

Abstract
Diabetic nephropathy is the leading cause of ESRD in high-income countries and a growing problem across the world. Vascular endothelial growth factor-A (VEGF-A) is thought to be a critical mediator of vascular dysfunction in diabetic nephropathy, yet VEGF-A knockout and overexpression of angiogenic VEGF-A isoforms each worsen diabetic nephropathy. We examined the vasculoprotective effects of the VEGF-A isoform VEGF-A165b in diabetic nephropathy. Renal expression of VEGF-A165b mRNA was upregulated in diabetic individuals with well preserved kidney function, but not in those with progressive disease. Reproducing this VEGF-A165b upregulation in mouse podocytes in vivo prevented functional and histologic abnormalities in diabetic nephropathy. Biweekly systemic injections of recombinant human VEGF-A165b reduced features of diabetic nephropathy when initiated during early or advanced nephropathy in a model of type 1 diabetes and when initiated during early nephropathy in a model of type 2 diabetes. VEGF-A165b normalized glomerular permeability through phosphorylation of VEGF receptor 2 in glomerular endothelial cells, and reversed diabetes-induced damage to the glomerular endothelial glycocalyx. VEGF-A165b also improved the permeability function of isolated diabetic human glomeruli. These results show that VEGF-A165b acts via the endothelium to protect blood vessels and ameliorate diabetic nephropathy.
Keywords: VEGF, VEGF-A, glomerulus, permeability, diabetes, diabetic nephropathy, albuminuria, glycocalyx, endothelial glycocalyx
Diabetes is the leading cause of ESRD in the industrialized world,1 is the leading cause of blindness in the working-age population,2 and diabetic nephropathy eventually affects up to 50% of patients with long-standing diabetes.3 Microvascular dysfunction contributes to the development and progression of each of these complications, with abnormal vascular hemodynamics and disturbed vascular permeability leading to clinically important excessive vessel leak, such as macular edema and albuminuria.4
In recent years, there has been increasing interest in identifying factors that are endogenously produced within the vasculature, are activated or inhibited in diabetes, and therefore could protect against diabetic vasculopathy5 and potentially be used alongside traditional treatments. These factors, such as activated protein C,6 modify the response of the vasculature to the environment, thereby protecting endothelial and other vascular cells from the diabetic milieu. Vascular endothelial growth factor-A (VEGF-A), a family of secreted glycoprotein isoforms, has been suggested as such an endogenous protective factor.5,7 VEGF-A levels are dysregulated and VEGF-A receptor pathways activated in many dysfunctional vascular beds in diabetes.5 In glomeruli, VEGF-A is predominantly produced by podocytes, and its actions on endothelial cells and podocytes have been described.8,9 Targeted, inducible genetic deletion of all VEGF-A isoforms from podocytes causes glomerular disease in healthy animals,10 and accelerated nephropathy in diabetic animals.7 Total glomerular VEGF-A levels decrease as diabetic nephropathy progresses in humans,11 and anti–VEGF-A antibodies induce proteinuria in humans.10 VEGF-A is therefore considered to provide an endogenous “protective” signal that prevents apoptosis of vascular wall cells and hence progression of diabetic nephropathy.
However, upregulating specific VEGF-A isoforms, such as VEGF-A165a, can also accelerate diabetic nephropathy.12 VEGF-A165a is a potent vasoactive agent, increasing vasodilation, vascular permeability, and angiogenesis,13 processes that can drive the adverse functional consequences of diabetic vasculopathy. An equilibrium between sufficient (protective) and excessive (damaging) levels of VEGF-A, or an appropriately timed VEGF-A response, may underlie this paradox. An alternative explanation, however, is that these protective and deleterious VEGF-A actions in diabetes could be specific to VEGF-A isoforms, wherein some VEGF-A isoforms (e.g., VEGF-A164a) are detrimental and others confer protection.14,15 An alternative VEGF-A isoform, VEGF-A165b, confers benefit in other microvascular disease states.16–18 We therefore tested the hypothesis that VEGF-A165b could act as a protective factor in diabetic nephropathy.
Results
VEGF-A165b Is Upregulated in Humans with Diabetic Nephropathy and Well Preserved Kidney Function
In 12 patients with biopsy-confirmed diabetic glomerulosclerosis, expression of exon 5/7/8-containing VEGF-A isoform (i.e., VEGF-A165a+VEGF-A165b) mRNA was significantly reduced compared with findings in five nondiabetic individuals (11.9±4.2 versus 80.0±42.3 pg/μg mRNA; P<0.05, unpaired t test). When diabetic nephropathy was categorized as “early” with well preserved kidney function and low-grade proteinuria (creatinine, 89±8 µmol/L; proteinuria, 1.0±0.3 g/L; n=5; Supplemental Table 1) or “late” with elevated creatinine and heavy proteinuria (serum creatinine, 242±10 µmol/L; proteinuria, 7.8±0.2g/L; n=7; Supplemental Table 1), clear differences in the expression of different VEGF isoforms were apparent (Figure 1). Total VEGF-A165a+VEGF-A165b mRNA expression progressively declined from “early” to “late” diabetic nephropathy, primarily because of decreased VEGF-A165a mRNA levels (Figure 1, A and B). However, while VEGF-A165b levels also decreased in “late” diabetic nephropathy (Figure 1C), VEGF-A165b levels significantly increased approximately 6-fold in “early” diabetic nephropathy. Thus, only those with “early” diabetic nephropathy exhibited an approximately 12-fold increase in VEGF-A165b as a proportion of total VEGF-A isoforms present in the kidney (Figure 1D). Similar changes were observed in just the glomerular compartment (Figure 1E) and at the protein level (Figure 1F). On the basis of these observations, we hypothesized that increasing glomerular VEGF-A165b protects against diabetic nephropathy.
Figure 1.
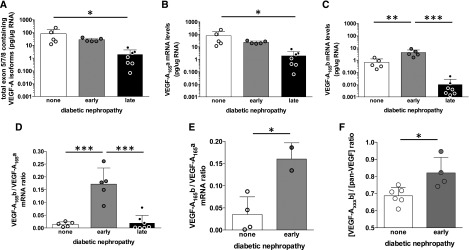
VEGF-A165b is upregulated in humans with early diabetic nephropathy and well preserved kidney function. (A) mRNA expression of exon 5/7/8 containing VEGF-A isoforms (i.e., those coding for 165 amino-acid proteins) determined by RT-qPCR relative to total RNA extracted in whole renal cortical tissue from kidneys unsuitable for transplantation from deceased donors without diabetes (“none”), donors with early diabetic nephropathy with well preserved kidney function (“early”), and kidney biopsy specimens from patients with diabetes and advanced nephropathy (“late”) (note logarithmic scale). (B) VEGF-A165a mRNA expression (note logarithmic scale). (C) VEGF-A165b expression (note logarithmic scale). (D) VEGF-A isoform expression ratio calculated as VEGF-A165b mRNA per unit total mRNA divided by VEGF-A165a mRNA per unit total mRNA. (E) Ratio of VEGF-A165a to VEGF-A165b mRNA in control and early diabetic samples in which it was possible to analyze mRNA from single glomeruli isolated by differential sieving. (F) Isoform-specific VEGF-A165b protein determined by ELISA as a proportion of total VEGF-A in whole kidney cortical tissue in kidney donors with diabetes and early nephropathy (“early”) and those without diabetes (“none”). Error bars, SEM. Multiple comparisons: one-way ANOVA. Two-group comparisons: unpaired t test. *P<0.05; **P<0.01; ***P<0.005, throughout.
Podocyte-Specific VEGF-A165b Overexpression Protects Against Diabetic Nephropathy
Streptozotocin (STZ)-induced diabetic wild-type and podocyte-specific VEGF-A165b–overexpressing mice (nephVEGF-A165b) (Figure 2A) demonstrated equivalent hyperglycemia (Figure 2B). Albuminuria increased 10.8-fold in diabetic wild-type mice but only 2.2-fold in diabetic nephVEGF-A165b mice, a change that was not statistically significantly different from findings in nondiabetic controls (Figure 2C). The early diabetes-related rise in creatinine clearance in diabetic wild-type mice was also attenuated in diabetic nephVEGF-A165b mice (Figure 2D). After 6 weeks of diabetes, VEGF-A165b–overexpressing mice also demonstrated less glomerular hypertrophy (Figure 2, E and F), less mesangial matrix expansion (Figure 2, G and H), and less glomerular basement membrane thickening (Figure 2, I and J).
Figure 2.

Podocyte-specific overexpression of VEGF-A165b ameliorates STZ-induced diabetic nephropathy. (A) Transgenic mice constitutively overexpressing human VEGF-A165b in podocytes under control of the nephrin promoter (blue; nephhVEGF-A165b) received injections of STZ or vehicle at 12 weeks of age. Plasma glucose (B), urine albumin-to-creatine ratio (C), and creatinine clearance (D) measured in nondiabetic wild-type, diabetic wild-type, nondiabetic transgenic, and diabetic transgenic mice 6 weeks after diabetes induction. n=5–10 mice per group. Error bars, SEM. *P<0.05, one-way ANOVA. (E and F) Glomerular area assessed in hematoxylin and eosin–stained kidney sections from 21 to 55 glomeruli from 3 mice per group. Scale bars: 20 µm. Error bars, SEM. *P<0.05, one-way ANOVA. (G and H) Periodic acid Schiff–stained kidney sections from each group of mice. Mesangial matrix expansion (arrows) measured as a proportion of total glomerular area in 45–60 glomeruli from 3 to 4 mice per group. Scale bars: 20 µm. Error bars, SEM. *P<0.05, one-way ANOVA. (I) Low- and high-magnification electron micrographs (scale bars: 1 µm and 250 nm, respectively) of glomerular capillaries and the glomerular capillary wall (GCW) from each group of mice. CL, capillary lumen; ec, endothelial cell; gbm, glomerular basement membrane; pfp, podocyte foot process; rbc, red blood cell; US, urinary space. (J) Glomerular basement membrane width calculated from 20 to 60 measurements in 8–16 glomerular capillaries per group. Error bars, SEM. *P<0.05, one-way ANOVA.
Given that podocyte-specific overexpression of murine VEGF-A164a (podVEGF-A164a) accelerates diabetic nephropathy19 and that nephVEGF-A165b overexpression can block inducible podVEGF-A164a–overexpression permeability changes in healthy mice,20 we hypothesized that nephVEGF-A165b effects would be sufficiently robust to prevent the more aggressive form of nephropathy in diabetic podVEGF-A164a mice. Wild-type, single- and double-transgenic nephVEGF-A165b/podVEGF-A164a mice (Figure 3A) all demonstrated equal hyperglycemia 5 weeks after streptozotocin. Albuminuria increased an additional 3-fold above diabetic wild-type littermate values after 1 week of VEGF-A165a induction in diabetic podVEGF-A164a mice, an effect completely blocked by co-overexpression of VEGF-A165a with VEGF-A165b (Figure 3B).
Figure 3.
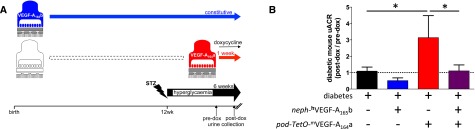
Podocyte-specific overexpression of VEGF-A165b reduces albuminuria in the dual insult of VEGF-A164a overexpression and STZ-induced diabetic nephropathy. (A) Heterozygous mice overexpressing human VEGF-A165b in podocytes (blue: nephhVEGF-A165b) were crossed with mice in which murine VEGF-A164a (mVEGF-A164a) overexpression in podocytes could be induced with doxycycline (dox): heterozygous podocin-rtTA:TetO-mVEGF-A164a mice (red: pod-TetO-mVEGF-A164a), to generate mice co-overexpressing mVEGF-A164a and hVEGF-A165b in podocytes. Offspring were treated with STZ at 12 weeks of age to induce diabetes in all groups. After 5 weeks of diabetes, all mice received doxycycline, inducing mVEGF-A164a overexpression in the two groups carrying the podocin-rtTA:TetO-mVEGF-A164a transgene. After 1 week of doxycycline treatment, urinary albumin-to-creatine ratio (uACR) was measured and compared with uACR obtained before doxycycline administration. (B) Change in uACR after 7 days of doxycycline treatment in diabetic mice with no transgenes, diabetic mice overexpressing hVEGF-A165b alone, diabetic mice overexpressing mVEGF-A164a alone, and diabetic mice co-overexpressing mVEGF-A164a and hVEGF-A165b. Dashed line represents no change in albuminuria during the 1-week doxycycline treatment period. n=3–6 mice per group. Error bars, SEM. *P<0.05, one-way ANOVA.
Systemic Injections of VEGF-A165b Protect against Diabetic Nephropathy
Systemic injections of recombinant human VEGF-A165b protein (rhVEGF-A165b) reduce angiogenesis-related cancer growth in mice without altering BP or renal function or eliciting adverse effects.21 We therefore assessed the efficacy of systemic rhVEGF-A165b injections in STZ-induced diabetes in DBA2J mice before and after the onset of diabetic nephropathy (Figure 4A). After 2 weeks of consistent hyperglycemia, low-dose 0.04µg/g rhVEGF-A165b injections were initiated and continued until an approved study endpoint occurred (significant weight loss). Equivalent blood glucose concentrations were observed in both treatment groups (Figure 4B). rhVEGF-A165b injections consistently reduced urinary albumin-to-creatinine ratios in diabetic mice, and this effect persisted throughout the entire period of hyperglycemia (Figure 4C).
Figure 4.

Systemic treatment with VEGF-A165b blocks progression of albuminuria and glomerular basement membrane thickening in STZ-induced diabetic nephropathy. (A) Diabetes was induced in wild-type DBA2J mice with STZ injection (nondiabetic control mice received buffer injection alone). After 2 consecutive weeks of hyperglycemia, mice received an 11-week course of biweekly intraperitoneal injections (blue arrows) of recombinant human VEGF-A165b (rhVEGF-A165b) (n=16) or vehicle (n=16). (B) Plasma glucose measured repeatedly in nondiabetic control, diabetic vehicle-treated, and 0.04 µg/g rhVEGF-A165b–treated mice. *P<0.05 versus both diabetic groups; "ns" indicates P>0.05 between diabetic groups; one-way ANOVA for both. (C) Urine albumin-to-creatinine ratio (uACR) was measured repeatedly in diabetic DBA2J mice receiving injections of vehicle (n=4) or 0.04 µg/g rhVEGF-A165b (n=4) for up to 11 weeks. *P<0.05, paired t test. (D) Diabetes was induced in wild-type DBA2J mice with STZ injection and vehicle or rhVEGF-A165b treatment after the onset of albuminuria. (E) uACR measured repeatedly in mice receiving biweekly injections of vehicle (n=10), 0.04 µg/g rhVEGF-A165b (n=10), and 0.2 µg/g rhVEGF-A165b (n=6) before organ harvest. Error bars, SEM. (F) Fold change in uACR (uACR at indicated time points, divided by uACR before initiation of treatment [arrow]). Dashed line represents no change in albuminuria after start of injections. Error bars, SEM. *P<0.05, two-way ANOVA. (G) Low- and high-magnification electron micrographs (scale bars: 1 μm and 500 nm, respectively) of glomerular capillaries and the glomerular capillary wall (GCW). CL, capillary lumen; ec, endothelial cell; gbm, glomerular basement membrane; pfp, podocyte foot process; rbc, red blood cell; US, urinary space. (H) Glomerular basement membrane (GBM) width measurements in indicated groups of diabetic mice. Error bars, SEM. *P<0.05, one-way ANOVA.
In a separate cohort of diabetic DBA2J mice, low-dose (0.04 µg/g) or high-dose (0.2 µg/g) systemic rhVEGF-A165b injections were initiated only after the onset of heavy albuminuria (12 weeks after STZ administration) (Figure 4D). Notably, albuminuria in these different cohort of mice receiving systemic injections was 5–20-fold greater than in the above groups. Again, vehicle- and rhVEGF-A165b–treated diabetic mice had equivalent blood glucose concentrations. Urinary albumin-to-creatinine ratios continued to rise in vehicle-treated diabetic DBA2J mice (Figure 4E). Despite having the highest urine albumin-to-creatinine ratios at the start of rhVEGF-A165b treatment, albuminuria did not increase in mice receiving 0.04 µg/g rhVEGF-A165b injections, and albuminuria decreased in mice receiving 0.2 µg/g rhVEGF-A165b (Figure 4, E and F). The rhVEGF-A165b–induced improvements in albuminuria were significant at both treatment doses (Figure 4F). rhVEGF-A165b injections also improved diabetes-induced glomerular basement membrane thickening (Figure 4, G and H) but not mesangial matrix expansion.
Systemic Injections of VEGF-A165b Improve Early Albuminuria but Not GFR in a Type 2 Diabetic Nephropathy Model
The global increase in diabetic nephropathy is predominantly due to type 2 diabetes. We therefore administered an 8-week round of systemic rhVEGF-A165b injections to obese db/db mice after the onset of hyperglycemia but before the onset of albuminuria or decreased GFR (Figure 5A). rhVEGF-A165b injections (0.2 µg/g) did not alter blood glucose or body weight (Figure 5B) but significantly reduced albuminuria (P<0.05) (Figure 5C). Despite this improvement in albuminuria, however, rhVEGF-A165b injections did not prevent the diabetes-related decline in creatinine clearance (Figure 5D) in these mice. As in the type 1 diabetes model, we also assessed the efficacy of rhVEGF-A165b injections started after the onset of both albuminuria and decreased creatinine clearance (Figure 5E). Albuminuria did not progress in these db/db mice after this time (Figure 5F), and rhVEGF-A165b injections initiated at this later time point did not modify the pattern of albuminuria (Figure 5F), prevent further loss of GFR (Figure 5G), or decrease mesangial matrix expansion (Figure 5H).
Figure 5.

Systemic treatment with VEGF-A165b blocks early albuminuria but not later features of diabetic nephropathy in a genetic model of type 2 diabetic nephropathy. (A) After the onset of hyperglycemia at 6 or 7 weeks of age, db/db mice received biweekly intraperitoneal injections of recombinant human VEGF-A165b (rhVEGF-A165b) or vehicle (blue arrows) for 8 weeks. (B) Blood glucose measurements in 6 lean and 20 db/db mice before and after receipt of twice-weekly vehicle or rhVEGF-A165b (0.2 µg/g) injections. Dashed line indicates start of intraperitoneal injections. Error bars, SEM. *P<0.05 compared with both groups of diabetic mice; "ns" indicates P>0.05 between groups of diabetic mice. (C) Urinary albumin-to-creatinine ratio (uACR) determined repeatedly during the 8-week treatment period, divided by uACR in the same animal before treatment (“baseline”) to calculate fold change in uACR. Dashed line represents no fold change in albuminuria since start of treatment. Error bars, SEM. *P<0.05, paired t test. (D) Creatinine clearance measured at the end of the 8-week injection period. Error bars, SEM. *P<0.05; "ns" indicates P>0.05; one-way ANOVA for both. (E) In a separate cohort of older mice, 5 weeks of biweekly intraperitoneal injections (blue arrows) of rhVEGF-A165b or vehicle were initiated in 14-week-old lean controls (n=5) and db/db mice (n=14) with long-standing hyperglycemia, established albuminuria, and decreased GFR. (F) Serial measurement of uACR in these older vehicle- and rhVEGF-A165b–treated db/db mice. Dashed line indicates start of intraperitoneal injections. Error bars, SEM. *P<0.05, paired t test. (G) At the end of the 5-week injection period, GFR measurements in these older vehicle- and rhVEGF-A165b–treated db/db and lean mice. Error bars, SEM. *P<0.05; "ns" indicates P>0.05; one-way ANOVA for both. (H) At the end of the 5-week injection period, mesangial matrix expansion (arrows) in periodic acid Schiff–stained kidney sections from each group of mice. Scale bar: 50 μm.
VEGF-A165b Acts via VEGFR-2 to Modify Glomerular Endothelium
Both podocytes and endothelial cells play important roles in the progression of diabetic nephropathy. Human podocytes exhibited substantial apoptosis when cultured in high glucose–containing medium, a response partially rescued by rhVEGF-A165b (Figure 6A). Human endothelial cells also demonstrated significant apoptosis in response to hyperglycemia, and rhVEGF-A165b completely blocked the hyperglycemia-induced endothelial cell apoptosis (Figure 6, B and C). The pan-VEGFR tyrosine kinase inhibitor PTK787 (Figure 6C) prevented the antiapoptotic effects of rhVEGF-A165b on hyperglycemic endothelial cells.
Figure 6.

VEGF-A165b reduces apoptosis, decreases glomerular permeability, and acts via VEGFR-2. (A) Apoptosis (caspase-3 activity) was determined in human podocytes in vitro exposed to normal (10 mM) or high (30 mM) cell culture glucose concentrations with or without recombinant human VEGF-A165b (rhVEGF-A165b). Experiments were done in triplicate. OD, optical density. Error bars, SEM. *P<0.05, one-way ANOVA. (B) Caspase-3 staining performed on human microvascular endothelial cells exposed to normal (5 mM) or high (30 mM) cell culture glucose concentrations in the presence or absence of rhVEGF-A165b. (C) Number of caspase-3–positive cells per high-powered field (hpf) compared between conditions. Experiments were done in quadruplicate. Error bars, SEM. ***P<0.001; "ns" indicates P>0.05; one-way ANOVA for both. (D) Glomerular water permeability (volume-corrected ultrafiltration coefficient: LPA/Vi) measured in glomeruli isolated from vehicle-injected nondiabetic (open bar), and STZ-injected diabetic rats (filled bars). Glomeruli were incubated for 1 hour in rhVEGF-A165b (1 nM), VEGF-A receptor-2 antagonist ZM323881 (10 µM), pan-VEGF-A receptor blocker PTK787 (100 nM), saline, or combinations thereof before LPA/Vi measurement. Error bars, SEM. Compared with diabetic: *P<0.05; "ns" indicates P>0.05. Compared with diabetic+rhVEGF-A165b: #P<0.05; "ns1" indicates P>0.05. One-way ANOVA for all. (E and F) Total and phosphorylated VEGFR-2 in glomeruli of nondiabetic wild-type and nephhVEGF-A165b mice (E) and in glomeruli from STZ-induced diabetic DBA2J mice treated with vehicle or rhVEGF-A165b (F). Controls without primary antibody are also shown. Scale bars: 20 μm. VEGFR-2 and phosporylated-VEGFR-2 by area relative to the whole glomerulus quantified for nondiabetic nephhVEGF-A165b versus littermates (G) and rhVEGF-A165b–treated diabetic mice versus vehicle-treated diabetic mice (H). Error bars, SEM. *P<0.05, one-way ANOVA.
To determine the functional significance of this VEGF-A165b-VEGFR-2 signaling, we measured glomerular water permeability in healthy and diabetic rat glomeruli in the presence of VEGF-receptor inhibitors (Figure 6D). Single glomeruli harvested from diabetic rat kidneys had higher volume-corrected glomerular ultrafiltration coefficient (LPA/Vi) than glomeruli from nondiabetic rats; this was normalized by blockade of all VEGF receptors (VEGFRs) with PTK787, but not by blockade of just VEGFR-2 with ZM323881, indicating that multiple VEGFRs contribute to glomerular permeability defects in diabetes. In keeping with effects in the intact animal, this diabetes-induced glomerular permeability change was reversed by rhVEGF-A165b. This normalization by rhVEGF-A165b was completely blocked by the selective VEGFR-2 inhibitor ZM323881, indicating a key role for VEGFR-2 in diabetic nephropathy in mediating these beneficial effects of VEGF-A165b.
Immunofluorescence studies demonstrated increased total and phosphorylated forms of VEGFR-2 in the glomeruli of both nephhVEGF-A165b–overexpressing mice (Figure 6, E and G), and more robustly in the glomeruli of STZ-induced diabetic mice treated with systemic rhVEGF-A165b injections (Figure 6, F and H). These latter findings confirm that systemic injections of VEGF-A165b had direct effects on VEGF-signaling pathways within glomerular cells. In keeping with previous transgenic studies,9 and dominant antiapoptotic effects of VEGF-A165b on endothelial cells, upregulated VEGFR-2 in nephhVEGF-A165b-overexpressing mice co-localized with platelet-endothelial cell adhesion molecule (PECAM-1) in glomerular endothelial cells (Figure 7, A and B), but not with nephrin (Figure 7, C and D). Thus, the dominant actions of acute incubation, local overexpression, and systemic treatment with VEGF-A165b appear to involve phosphorylation of VEGFR-2 in the glomerular endothelium.
Figure 7.

VEGF-A165b stimulates VEGFR-2 in glomerular endothelial cells in vivo. (A) Sections of kidney tissue from wild-type and nephhVEGF-A165b mice showing glomeruli costained for VEGFR-2 (green), and the endothelial cell marker PECAM-1 (red), and overlaid images showing colocalization (yellow). (B) High magnification of a single glomerular capillary loop showing VEGFR-2 expression and endothelial cell PECAM-1 colocalize. (C) Sections of kidney tissue from wild-type and nephhVEGF-A165b mice showing glomeruli costained for VEGFR-2 (green) and the podocyte marker nephrin (red). (D) High magnification of a single glomerular capillary loop showing that VEGFR-2 expression and podocyte nephrin do not colocalize.
VEGF-A165b Restores Glomerular Endothelial Glycocalyx in Diabetic Nephropathy
VEGF-A165b–induced VEGFR-2 activation in the glomerular endothelium raises the question of how glomerular endothelial modifications could restore normal glomerular function in diabetes. Electron micrographs (Figure 8A) revealed decreased glomerular endothelial fenestration in STZ-injected diabetic vehicle-treated DBA2J mice, but this was not reversed by rhVEGF-A165b treatment (Figure 8B). Direct imaging of the glomerular endothelial glycocalyx (Figure 8A) demonstrated a 50% reduction in glycocalyx depth in vehicle-treated diabetic mice (P<0.05) (Figure 8C). In keeping with the observed improvements in albuminuria, rhVEGF-A165b injections restored glomerular endothelial glycocalyx depth in diabetic mice to levels observed in healthy nondiabetic control animals (Figure 8C). Loss of endothelial glycocalyx in diabetes and restoration by rhVEGF-A165b was confirmed by confocal imaging of viable, unfixed glomeruli in STZ-injected diabetic rats (Figure 9).
Figure 8.

VEGF-A165b restores the glomerular endothelial glycocalyx in early STZ-induced diabetic nephropathy. Six weeks after STZ (or buffer) injections to induce diabetes (or normoglycemia) in DBA2J mice, mice received biweekly intraperitoneal injections of vehicle or rhVEGF-A165b for 4 weeks. Animals were cardiac perfusion fixed and renal cortices were processed for transmission electron microscopy. (A) Glomeruli were imaged at low magnification, the glomerular capillary wall at higher magnification, and endothelial cell surface at high magnification using transmission electron microscopy. e-glx, endothelial glycocalyx; ec, endothelial cell; fen, endothelial fenestra; gbm, glomerular basement membrane; gc, glomerular capillary; pfp, podocyte foot process; us, urinary space. (B) Endothelial fenestral density and (C) glycocalyx depth covering the glomerular endothelial cell surface quantified in electron microscopic images from vehicle-treated nondiabetic, vehicle-treated diabetic, and rhVEGF-A165b–treated diabetic animals. Error bars, SEM. n=3 mice per group. *P<0.05; "ns" indicates P>0.05; one-way ANOVA for both.
Figure 9.

VEGF-A165b restores glomerular endothelial glycocalyx acutely in diabetic rat glomeruli. Kidneys were perfused in vivo with cell membrane label (R18; red) and glycocalyx label (Alexa Fluor-488–wheat germ agglutinin lectin; green), and then glomeruli were isolated and treated with vehicle or VEGF-A165b for 1 hour (exactly as per physiology experiments) before imaging with confocal microscopy. (A) whole glomerulus; scale bar: 20 μm. (B) Glomerular capillaries (gc) with endothelial cell (ec) bodies; scale bar: 5 μm. (C) High-magnification image (scale bar: 1 μm) of a vehicle-treated glomerular capillary from a healthy animal, demonstrating glomerular endothelial glycocalyx (GLX) lining luminal surface of endothelial cell body. gcw, glomerular capillary wall. (D) High-magnification image (scale bar: 1 μm) of a vehicle-treated glomerular capillary from a diabetic animal. Note absent glomerular endothelial glycocalyx (absent GLX) lining the luminal surface of endothelial cell body. (E) High-magnification image (scale bar: 1μm) of a VEGF-A165b–treated glomerular capillary from a diabetic animal. Note restoration of glomerular endothelial glycocalyx. (F) Mean±SEM glomerular endothelial glycocalyx depth in five glomeruli in each of three animals per group. *P<0.05, one-way ANOVA.
VEGF-A165b Improves Human Diabetic Glomerular Function
Animal models of diabetic nephropathy exhibit substantial differences from human disease. The functional benefits of VEGF-A165b observed in multiple diabetic animal models were therefore tested on diabetic human glomeruli as well. In humans, rats, and mice, LPA/Vi was significantly higher in glomeruli harvested from diabetic kidneys (all P<0.05 versus nondiabetic controls) (Figure 10A). In addition, long-term podocyte-specific overexpression of hVEGF-A165b (mice) or acute incubation with rhVEGF-A165b (rats and humans) reversed this diabetes-related increase in glomerular permeability (all P<0.05 versus vehicle-incubated diabetic glomeruli) and restored glomerular permeability to levels observed in nondiabetic controls (Figure 10A). In paired measurements of diabetic human glomeruli permeability before and after exposure to rhVEGF-A165b, incubation with rhVEGF-A165b also reduced glomerular LPA/Vi to normal values (Figure 10B).
Figure 10.

VEGF-A165b normalizes permeability of diabetic mouse, rat, and human glomeruli. (A) Glomeruli were harvested from 6 diabetic (STZ-injected) wild-type, 4 nondiabetic wild-type, and 8 diabetic (STZ) nephhVEGF-A165b mice (102 glomeruli); from 3 diabetic (STZ) and 3 nondiabetic rats (74 glomeruli); and from untransplantable kidneys from 3 nondiabetic and 3 diabetic human kidney donors (54 glomeruli). Glomerular water permeability (volume-corrected ultrafiltration coefficient: LPA/Vi) was measured in individual glomeruli following exposure to constitutively overexpressed hVEGF-A165b (mice) or 1-hour incubation in vehicle or rhVEGF-A165b (rat and human glomeruli). Error bars, SEM. *P<0.05; "ns" indicates P>0.05; one-way ANOVA for both. (B) Glomerular LPA/Vi was measured twice in 3 diabetic human glomeruli, before (black) and after (blue) 1-hour incubation in rhVEGF-A165b. *P<0.05, paired t test.
Discussion
We show here that increasing VEGF-A165b levels effectively improves several functional and histologic features of diabetic nephropathy. It can do so locally (through podocyte-specific overexpression) and systemically (via repeated intraperitoneal injections). It can do so in the context of additional upregulation of VEGF-A164a and in nephropathy-complicating models of both type 1 and type 2 diabetes, and it can do so both before and after the onset of albuminuria. We further demonstrate that VEGF-A165b acts on glomerular permeability through VEGFR-2 in glomerular endothelial cells and reverses diabetes-induced damage to the glomerular endothelial glycocalyx. We also demonstrate changes in the balance of VEGF-Axxxa/VEGF-Axxxb isoforms in human diabetic nephropathy, show that glomerular permeability is increased in diabetic human glomeruli, and show that the direct effects of VEGF-A165b on the glomerular capillary wall observed in animal models of diabetic nephropathy are reproduced at a functional level in diabetic human glomeruli. Thus, it appears that VEGF-A165b can provide the endogenous protective actions of VEGF-A in diabetic nephropathy, without driving the deleterious consequences that appear to be mediated via VEGF-A165a in diabetic nephropathy.
Despite improved control of systemic parameters in diabetes, such as hyperglycemia, hypertension, and hyperlipidemia, diabetic nephropathy remains the leading cause of ESRD across the developed world and no specific treatment for diabetic nephropathy is available.1 Recent developments have focused on harnessing endogenous vasculoprotective factors that could provide additional and complementary benefits for patients with diabetes.5 VEGF-A has long been considered a critical component of the vascular complications of diabetes. VEGF-A is generated as two families of isoforms according to alternative splicing of the terminal exon, exon 8.22 The canonical, proangiogenic isoforms, such as VEGF-A165a (VEGF-A164a in the mouse), are matched by a family of antagonistic isoforms (e.g., VEGF-A165b, generically termed VEGF-Axxxb, where xxx is the number of amino acids they encode). These VEGF-Axxxb isoforms match the prosurvival and antiapoptotic effects of angiogenic VEGF-A isoforms but do not drive angiogenesis, do not cause sustained increases in permeability, do not cause vasodilatation, and can prevent the angiogenic and pro-permeability effects of VEGF-A165a.20,23 VEGF-A164a is not an endogenous renoprotective factor in diabetes because overexpressing VEGF-A164a in diabetic mice accelerates nephropathy.19 Our studies demonstrate that the VEGF-A165b isoform confers benefit in diabetic nephropathy. This clarifies the previous paradox that both VEGF overexpression and depletion are detrimental in diabetic nephropathy. It is therefore not just a timely and appropriate level of total VEGF-A that regulates glomerular and kidney function in diabetic nephropathy but an appropriate balance of isoforms, and specifically sufficient levels of the protective VEGF-A165b isoform.
This demonstration that VEGF-A165b is a protective isoform in kidney disease is consistent with a series of reports that nonspecific inhibition of all VEGF isoforms, for example with anti-VEGF antibodies, results in proteinuria and overt renal disease.10 Such strategies will deplete both VEGF-A isoform families, thereby removing the protective actions of VEGF-A165b from the glomerular capillary wall and eliciting renal dysfunction and disease. Similar isoform-specific benefits of VEGF-A165b have also been reported in systemic sclerosis17 and Denys-Drash syndrome,18 ocular disease,16,24 and cancer.25 We show here that diabetic patients with well preserved kidney function have increased VEGF-A165b levels relative to VEGF-A165a. Transgenic podocyte-specific VEGF-A165b overexpression is the most analogous experimental model of this human situation, and it is noteworthy that the local upregulation of VEGF-A165b was the most effective approach in preventing multiple physiologic and histologic changes of diabetic nephropathy. We hypothesize that patients capable of upregulating VEGF-A165b relative to VEGF-A165a, via the modifiable machinery that regulates alternative splicing of the VEGF-A gene,23,26–29 may be protected from progressive nephropathy in diabetes. How to manipulate this alternative splicing mechanism for therapeutic benefit requires further exploration.
VEGFR-2 mediates the major actions of VEGF-A family members, is indispensable for normal glomerular function,9 and mediates the beneficial effects of VEGF-A165b observed here. VEGF-A165b and VEGF-A165a bind VEGFR-2 with equal affinity but elicit different tyrosine residue phosphorylation patterns, different VEGFR-2/neuropilin-1 heterodimerization patterns,30,31 and different downstream signaling pathways,30 resulting in different migration, proliferation, and cytoprotection behaviors in endothelial (and other) cell types.22,24,31 The observation that selective VEGFR-2 blockade in diabetic animals worsened albuminuria and endothelial cell apoptosis32 is consistent with blocking beneficial endogenous VEGF-A165b signaling in rodents. Despite appropriate positive controls, however, the coding sequence of the rodent homolog of VEGF-A165b had not been identified33,34 until very recently.35 In addition, the coding sequences for exon 8 of VEGF-A165a (exon 8a) and VEGF-A165b (exon 8b) are overlapping, hitherto preventing generation of a VEGF-A165b knockout mouse model.
Importantly, systemic injections of VEGF-A165b in the type 1 diabetes model also decreased heavy albuminuria and improved some of the histologic features of diabetic nephropathy, even when treatment was started after the onset of nephropathy, as might be required in clinical practice. The importance of defects in glomerular permeability in animal models of early albuminuric diabetic nephropathy is contested.36,37 These studies confirm that at least some facets of glomerular structure (endothelial glycocalyx) and permeability (ultrafiltration coefficient) are abnormal in early albuminuric diabetic nephropathy and that reversing these glomerular defects through VEGF-A165b administration improves glomerular structure/function, albuminuria, and histology. This emphasizes the important link between changes in glomerular function and albuminuria in early diabetic nephropathy, without excluding the potential importance of diabetes-induced changes in other parts of the nephron.36–38 In the type 2 diabetes db/db mouse model, VEGF-A165b injections reduced early albuminuria but did not preserve GFR or modify outcomes when initiated after the onset of nephropathy. Whether greater VEGF-A165b efficacy in type 1 diabetes models represents differences in the disease time course,4 pharmacokinetics, or validity of the animal model39,40 is not clear. In this type 2 diabetes model, however, albuminuria was relatively low in all animals, and after 3 months it regressed rather than progressed with time.
VEGFR-2 activated by VEGF-A165b was localized to the endothelium. Decreased glomerular endothelial fenestrations, as observed here, have also been reported in diabetic humans, and the possibility of an associated reduction in water permeability was considered.41 In fact, the opposite is the case: Water permeability is increased in diabetic human glomeruli. Moreover, VEGF-A165b restored normal water permeability and decreased albuminuria but did not modify fenestral density, suggesting that an alternative endothelial structure may be physiologically important and amenable to regeneration in diabetic nephropathy. The endothelial glycocalyx is a key regulator of the amount of water and solutes that can cross vessel walls throughout the microcirculation, as well as regulating leukocyte adhesion and endothelial responses to mechanical stimuli.42 The techniques used here confirm43 that glomerular endothelial glycocalyx can be imaged directly in functioning glomeruli and coupled with physiologic measurements, complementing techniques involving tissue fixation and indirect estimates of glycocalyx depth by exclusion. The observed decrease in glomerular endothelial glycocalyx depth using two different methods is consistent with albuminuria and increased water permeability in these two animal models of diabetic nephropathy, and matches glomerular permeability defects (Figure 10) and systemic endothelial glycocalyx defects44,45 in diabetic humans. In addition, both acute and chronic restoration of the endothelial glycocalyx by VEGF-A165b are also compatible with the observed improvements in both water permeability and albuminuria. The time course of acute VEGF-A165b–induced changes in glomerular structure and function match the time course of VEGF-induced changes in glomerular permeability,15,46,47 and both growth factor–induced48 and pharmacologic49 restoration of endothelial glycocalyx indicate that turnover and restoration of endothelial glycocalyx can be rapid.
Importantly, we believe this is also the first demonstration that long-term restoration of the endothelial glycocalyx is both feasible and associated with improved long-term outcomes. Because the endothelial glycocalyx regulates mechanotransduction, leukocyte adhesion, and nitric oxide bioavailability,50 all of which are abnormal in diabetic microvessels, restoring the endothelial glycocalyx may improve multiple vascular abnormalities in diabetes. Identifying the causal nature of the relationship provides an important direction for future work that may have implications for vasculoprotection in a broad range of renal and other diseases.
In summary, we show here that VEGF-A165b is upregulated in diabetic patients whose kidney function is well preserved and that when VEGF-A165b is administered through a variety of routes, it confers functional and histologic benefit in a series of animal models of diabetic nephropathy. VEGF-A165b also effectively restores the endothelial glycocalyx in diabetic nephropathy and improves the permeability function of glomeruli from diabetic humans. VEGF-A165b therefore appears to exert important protective actions in diabetic nephropathy.
Concise Methods
All experiments were conducted in accordance with United Kingdom legislation and local ethical committee approval. Studies on human kidney tissue were approved by National and Local Research Ethics Committees (Institutional Ethical Committee, Leiden University Medical Centre, The Netherlands; South West–Central Bristol National Health Service Research Ethics Committee, United Kingdom; East Midlands–Leicester National Health Service Research Ethics Committee, United Kingdom), and conducted in accordance with the Declaration of Helsinki. Human renal cortex samples were harvested from kidneys offered for transplantation but were technically unsuitable for implantation from donors without diabetes and those with early diabetic nephropathy, and for the late nephropathy group from renal biopsy specimens harvested for clinical reasons. Single glomeruli were harvested by differential sieving for molecular and physiology studies. Animal studies were approved by University of Bristol research ethics committee and conducted in accordance with legislation in the United Kingdom.
Isoform-Specific RT–Quantitative PCR
Total RNA (RNA) was isolated from human kidney tissues with Trizol (Invitrogen) and cleaned up with RNeasy Micro Kit (Qiagen) as per the manufacturer’s suggestion. RNA, 0.5–1 μg, was reverse transcribed into cDNA with QuantiTect Reverse Transcription Kit (Qiagen). cDNA was subjected to quantitative PCR (qPCR) with forward primer 5′- GAGCAAGACAAGAAAATCCC -3′ that spans exon 5 and 7 and reverse primer 5′- CCTCGGCTTGTCACATCTG -3′ spanning exon 7 and 8a to amplify VEGF-A165a, and with forward primer 5′- GAGCAAGACAAGAAA ATCCC -3′ and reverse primer 5′- GTGAGAGATCTGCAAGTACG -3′ spanning exon 7 and 8b to amplify VEGF-A165b. Each reaction contained 10 μl of SYBR green master (from Qiagen), 400 nM forward and reverse primers, 2 μl plasmid DNA or sample cDNA, and water to 20 μl. qPCR amplification was initiated at 95°C for 15 minutes, 45 cycles at 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, followed by final extension at 72°C for 10 minutes. For VEGF-A165a qPCR, both standards (10−2 to 10−6 ng of pcDNA3-VEGF-A165a) and samples were repeated in duplicate. Amplification of known amounts of plasmid pcDNA3-VEGF-A165a DNA rendered the formation of a standards curve of DNA amount (x axis) against cycle threshold (Ct) values (y axis). The amount of VEGF-A165a mRNA expression was extrapolated by comparing sample Ct value with the standards curve from plasmid DNA amplification. Interference for VEGF-A165a qPCR from VEGF-A165b was calculated from Ct values obtained from the amplification of VEGF-A165a signal from known concentrations of plasmid pcDNA3-VEGF-A165b (10−2 to 10−4 ng). The interference was <1/1000. For VEGF-A165b qPCR, a standard curve was formed from amplification of known amounts of plasmid pcDNA3-VEGF-A165b DNA (10−2 to 10−6 ng). The amount of VEGF-A165b in samples was extrapolated by comparing sample Ct value with the standards curve from plasmid DNA amplification. Interference for VEGF-A165b qPCR from VEGF-A165 was calculated from Ct values obtained from the amplification of VEGF-A165b signal from known concentrations of plasmid pcDNA3-VEGF-A165 (10−2 to 10−4 ng). The interference was <1/3000.
ELISA of Pan-VEGF and VEGF-A165b
Protein was isolated from human kidney tissues after being homogenized in RIPA buffer (Sigma-Aldrich) and quantified with Bio-Rad assay. Pan-VEGF-A and VEGF-A165b detection were followed with the manufacturer’s suggestion (Duoset human VEGF-A, catalog no. DY293; R&D Systems, and Duoset human VEGF-A165b, catalog no. DY3045; R&D Systems). Briefly, 0.08 µg of capture antibody diluted in 1× PBS (pH, 7.4) was adsorbed onto each well of a 96-well plate (Immulon 2HB; Thermo Fisher Scientific, Basingstoke, UK) overnight at room temperature. The plate was washed three times between each step with 1xPBS-Tween (0.05%). After blocking with 100 μl of 1% BSA in PBS for 1 hour at 37°C, 100 μl of recombinant human VEGF-A165a for VEGF-A ELISA, or recombinant human VEGF-A165b for VEGF-A165b ELISA, diluted in 1% BSA in PBS (ranging from 62.5 pg/ml to 4 ng/ml) or protein samples were added to each well. After incubation for 1 hour at 37°C with shaking and three washes, 100 µl of detection antibody at 0.05 µg/ml was added to each well, and the plate was left for 1 hour at 37°C with shaking. Next, 100 µl of streptavidin-horseradish peroxidase (R&D Systems) at 1:200 dilution in 1% BSA in PBS was added, the plate was left at room temperature for 20 minutes, and 100 µl of O-phenylenediamine dihydrochloride solution per well (Substrate reagent pack DY-999; R&D Systems) was added, protected from light and incubated for 20 minutes at room temperature. The reaction was stopped with 50 µl/well 1 M H2SO4, and absorbance was read immediately in the Opsys MR 96 well plate reader at 492 nm, with control reading at 460 nm.
Animal Studies
Transgenic mice were obtained from in-house colonies. Adult male mice were used for all mouse studies, and wild-type littermate controls were used for comparison. Details of the transgenic strategies for the nephhVEGF-A165b and pod-rTta:TetO-mVEGF-A164a mice are provided in detail elsewhere.15,20 pod-rTta:TetO-mVEGF-A164a mice were generated by crossing podocin-rTtA and TetO-VEGF-A164a mice supplied by Professor Susan Quaggin with permission from Jeffrey Kopp (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD) and Jeff Whitsett (Children’s Hospital Medical Center, Cincinnati, OH). Overexpression of human VEGF-A165b in podocytes (nephhVEGF-A165b) was constitutive. Overexpression of murine VEGF-A164a (mVEGF-A164a) in podocytes in pod-rTta:TetO-mVEGF-A164a mice was induced by adding doxycycline (2 mg/ml) in light-protected drinking water containing 5% w/v sucrose that was changed twice per week.
nephhVEGF-A165b were generated on a C57Bl6 background, and diabetes induction with a moderate-dose STZ regimen (100 mg/kg for 3 consecutive days) was therefore required to elicit nephropathy in the littermate controls. After crossing nephhVEGF-A165b mice with pod-rTta:TetO-mVEGF-A164a mice (mixed strain background), the low-dose Animal Models of Diabetic Complications Consortium (AMDCC) STZ protocol (50 mg/kg for 5 consecutive days) was used. DBA2J mice were also treated with the low-dose AMDCC streptozotocin protocol51 to induce diabetes and associated nephropathy. db/db mice (BKS.Cg-+Leprdb/+Leprdb/OlaHsd) were purchased from Harlan Laboratories, UK, and fed a standard rodent diet. Diabetes was induced in rats with a single intravenous injection of STZ (45 mg/kg).52 All animals were fasted for 4–6 hours before administration of STZ. Recombinant human VEGF-A165b (rhVEGF-A165b) for intraperitoneal injection was generated as previously described,30 and was administered after confirmation of two consecutive readings of hyperglycemia (>16 mmol).
Body weight and blood glucose (ACCU-CHEK Aviva, Roche) were monitored weekly after STZ administration in mice, 1 week after administration of STZ in rats, and starting from 6 weeks of age in db/db mice. Urine samples were collected by overnight housing in metabolic cages. Urinary albumin was quantified with albumin ELISA (Bethyl Laboratories, Inc.), and creatinine was measured using an enzymatic spectrophotometric assay (Konelab T-Series 981845; Thermo Fisher Scientific). Urinary albumin-to-creatinine ratio was calculated as the quotient of albumin concentration and creatinine concentration. Creatinine clearance was calculated from the volume of urine produced in a timed collection, creatinine concentration in the same sample, and creatinine concentration in blood harvested at culling. GFR was determined with FITC-inulin injection in conscious mice before culling, in accordance with the AMDCC protocol.53
Kidney cortex samples harvested immediately postmortem were flash frozen, and 5- to 10-μm sections were cut using a rotary microtome. Sections were mounted onto glass slides and stained with hematoxylin and eosin using standard techniques. Images were captured using a DCN-100 digital imaging system (Nikon Instruments, Surrey, UK). Glomerular area was calculated from the glomerular outline. Separate sections were stained with 0.5% periodic acid, Schiff reagent, and Mayer hematoxylin. The proportion of the glomerulus positive for periodic acid-Schiff stain was determined according to AMDCC protocols using Adobe Photoshop software.
For standard transmission electron microscopy, animals were flushed by transcardiac perfusion with Ringer solution and then fixed with 2.5% glutaraldehyde (±1% Alcian blue for endothelial glycocalyx labeling), and small pieces (0.5- to 1-mm diameter) of kidney cortex studies were rapidly excised and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (4°C), washed in 0.1 M cacodylate buffer, postfixed in 1% osmium tetroxide, and washed in distilled water. Tissues were ethanol-dehydrated and embedded in an Araldite (Agar Scientific). Sections 50 to 100 nm thick were stained with 3% (aqueous) uranyl acetate and Reynolds lead citrate solution. Digital micrographs were taken on a Phillips 100CS microscope or Tecnai T12 microscope at ×940, ×1250, and ×6200. Images of the glomerular capillary wall were blinded, and detailed measurements were taken randomly using a predefined algorithm using Adobe Photoshop. A fixed digital grid was superimposed over the electron micrograph, and glycocalyx depth was measured at points of intersection between these fixed gridlines and the image of the glomerular capillary wall. In total, 1696 randomized measurements of glycocalyx depth in 179 glomerular capillaries from nine animals across the three experimental groups were made using this algorithm by a blinded observer, and all analyses were completed before unblinding. Endothelial fenestrae were also counted in the same images, and fenestral density was calculated as the percentage of glomerular basement membrane filtration surface covered by endothelial fenestrae.
Kidney samples for immunofluorescence studies were formalin-fixed and embedded in wax. Five-micrometer sections were mounted onto glass slides and left to dry overnight at 37°C. Sections were then dewaxed and rehydrated before staining using anti-VEGFR-2 1:50 (Cell Signaling Technology) and anti–P-VEGFR-2 (Y-951) 10 μg/ml (Santa Cruz Biotechnology). Primary antibodies were left to incubate at 4°C overnight before incubating with the appropriate fluorescent secondary. Before imaging, the sections were also stained for Hoechst and then viewed on the fluorescent microscope. The area of staining for VEGFR-2/p-VEGFR-2 was normalized to glomerular area to calculate the percentage glomerular area covered by staining. For colocalization studies, fresh-frozen 5-μm sections were embedded in optimal cutting temperature compound. Sections were fixed in 4% paraformaldehyde before costaining with anti–VEGFR-2 1:50 (Cell Signaling Technology) and either antinephrin 10 μg/ml (2B Scientific/Acris) or anti–PECAM-1 10 μg/ml (Abcam, Inc.). Z-stack confocal microscopy imaging was used to assess colocalization.
For confocal imaging of the glomerular endothelial glycocalyx in functioning glomeruli avoiding fixation artifact, kidneys were perfused in vivo with R18 (cell membrane label) and Alexa Fluor-488–labeled wheat germ agglutinin lectin (labels N-acetyl glucosamine [a component of heparan sulfate and hyaluronan glycosaminoglycans] and N-acetyl neuraminic acid [a major sialic acid present on the endothelial cell surface] glycosaminoglycans on the endothelial cell surface). Glomeruli were isolated by differential sieving, incubated in vehicle or 1 nM rhVEGF-A165b to match glomerular physiology measurements, and imaged with confocal microscopy. Glomerular endothelial glycocalyx depth was measured as the distance between peak fluorescent signal from the cell membrane over the endothelial cell body, and from the overlying glomerular endothelial glycocalyx; measurements were taken over the endothelial cell body to eliminate interference from labeled glycosaminoglycans within the glomerular basement membrane beneath attenuated and fenestrated endothelial areas elsewhere in the glomerular capillary wall.
Cell Culture Studies
Caspase 3 activity was measured in conditionally immortalized human podocytes54 using the CaspACE assay kit (Promega) according to the manufacturer’s indications. Briefly, cells were lysed and incubated with the Asp-Glu-Val-Asp-p-Nitroaniline (DEVD-pNA) colorimetric substrate, which, upon cleavage by caspase 3, results in free pNA. Free pNA produces a yellow color monitored at 405 nm and proportional to the caspase activity. Absorption at 405 nm is normalized on the amount of total protein in samples.
Human umbilical vein endothelial cells were isolated from umbilical cords obtained with consent under ethical approval and cultured (up to passage 6) until they reached 80% confluence. These cells were then treated with normal EBM2 media (5 mM glucose; Lonza) or with media that contained an additional 30 mM glucose for hyperglycemic conditions. Osmotic control was performed replacing 30 mM glucose with 30 mM mannitol. Drug groups used combinations of the following: vehicle (PBS; n=4 per group), 2.5 nM VEGF-A165b (n=4 per group), and/or 200 nM PTK787 (vandetanib55). The number of activated caspase-3 cells was totaled from five random fields of view per well. These were incubated for 24 hours and were processed for polyclonal rabbit activated caspase-3 (apoptotic cell death marker, 1 in 500; New England Biolabs) staining and Hoechst staining. Negative controls were performed with no primary antibody or cleaved caspase-3 blocking peptide (New England Biolabs).
Glomerular Permeability Assay
Glomeruli were harvested from mouse, rat, or human cortical renal tissue by differential sieving, and the glomerular ultrafiltration coefficient (LPA: hydraulic conductivity-area product) was measured using a validated oncometric assay as previously described.46 In brief, individual glomeruli were mounted on a holding pipette within a flow-controlled observation chamber by gentle aspiration. The perifusate surrounding the glomerulus was rapidly exchanged from incubating solution (1% BSA in HEPES Ringer solution) to 8% BSA-HEPES Ringer solution, thereby increasing the oncotic pressure outside the glomerular capillary wall relative to the glomerular capillary lumen. The consequent flux of water out of the glomerular capillaries results in a rapid reduction in glomerular volume, which was recorded continuously on video camera and analyzed offline using Fiji software56 with in-house macros (developed by K. Arkill). Linear regression analysis was used to calculate the initial (<0.1 second) rate of reduction of glomerular volume (JV) in response to the applied oncotic pressure gradient (ΔΠ), the quotient of which describes glomerular ultrafiltration coefficient (LPA). For incubation studies, glomeruli were incubated for 1 hour with appropriate agents: 1 nM rhVEGF-A165b30; 10 μM ZM 323881 (Calbiochem; EMD Millipore); 100 nM PTK 787 (Novartis Pharma, Basel, Switzerland). Human glomeruli are significantly larger than rodent glomeruli (P<0.05) (Supplemental Figure 1A) and therefore present a greater surface area (A) for fluid exchange across the glomerular capillary wall. Glomeruli harvested from healthy humans had substantially higher ultrafiltration coefficient (LPA) values than rats and mice (P<0.05) (Supplemental Figure 1B). When corrected for glomerular volume (Vi), human and rat glomeruli had identical normalized ultrafiltration coefficient values (LPA/Vi) (Supplemental Figure 1C) but had significantly lower LPA/Vi than mouse glomeruli (both P<0.05) (Supplemental Figure 1C).
Disclosures
S.J. Harper, D.O. Bates, L. Donaldson, A.H. Salmon, S. Oltean, and M. Gammons are inventors on patents involving VEGF-A165b.
Supplementary Material
Acknowledgments
We would like to thank kidney donors and their families for consenting to research on donated organs that were not suitable for transplantation, and to transplant coordinators in Bristol and other centers for enabling these studies.
This work was supported by grants from the Medical Research Council (G0802829 fellowship to A.H.J.S. and G10002073, GR0600920 projects to S.J.H., D.O.B., A.H.J.S.), Kidney Research UK (RP18/2010 to A.H.J.S., D.O.B., S.J.H., ST5/2012 to A.H.J.S., R.R.F., S.C.S., RP45/2013 to A.H.J.S., R.R.F., S.C.S., S.J.H.), British Heart Foundation (FS/05/114/19959 studentship to S.J.H. and A.H.J.S., FS/10/017/28249 fellowship to R.R.F., project PG08/022/21636 to D.O.B., S.J.H. and A.H.J.S., PG/08/059/25335 to C.R.N. and K.A.), Wellcome Trust (project 079736 to D.O.B. and S.J.H.), Diabetes UK (RJ5522 to L.F.D. and D.O.B.), BBSRC project grant BB//J007293/1 (S.O. and D.O.B.), Swiss National Science Foundation (grant 31003A-130463), Oncosuisse (grant OC2 01200-08-2007), NOVARTIS Stiftung für medizinischbiologische Forschung (grant 10C61) to K.B.H., and Richard Bright VEGF Research Trust (to A.R. and A.H.J.S.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014040350/-/DCSupplemental.
References
- 1.Gilbertson DT, Liu J, Xue JL, Louis TA, Solid CA, Ebben JP, Collins AJ: Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J Am Soc Nephrol 16: 3736–3741, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Frank RN: Diabetic retinopathy. N Engl J Med 350: 48–58, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Abbott CA, Malik RA, van Ross ER, Kulkarni J, Boulton AJ: Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 34: 2220–2224, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forbes JM, Cooper ME: Mechanisms of diabetic complications. Physiol Rev 93: 137–188, 2013 [DOI] [PubMed] [Google Scholar]
- 5.Rask-Madsen C, King GL: Kidney complications: Factors that protect the diabetic vasculature. Nat Med 16: 40–41, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP: Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 13: 1349–1358, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Sivaskandarajah GA, Jeansson M, Maezawa Y, Eremina V, Baelde HJ, Quaggin SE: Vegfa protects the glomerular microvasculature in diabetes. Diabetes 61: 2958–2966, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster RR, Hole R, Anderson K, Satchell SC, Coward RJ, Mathieson PW, Gillatt DA, Saleem MA, Bates DO, Harper SJ: Functional evidence that vascular endothelial growth factor may act as an autocrine factor on human podocytes. Am J Physiol Renal Physiol 284: F1263–F1273, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE: Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21: 1691–1701, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE: VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baelde HJ, Eikmans M, Lappin DW, Doran PP, Hohenadel D, Brinkkoetter PT, van der Woude FJ, Waldherr R, Rabelink TJ, de Heer E, Bruijn JA: Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int 71: 637–645, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Saito D, Maeshima Y, Nasu T, Yamasaki H, Tanabe K, Sugiyama H, Sonoda H, Sato Y, Makino H: Amelioration of renal alterations in obese type 2 diabetic mice by vasohibin-1, a negative feedback regulator of angiogenesis. Am J Physiol Renal Physiol 300: F873–F886, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Bates DO: Vascular endothelial growth factors and vascular permeability. Cardiovasc Res 87: 262–271, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathieson PW: How much VEGF do you need? J Am Soc Nephrol 17: 602–603, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Qiu Y, Ferguson J, Oltean S, Neal CR, Kaura A, Bevan H, Wood E, Sage LM, Lanati S, Nowak DG, Salmon AH, Bates D, Harper SJ: Overexpression of VEGF165b in podocytes reduces glomerular permeability. J Am Soc Nephrol 21: 1498–1509, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hua J, Spee C, Kase S, Rennel ES, Magnussen AL, Qiu Y, Varey A, Dhayade S, Churchill AJ, Harper SJ, Bates DO, Hinton DR: Recombinant human VEGF165b inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci 51: 4282–4288, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M: Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res 109: e14–e26, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Schumacher VA, Jeruschke S, Eitner F, Becker JU, Pitschke G, Ince Y, Miner JH, Leuschner I, Engers R, Everding AS, Bulla M, Royer-Pokora B: Impaired glomerular maturation and lack of VEGF165b in Denys-Drash syndrome. J Am Soc Nephrol 18: 719–729, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Veron D, Bertuccio CA, Marlier A, Reidy K, Garcia AM, Jimenez J, Velazquez H, Kashgarian M, Moeckel GW, Tufro A: Podocyte vascular endothelial growth factor (Vegf₁₆₄) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia 54: 1227–1241, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oltean S, Neal CR, Mavrou A, Patel P, Ahad T, Alsop C, Lee T, Sison K, Qiu Y, Harper SJ, Bates DO, Salmon AH: VEGF165b overexpression restores normal glomerular water permeability in VEGF164-overexpressing adult mice. Am J Physiol Renal Physiol 303: F1026–F1036, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rennel ES, Hamdollah-Zadeh MA, Wheatley ER, Magnussen A, Schüler Y, Kelly SP, Finucane C, Ellison D, Cebe-Suarez S, Ballmer-Hofer K, Mather S, Stewart L, Bates DO, Harper SJ: Recombinant human VEGF165b protein is an effective anti-cancer agent in mice. Eur J Cancer 44: 1883–1894, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ: VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res 62: 4123–4131, 2002 [PubMed] [Google Scholar]
- 23.Harper SJ, Bates DO: VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 8: 880–887, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magnussen AL, Rennel ES, Hua J, Bevan HS, Beazley Long N, Lehrling C, Gammons M, Floege J, Harper SJ, Agostini HT, Bates DO, Churchill AJ: VEGF-A165b is cytoprotective and antiangiogenic in the retina. Invest Ophthalmol Vis Sci 51: 4273–4281, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rennel E, Waine E, Guan H, Schüler Y, Leenders W, Woolard J, Sugiono M, Gillatt D, Kleinerman E, Bates D, Harper S: The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br J Cancer 98: 1250–1257, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oltean S, Gammons M, Hulse R, Hamdollah-Zadeh M, Mavrou A, Donaldson L, Salmon AH, Harper SJ, Ladomery MR, Bates DO: SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem Soc Trans 40: 831–835, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, Symonds KE, Nowak DG, Royer-Pokora B, Saleem MA, Hagiwara M, Schumacher VA, Harper SJ, Hinton DR, Bates DO, Ladomery MR: WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20: 768–780, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G, Hagiwara M, Harper SJ, Woolard J, Ladomery MR, Bates DO: Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J Biol Chem 285: 5532–5540, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, Churchill AJ, Ladomery MR, Harper SJ, Bates DO: Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci 121: 3487–3495, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cébe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, Ballmer-Hofer K: A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci 63: 2067–2077, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L: Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 68: 4683–4692, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Kim HW, Lim JH, Kim MY, Chung S, Shin SJ, Chung HW, Choi BS, Kim YS, Chang YS, Park CW: Long-term blockade of vascular endothelial growth factor receptor-2 aggravates the diabetic renal dysfunction associated with inactivation of the Akt/eNOS-NO axis. Nephrol Dial Transplant 26: 1173–1188, 2011 [DOI] [PubMed] [Google Scholar]
- 33.Bates DO, Mavrou A, Qiu Y, Carter JG, Hamdollah-Zadeh M, Barratt S, Gammons MV, Millar AB, Salmon AH, Oltean S, Harper SJ: Detection of VEGF-A(xxx)b isoforms in human tissues. PLoS ONE 8: e68399, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris S, Craze M, Newton J, Fisher M, Shima DT, Tozer GM, Kanthou C: Do anti-angiogenic VEGF (VEGFxxxb) isoforms exist? A cautionary tale. PLoS ONE 7: e35231, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kikuchi R, Nakamura K, MacLauchlan S, Ngo DT, Shimizu I, Fuster JJ, Katanasaka Y, Yoshida S, Qiu Y, Yamaguchi TP, Matsushita T, Murohara T, Gokce N, Bates DO, Hamburg NM, Walsh K: An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease [published online ahead of print November 2, 2014]. Nat Med 10.1038/nm.3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rippe C, Rippe A, Torffvit O, Rippe B: Size and charge selectivity of the glomerular filter in early experimental diabetes in rats. Am J Physiol Renal Physiol 293: F1533–F1538, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Russo LM, Sandoval RM, Campos SB, Molitoris BA, Comper WD, Brown D: Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J Am Soc Nephrol 20: 489–494, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomson SC, Vallon V, Blantz RC: Kidney function in early diabetes: The tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol 286: F8–F15, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Breyer MD: Translating experimental diabetic nephropathy studies from mice to men. Contrib Nephrol 170: 156–164, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Tesch GH, Lim AK: Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. Am J Physiol Renal Physiol 300: F301–F310, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, Lovato T, Richardson M, Myers BD, Nelson RG: Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 82: 1010–1017, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salmon AH, Satchell SC: Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J Pathol 226: 562–574, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Salmon A, Ferguson J, Burford J, Gevorgyan A, Nakano D, Harper S, Bates D, Peti-Peterdi J: Widespread loss of endothelial glycocalyx links albuminuria and systemic vascular dysfunction. J Am Soc Nephrol 23: 1339–1350, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broekhuizen LN, Lemkes BA, Mooij HL, Meuwese MC, Verberne H, Holleman F, Schlingemann RO, Nieuwdorp M, Stroes ES, Vink H: Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 53: 2646–2655, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JA, Ince C, Holleman F, Diamant M, Heine RJ, Hoekstra JB, Kastelein JJ, Stroes ES, Vink H: Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 55: 1127–1132, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Salmon AH, Neal CR, Bates DO, Harper SJ: Vascular endothelial growth factor increases the ultrafiltration coefficient in isolated intact Wistar rat glomeruli. J Physiol 570: 141–156, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satchell SC, Anderson KL, Mathieson PW: Angiopoietin 1 and vascular endothelial growth factor modulate human glomerular endothelial cell barrier properties. J Am Soc Nephrol 15: 566–574, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Salmon AH, Neal CR, Sage LM, Glass CA, Harper SJ, Bates DO: Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc Res 83: 24–33, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, Welsch U, Becker BF: Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology 107: 776–784, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Weinbaum S, Tarbell JM, Damiano ER: The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 9: 121–167, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Breyer MD, Böttinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K, AMDCC : Mouse models of diabetic nephropathy. J Am Soc Nephrol 16: 27–45, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Palm F, Ortsäter H, Hansell P, Liss P, Carlsson PO: Differentiating between effects of streptozotocin per se and subsequent hyperglycemia on renal function and metabolism in the streptozotocin-diabetic rat model. Diabetes Metab Res Rev 20: 452–459, 2004 [DOI] [PubMed] [Google Scholar]
- 53.Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD: Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol 286: F590–F596, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P: A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Whittles CE, Pocock TM, Wedge SR, Kendrew J, Hennequin LF, Harper SJ, Bates DO: ZM323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation 9: 513–522, 2002 [DOI] [PubMed] [Google Scholar]
- 56.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A: Fiji: An open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.