Pure Chemical (Tokyo, Japan) and used as a cellular damage inducer in vitro. Leptomycin B was obtained from LC Laboratories (Woburn, MA). PS was purchased from Sigma-Aldrich.
Polyclonal rabbit anti-SIRT1 antibody (1:300; 07–131), monoclonal mouse anticortactin antibody (1:300; 05–180; clone 4F11), polyclonal rabbit antiacetyl cortactin antibody (1:100; 09–881), and polyclonal rabbit antiacetyl histone H3 antibody (1:5000; 06–599) were purchased from Millipore (Billerica, MA) and used for WB analysis, immunoprecipitation, or immunostaining. Polyclonal rabbit anticortactin antibody (1:30; 3502) was purchased from Cell Signaling Technology (Beverly, MA), and monoclonal rabbit anticortactin antibody (1:100; ab81208; clone EP1922Y) was obtained from Abcam, Inc. (Cambridge, UK) and used for confirmation of the results of immunostaining using the monoclonal mouse anticortactin antibody (EMD Millipore) mentioned above. Polyclonal rabbit anti–WT-1 antibody (1:200; sc-192), polyclonal rabbit antisynaptopodin antibody (1:200; sc-50459), polyclonal goat antinephrin antibody (1:100; sc-19000), and monoclonal mouse antihistone H1 antibody (1:100; sc-8030) were obtained from Santa Cruz Biotechnology and used for WB analysis or immunofluorescence study. Polyclonal rabbit antiactin antibody (1:1000; A2066), polyclonal rabbit antinitrotyrosine antibody (1:300; N0409), Texas Red-conjugated polyclonal rabbit anti-sheep antibody (1:20; SAB3700695), and monoclonal mouse anti–α-tubulin antibody (1:1000; T6199; clone DM1A) were purchased from Sigma-Aldrich. Monoclonal rat anti-mouse F4/80 antibody (1:400; RM2900) was obtained from Caltag Laboratories (Burlingame, CA). As a secondary antibody for immunohistochemistry, biotinylated goat anti-rabbit IgG antibody (1:1000; BA-1000) and biotinylated goat anti-rat IgG (H+L) antibody (1:400; BA-9400) were obtained from Vector Laboratories (Burlingame, CA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (1:10,000; 170–6516; Bio-Rad, Hercules, CA), HRP-conjugated goat anti-rabbit IgG antibody (1:10,000; 170–6515; Bio-Rad), or HRP-conjugated donkey anti-goat IgG antibody (1:500; sc-2020; Santa Cruz Biotechnology) was used as a secondary antibody for WB analysis. For immunofluorescence study, FITC-conjugated polyclonal swine anti-rabbit Ig antibody (1:20; F0205; Dako, Carpinteria, CA), Texas Red-conjugated goat anti-rabbit IgG (H+L) antibody (1:20; T2767; Invitrogen, Eugene, OR), Alexa Fluor 546-conjugated goat anti-mouse IgG (H+L) antibody (1:200; A11003; Invitrogen), or FITC-conjugated goat anti-mouse IgG antibody (1:200; 1070–02; Southern Biotechnology Associates, Birmingham, AL) was used as a secondary antibody. Alexa Fluor 488 phalloidin (1:40) was purchased from Invitrogen and used for detection of actin fiber in vitro. Hoechst 33258 (1:10,000; Sigma-Aldrich) was used for the detection of nucleus.
Animal Experiments
SIRT1pod−/− mice were established by crossing podocin-Cre mice63 on C57BL/6J background with Sirt1flox/flox mice on the same background as described previously (Takikawa A. et al., unpublished data). The littermates of SIRT1pod−/− mice were examined as control wild-type mice. To evaluate the basal state of SIRT1pod−/− mice, we measured body weight, BP, pulse rate, BUN, and U-alb/cre in male SIRT1pod−/− mice and wild-type mice at 13–14 weeks of age.
For glomerular disease induction, male SIRT1pod−/− and wild-type mice were administered NTS (0.7 ml/kg) at 13–14 weeks of age. NTS was produced by the immunization of sheep with isolated rat glomeruli (12 mg in 4 ml CFA) five times every 2 weeks. The obtained antiserum was used for induction of podocyte injury. NTS is known to have reactivity to podocyte antigens in addition to the glomerular basement membrane.64 Two or seven days after administration, serum and urine (24 hours) were collected to measure BUN and U-alb/cre. The experimental mice were then euthanized, and the kidneys were removed for histologic analysis or isolation of glomeruli.
Perfusion of PS was performed according to the previously described method65 with some modification. Briefly, both kidneys were perfused in situ through the left ventricle at an infusion rate of 9 ml/min. Kidneys were first perfused with HBSS at 37°C for 2 minutes and subsequently perfused with PS (4 mg/ml in HBSS) at 37°C for 15 minutes. Soon after perfusion, the kidneys were removed and fixed for electron microscopy.
Isolation of Glomeruli
Glomeruli were isolated using a previously described method66 with some modification. Briefly, we placed a catheter into the aorta of the mouse and perfused magnetic Dynabeads (Invitrogen) through the catheter. After removal, kidneys were minced into small pieces, digested by collagenase and DNase, and filtered. After washing several times, the glomeruli were collected using a magnet. The purity of glomeruli was confirmed to be >95% in each sample by phase-contrast microscopy.
Histologic Analyses and Immunohistochemistry
Glomerular injury was evaluated by optical microscopy of formalin-fixed sections (3-μm thickness) stained with periodic acid–Schiff reagent. For quantitative comparison, 100 glomeruli were observed, and the number of damaged glomeruli, such as those with crescent formation or tuft necrosis, was counted. The ratio of injured glomeruli to all glomeruli was calculated for every group of mice. For quantitative analysis of tubular casts, the periodic acid–Schiff staining sections were observed, and the area of casts in randomly chosen 15 high-power fields was measured in each mouse.
In immunohistochemical analysis, kidneys were fixed in formalin solution or methyl Carnoy’s solution and embedded in paraffin. Sections of 3-μm thickness were incubated with primary antibody for 12 hours at 4°C, biotinylated secondary antibodies for 1 hour, and HRP-conjugated avidin D (1:2000; A-2004; Vector Laboratories) for 30 minutes. Color was developed by incubation with diaminobenzidine and H2O2 (Wako Pure Chemical) at 37°C. For quantitative analysis of infiltrated macrophages, methyl Carnoy’s solution-fixed sections were stained using anti-mouse F4/80 antibody as the primary antibody, and F4/80-positive cells were counted in 30 glomeruli in each mouse.
Immunofluorescence Study
Kidneys were removed from mice, embedded in O.C.T. compound (Sakura Finetek Japan, Tokyo, Japan), and frozen on dry ice. The frozen block was cut into 4-μm sections, which were then fixed in methanol:acetone (1:1) and incubated with primary antibody for 12 hours. They were subsequently incubated with fluorescence-conjugated secondary antibody and/or fluorescence-conjugated probes for 1 hour in dark conditions. For quantitative analysis of fluorescence intensity, fluorescence-positive area in 30 glomeruli was measured in each section by National Institutes of Health ImageJ software (National Institutes of Health, Bethesda, MD).
Electron Microscopy
For electron microscopic examination, kidney tissue was fixed in 2.5% glutaraldehyde solution in phosphate buffer (pH 7.4), postfixed with 1% osmium tetroxide, dehydrated, and embedded in Epok 812. Ultrathin sections were stained with uranyl acetate and lead citrate and then examined with an electron microscope (model H7100; Hitachi Corp., Tokyo, Japan). For quantitative analysis of FP effacement, the width of the FP along the glomerular basement membrane was measured in 20 randomly chosen fields (×20,000) in each section.
Cell Culture
Conditionally immortalized murine podocytes were generated and characterized as described previously.67 The cells were grown in RPMI 1640 media (Nissui Pharmaceutical, Tokyo, Japan) containing 10% FBS (Thermo Fisher Scientific, Waltham, MA), penicillin (50 units/ml), streptomycin (50 μg/ml), sodium pyruvate (1 mmol/L; Life Technologies, Carlsbad, CA), Hepes buffer (10 mmol/L; Sigma-Aldrich), and sodium bicarbonate (0.075%; Nissui Pharmaceutical). To passage cells, podocytes were grown under permissive conditions (33°C in the presence of IFN-γ; 50 units/ml). For podocytes to acquire differentiation and quiescence resembling the in vivo phenotype, cells were grown under restrictive conditions at 37°C in 95% air/5% CO2 without IFN-γ for >11 days. All experiments were performed using podocytes under growth-restricted, differentiated conditions.
WB Analyses and Immunoprecipitation
For WB analysis, cultured cells or isolated glomeruli were lysed by the addition of lysis buffer containing 50 mM Tris-buffer (pH 8.0), 100 mM NaCl, 5 mM EDTA, 1% NP40, and 1% Triton X-100. Protein concentration was measured with a DC protein kit (Bio-Rad). SDS sample buffer containing 0.35 M Tris-HCl (pH 6.8), 10% SDS, 36% glycerol, 5% β-mercaptoethanol, and 0.012% bromophenol blue was added to the lysate. Cytoplasmic or nuclear extract of the cells was obtained using an NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific), and the SDS sample buffer was added to the extract solution. The proteins were separated by 8% or 12% SDS polyacrylamide gels. After electrophoresis, the proteins were transferred onto a polyvinylidene difluoride transfer membrane (GE Healthcare, Buckinghamshire, UK) in a Tris-glycine transfer buffer (48 mM Tris-buffer, 39 mM glycine, 0.05% SDS, and 10% methanol). Membranes were incubated in primary and secondary antibodies, and an ECL Plus Western Blotting System (GE Healthcare) was used for detection. Reproducibility was confirmed in at least three independent experiments, and representative data are presented in the figures. The intensity of bands was quantified using the National Institutes of Health ImageJ software.
For immunoprecipitation, the cells were lysed by Triton cell lysis buffer containing 1 M Tris-buffer (pH 7.4), 100 mM NaCl, 5 mM EDTA, 10 mM sodium pyrophosphate (Sigma-Aldrich), 200 μM Na3VO4 (Wako Pure Chemical), and 1% Triton X-100; 1 ml cell extract containing 300–400 μg protein was incubated with 5–10 μg target antibody or control IgG overnight at 4°C and subsequently incubated with 30 μg protein G sepharose (GE Healthcare) for 2 hours at 4°C. Proteins bound to the beads were eluted by boiling at 95°C for 5 minutes, and the extract solution was used for WB analysis.
Immunocytochemistry
In experiments using SIRT1 inhibitors, podocytes were incubated in media with EX-527 (100 μM), cambinol (50 μM), NAM (10 mM), or vehicle (ethanol) for 24 hours. The media were then exchanged with media containing H2O2 (300 μM) or vehicle, and incubation was continued for 24 hours to expose cells to mild oxidative stress. In experiments using the SIRT1 activator resveratrol, podocytes were incubated in media with resveratrol (200 μM) for 3 hours and then incubated in media with H2O2 (700 μM) for 1 hour to cause strong oxidative stress. In experiments using nuclear export inhibitor leptomycin B, podocytes were incubated in media with leptomycin B (20 nM) for 12 hours and subsequently assessed by immunofluorescence analysis. PS treatment experiment was performed according to the method described previously68 with some modification. Podocytes were incubated in media with EX-527 (100 μM) or vehicle (ethanol) for 24 hours. The media were then exchanged with the media containing PS (300 µg/ml) or vehicle, and the cells were further incubated for 80 minutes.
To assess actin fibers and cellular morphology, cells were fixed in 4% paraformaldehyde solution, permeabilized in 0.3% Triton X solution, and incubated with Alexa Fluor 488 phalloidin (1:40 dilution) in darkness. To detect other molecules, fixed and permeabilized cells were incubated with primary antibodies for 12 hours and subsequently incubated with fluorescence-conjugated secondary antibody for 1 hour in darkness. A BZ-9000 fluorescence microscope (Keyence, Osaka, Japan) was used for observation.
Scoring of Actin Cytoskeleton Derangement
For semiquantitative analysis of actin cytoskeleton derangement, we performed a scoring study according to the previously described methods68 with some modification. The area with disorganized F-actin stained by Alexa Fluor 488 phalloidin was defined as the deranged area. Each podocyte was scored on a scale ranging from one to four on the basis of the ratio of the deranged area (score=1, 0%–25%; score=2, 25%–50%; score=3, 50%–75%; score=4, 75%–100%) in a blinded manner by three independent investigators. At least 100 cells in 10 randomly chosen high-power fields were tested, and the mean scores for the deranged area in those cells were calculated. In addition, the ratios of cells with severe derangement of actin cytoskeleton (score=4) were also determined.
Scratch Assay
Podocytes were seeded and differentiated in six-well plates. Each well was scratched with a sterile 200-μl pipette tip and incubated in medium with an SIRT1 inhibitor or vehicle (ethanol). After 3 days of incubation, the migration of cells in each stimulation group was observed. Cells were photographed under phase-contrast microscopy on days 0 and 3 after scratching, and the number of cells that migrated into the scratched area was counted. The results were confirmed by three independent experiments for each SIRT1 inhibitor.
RNA Isolation and Real-Time Quantitative PCR
RNA extraction and real-time quantitative PCR were performed as described previously.69 Primer sequences were as follows: mouse Sirt1, forward: 5′-AGAACCACCAAAGCGGAAAA-3′ and reverse: 5′-AATCCCACAGGAGACAGAAACC-3′; mouse cortactin, forward: 5′-AGGTGCCATCTGCCTATCA-3′ and reverse: 5′-TCTCGGCTTCTGCCTTC-3′; and mouse β-actin (internal control), forward: 5′-CATCGTGGGCCGCTCTA-3′ and reverse: 5′-CACCCACATAGGAGTCCTTCTG-3′.
siRNA Transfection
Sirt1 siRNAs (sc-40987) was purchased from Santa Cruz Biotechnology, and cortactin siRNA (SI02666629) was purchased from QIAGEN (Hilden, Germany). Transfection was performed with HiPerFect Transfection Reagent (QIAGEN) in murine podocytes according to the manufacturer’s protocols. At 72 hours after transfection, siRNA-treated cells were processed for additional experiments. Stealth RNAi Negative Control Med GC Duplex 2 (Invitrogen) was used as a negative control.
Statistical Analyses
All data are reported as means±SEMs. Data for two groups were analyzed with the unpaired t test, except for the scoring data of actin cytoskeleton derangement, which were analyzed with the Mann–Whitney test (nonparametric analysis). Data for more than two groups were compared by ANOVA with the Bonferroni post hoc test. Differences with a P value<0.05 were considered significant. GraphPad Prism software, version 5.04 for Windows (GraphPad Software, San Diego, CA) was used for data analyses.
Study Approval
All animal procedures were conducted in accordance with the guidelines for the care and use of laboratory animals approved by the University of Tokyo Graduate School of Medicine (M-P12–66).
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Dr. Katsuhiro Asanuma for technical comments on protamine sulfate perfusion.
This work was supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research 24390213 (to M.N.), 24591189 (to T.W.), and 25461207 (to R.I.). The study was also supported by Japanese Association of Dialysis Physicians Grant 2012-05 (to R.I.), Kyowa Hakko Kirin Co., Ltd. (R.I.), and Charitable Trust Araki Memorial Foundation for Medical and Biochemical Research.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014030289/-/DCSupplemental.
References
- 1.Reiser J, Gupta V, Kistler AD: Toward the development of podocyte-specific drugs. Kidney Int 77: 662–668, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Agati VD, Kaskel FJ, Falk RJ: Focal segmental glomerulosclerosis. N Engl J Med 365: 2398–2411, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Wiggins RC: The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Bariéty J, Bruneval P, Meyrier A, Mandet C, Hill G, Jacquot C: Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int 68: 1109–1119, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Besse-Eschmann V, Le Hir M, Endlich N, Endlich K: Alteration of podocytes in a murine model of crescentic glomerulonephritis. Histochem Cell Biol 122: 139–149, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Guo J-K, Menke AL, Gubler M-C, Clarke AR, Harrison D, Hammes A, Hastie ND, Schedl A: WT1 is a key regulator of podocyte function: Reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet 11: 651–659, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Oh J, Reiser J, Mundel P: Dynamic (re)organization of the podocyte actin cytoskeleton in the nephrotic syndrome. Pediatr Nephrol 19: 130–137, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Reiser J, Sever S: Podocyte biology and pathogenesis of kidney disease. Annu Rev Med 64: 357–366, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guarente L: Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med 364: 2235–2244, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Baur JA: Biochemical effects of SIRT1 activators. Biochim Biophys Acta 1804: 1626–1634, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P: The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134: 329–340, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U: SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134: 317–328, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J: AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakagawa T, Guarente L: Sirtuins at a glance. J Cell Sci 124: 833–838, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW: Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J: Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127: 1109–1122, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB: Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123: 437–448, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA: The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3: e2020, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM: Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 281: 21745–21754, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX: Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 32: 11–20, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Planavila A, Iglesias R, Giralt M, Villarroya F: Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res 90: 276–284, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Hao C-M, Haase VH: Sirtuins and their relevance to the kidney. J Am Soc Nephrol 21: 1620–1627, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D: Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin Sci (Lond) 124: 153–164, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nangaku M, Izuhara Y, Usuda N, Inagi R, Shibata T, Sugiyama S, Kurokawa K, van Ypersele de Strihou C, Miyata T: In a type 2 diabetic nephropathy rat model, the improvement of obesity by a low calorie diet reduces oxidative/carbonyl stress and prevents diabetic nephropathy. Nephrol Dial Transplant 20: 2661–2669, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Mattagajasingh I, Kim C-S, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K: SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 104: 14855–14860, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern JS, Gades MD, Wheeldon CM, Borchers AT: Calorie restriction in obesity: Prevention of kidney disease in rodents. J Nutr 131: 913S–917S, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH: Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A 105: 9793–9798, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA: Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324: 1289–1293, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Jung YJ, Lee JE, Lee AS, Kang KP, Lee S, Park SK, Lee SY, Han MK, Kim DH, Kim W: SIRT1 overexpression decreases cisplatin-induced acetylation of NF-κB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem Biophys Res Commun 419: 206–210, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M, Skorecki K, Hayashi K, Itoh H: Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem 285: 13045–13056, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, Koya D: Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120: 1043–1055, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC, Breyer MD, Hao CM: Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest 120: 1056–1068, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isono M, Isshiki K, Uzu T, Kashiwagi A, Koya D: Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic Biol Med 40: 2175–2182, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isshiki K, Isono M, Uzu T, Guarente L, Kashiwagi A, Koya D: SIRT1 inhibits transforming growth factor beta-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J Biol Chem 282: 151–158, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Chuang PY, Dai Y, Liu R, He H, Kretzler M, Jim B, Cohen CD, He JC: Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS ONE 6: e23566, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C, Ding G, Liu B, Yang T, Zhang A: Activation of peroxisome proliferator-activated receptor-γ coactivator 1α ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int 82: 771–789, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, Hosoya K, Komatsu M, Kaneko Y, Kanda T, Kubota E, Tokuyama H, Hayashi K, Guarente L, Itoh H: Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med 19: 1496–1504, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkbride KC, Sung BH, Sinha S, Weaver AM: Cortactin: A multifunctional regulator of cellular invasiveness. Cell Adhes Migr 5: 187–198, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pollard TD, Borisy GG: Cellular motility driven by assembly and disassembly of actin filaments. Cell 112: 453–465, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Rigothier C, Auguste P, Welsh GI, Lepreux S, Deminière C, Mathieson PW, Saleem MA, Ripoche J, Combe C: IQGAP1 interacts with components of the slit diaphragm complex in podocytes and is involved in podocyte migration and permeability in vitro. PLoS ONE 7: e37695, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mundel P, Reiser J: Proteinuria: An enzymatic disease of the podocyte? Kidney Int 77: 571–580, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang X-J, Dent SR, Yao T-P, Lane WS, Seto E: HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell 27: 197–213, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Zhang M, Dong H, Yong S, Li X, Olashaw N, Kruk PA, Cheng JQ, Bai W, Chen J, Nicosia SV, Zhang X: Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene 28: 445–460, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Kaluza D, Kroll J, Gesierich S, Yao T-P, Boon RA, Hergenreider E, Tjwa M, Rössig L, Seto E, Augustin HG, Zeiher AM, Dimmeler S, Urbich C: Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J 30: 4142–4156, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichimura K, Kurihara H, Sakai T: Actin filament organization of foot processes in rat podocytes. J Histochem Cytochem 51: 1589–1600, 2003 [DOI] [PubMed] [Google Scholar]
- 46.Tryggvason K, Patrakka J, Wartiovaara J: Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354: 1387–1401, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Chiang C-K, Inagi R: Glomerular diseases: Genetic causes and future therapeutics. Nat Rev Nephrol 6: 539–554, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P: Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 8: 485–491, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Min SW, Sohn PD, Cho SH, Swanson RA, Gan L: Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front Aging Neurosci 5: 53, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu C, Thompson CB: Metabolic regulation of epigenetics. Cell Metab 16: 9–17, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martínez-Redondo P, Vaquero A: The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer 4: 148–163, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh SK, Williams CA, Klarmann K, Burkett SS, Keller JR, Oberdoerffer P: Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. J Exp Med 210: 987–1001, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Reynolds AB, Kanner SB, Vines RR, Parsons JT: Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol Cell Biol 11: 5113–5124, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu H, Parsons JT: Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol 120: 1417–1426, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buday L, Downward J: Roles of cortactin in tumor pathogenesis. Biochim Biophys Acta 1775: 263–273, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Weaver AM: Cortactin in tumor invasiveness. Cancer Lett 265: 157–166, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothschild BL, Shim AH, Ammer AG, Kelley LC, Irby KB, Head JA, Chen L, Varella-Garcia M, Sacks PG, Frederick B, Raben D, Weed SA: Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res 66: 8017–8025, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi T, Notoya M, Shinosaki T, Kurihara H: Cortactin interacts with podocalyxin and mediates morphological change of podocytes through its phosphorylation. Nephron, Exp Nephrol 113: e89–e96, 2009 [DOI] [PubMed] [Google Scholar]
- 59.Zhao J, Bruck S, Cemerski S, Zhang L, Butler B, Dani A, Cooper JA, Shaw AS: CD2AP links cortactin and capping protein at the cell periphery to facilitate formation of lamellipodia. Mol Cell Biol 33: 38–47, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadoul K, Wang J, Diagouraga B, Khochbin S: The tale of protein lysine acetylation in the cytoplasm. J Biomed Biotechnol 2011: 970382, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lan F, Cacicedo JM, Ruderman N, Ido Y: SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem 283: 27628–27635, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hering H, Sheng M: Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23: 11759–11769, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB: Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39–42, 2003 [DOI] [PubMed] [Google Scholar]
- 64.Chugh S, Yuan H, Topham PS, Haydar SA, Mittal V, Taylor GA, Kalluri R, Salant DJ: Aminopeptidase A: A nephritogenic target antigen of nephrotoxic serum. Kidney Int 59: 601–613, 2001 [DOI] [PubMed] [Google Scholar]
- 65.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P: Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J Clin Invest 115: 1188–1198, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsusaka T, Asano T, Niimura F, Kinomura M, Shimizu A, Shintani A, Pastan I, Fogo AB, Ichikawa I: Angiotensin receptor blocker protection against podocyte-induced sclerosis is podocyte angiotensin II type 1 receptor-independent. Hypertension 55: 967–973, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wada T, Pippin JW, Terada Y, Shankland SJ: The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int 68: 1618–1629, 2005 [DOI] [PubMed] [Google Scholar]
- 68.Schaldecker T, Kim S, Tarabanis C, Tian D, Hakroush S, Castonguay P, Ahn W, Wallentin H, Heid H, Hopkins CR, Lindsley CW, Riccio A, Buvall L, Weins A, Greka A: Inhibition of the TRPC5 ion channel protects the kidney filter. J Clin Invest 123: 5298–5309, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chiang C-K, Tanaka T, Inagi R, Fujita T, Nangaku M: Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab Invest 91: 1564–1571, 2011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.