

Abstract
The direct α-heteroarylation of tertiary amines has been accomplished via photoredox catalysis to generate valuable benzylic amine pharmacophores. A variety of five-and six-membered chloroheteroarenes are shown to function as viable coupling partners for the α-arylation of a diverse range of cyclic and acyclic amines. Evidence is provided for a homolytic aromatic substitution mechanism, in which a catalyticallygenerated α-amino radical undergoes direct addition to an electrophilic chloroarene.
Introduction
Heterocycles and heteroaromatics are ubiquitous components of biological systems and pharmaceutical research. These invaluable structural motifs are broadly employed in the development of medicinal agents due to their capacity to (i) drive therapeutic potency, (ii) lower drug lipophilicity, (iii) increase aqueous solubility, and (iv) reduce the inhibition of cytochrome P450s.1 Indeed, at the present time heteroaromatics feature in approximately half of the top 200 pharmaceuticals sold worldwide.2 It is not surprising therefore that coupling reactions which generically connect heterocyclic moieties to other molecular fragments are now mainstay transformations in medicinal agent synthesis. For example, couplings between heterocycles and amines via C–N bond-forming reactions, either via nucleophilic aromatic substitution (SNAr)3 or transition metal-catalyzed approaches,4 have been extensively studied and applied (Fig. 1, eqn (1)).5 In contrast, fragment coupling methods that unite heteroaromatic units with the amine α-carbon position (Fig. 1, eqn (2)) are relatively unknown, despite the opportunity to access medicinal agents with similar physiological properties and with proven clinical utility (e.g. Veliparib,6 Pradaxa,7 and Benthiavalicarb8). While important strategies for amine α-arylation have been reported by Dieter,9 Campos,10 Nakamura,11 and others,12 a generic yet mild α-arylation protocol remains largely elusive.
Fig. 1.
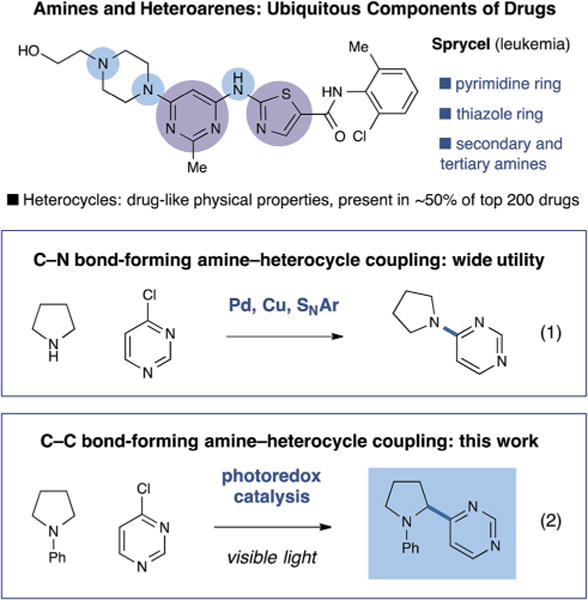
Significance and approaches to amine–heteroaromatic coupling.
Visible light photoredox catalysis has emerged in recent years as a powerful platform for the development of new reactions in organic synthesis.13 In this context, we have recently demonstrated that photoredox catalysis can be employed to mediate the α-arylation of amines using electron-deficient benzonitrile coupling partners.14 In this protocol, activation of the amine α-C–H bond is achieved via single-electron oxidation followed by α-deprotonation to generate the critical α-amino radical 1 (Scheme 1).15 Concomitant single-electron reduction of an accompanying benzonitrile provides a stabilized radical anion, which undergoes selective hetero radical–radical coupling with the neutral radical species 1 to forge the desired α-amino arylation products. Taking inspiration from the Minisci reaction,16 we recently questioned whether a photoredoxbased amine α-arylation could be achieved by the addition of an α-amino radical 1 to a ground-state, neutral arene via a homolytic aromatic substitution pathway.17–19 This approach would constitute an alternative catalytic mechanism that would obviate the need for cyanoarenes and the accompanying radical anion pathway. Herein, we report the successful execution of this plan and describe a new coupling mechanism that employs chloroheteroarenes for the direct α-arylation of amines.
Scheme 1.
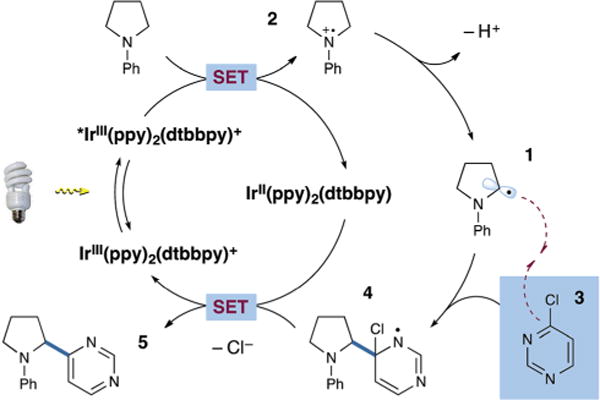
Proposed arylation via homolytic aromatic substitution.
Design plan
As shown in Scheme 1, our proposed mechanism called for a sufficiently oxidizing photocatalyst, e.g. iridium complex Ir(ppy)2(dtbbpy)PF6 (ppy = 2-phenylpyridine, dtbbpy = 4,4′-ditert-butyl-2,2′-bipyridine, vs. SCE)20 to oxidize the amine substrate via a single electron transfer (SET) mechanism vs. SCE for N,N-dimethylaniline).21 This electron transfer step would lead to a reduced iridium complex IrII(ppy)2(dtbbpy) with concomitant formation of the amine radical cation 2, a species that may be deprotonated at the α-C–H position to generate the α-amino radical 1.22 Addition of this neutral radical species 1 to a highly electrophilic heteroarene 3 via homolytic aromatic substitution was expected to provide the radical σ-complex 4, which may readily undergo single-electron reduction ( vs. SCE for the hydroxycyclohexadienyl radical)23,24 followed by loss of an anionic leaving group (Cl−) to provide the α-heteroaryl amine product 5. This reduction step would simultaneously complete the photoredox cycle (Ir(II) to Ir(III)) via reconstitution of the Ir(ppy)2(dtbbpy)PF6 catalyst. Importantly, the incorporation of an anionic leaving group in the arene coupling partner would render this process redox neutral, obviating the need to employ either stoichiometric oxidants or reductants, while only generating HX as a reaction byproduct. Moreover, we anticipated that this strategy would enable the coupling of a diverse array of heteroaromatic rings while exhibiting complementary scope to our previously reported cyanoarene radical anion pathway.
Results and discussion
We initiated our α-amino arylation investigation by examining the utility of various 2-halo-benzothiazoles given their utility as potent electrophiles in two-electron SNAr chemistry. While attempts to employ the cyano-substituted benzothiazole met with little success, we were delighted to find that bromo-and chloro-leaving groups provided the product of α-heteroarylation in useful yields using tris(2-phenylpyridinato-C2,N)iridium(III) [fac-Ir(ppy)3] as the photocatalyst (Table 1, entries 3 and 4, 50 and 56% yield). Next, evaluation of a variety of visible light photocatalysts revealed higher efficiencies could be obtained with the heteroleptic iridium complex Ir(ppy)2(dtbbpy)PF6 in the presence of 2-chlorobenzothiazole (entry 6, 75% yield). Moreover, lowering the photocatalyst and amine loadings did not lead to diminished reaction yields (entries 7 and 8). Intriguingly, the addition of water was found to improve the overall efficiency of this arylation protocol, with ten equivalents providing optimal yields (entry 10, 87% yield).25 Finally, while a low yield of product was obtained upon visible light irradiation in the absence of photocatalyst (entry 11),26 no reaction was observed in the absence of either light or base (entries 12 and 13).
Table 1.
Evaluation of arene coupling partners and photocatalysts

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Leaving group (X) | Amine (equiv.) | Photocatalyst (mol%) | Yielda (%) |
| 1 | CN | 3 | Ir(ppy)3 (1 mol%) | 4% |
| 2 | I | 3 | Ir(ppy)3 (1 mol%) | 5% |
| 3 | Br | 3 | Ir(ppy)3 (1 mol%) | 50% |
| 4 | Cl | 3 | Ir(ppy)3 (1 mol%) | 56% |
| 5 | F | 3 | Ir(ppy)3 (1 mol%) | 16% |
| 6 | Cl | 3 | Ir(ppy)2(dtbbpy)PF6 (1 mol%) | 75% |
| 7 | Cl | 3 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 78% |
| 8 | Cl | 1.5 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 77% |
| 9b | Cl | 1.5 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 84% |
| 10c | Cl | 1.5 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 87% |
| 11 | Cl | 1.5 | None | 11% |
| 12d | Cl | 1.5 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 0% |
| 13e | Cl | 1.5 | Ir(ppy)2(dtbbpy)PF6 (0.5 mol%) | 0% |
Yield after 24 h determined by 1H NMR analysis of crude reaction mixture with internal standard. Reactions performed with 2 equiv. NaOAc and 0.25 M DMA.
With 3 equiv. H2O.
With 10 equiv. H2O, isolated yield.
Performed in the absence of light.
Performed in the absence of NaOAc.
With respect to the scope of the arene component in this coupling reaction, we have found that a range of five-membered heteroaryl chlorides function as suitable substrates (Table 2). For example, a variety of benzoxazole, benzothiazole, and benzimidazole-derived scaffolds may be installed in high yields (entries 1–5 and 7, 87–93% yield). Moreover, monocyclic imidazole and thiazole substrates are well-tolerated (entries 6, 9, and 11, 77–82% yield). The nature of the 6,5-heteroaromatic may also be broadly varied, as exemplified by the installation of the imidazopyridine and caffeine ring systems (entries 8 and 10, 94 and 81% yield). Furthermore, five-membered heteroaromatic rings that incorporate three heteroatoms may be utilized, as highlighted by the coupling of a 1,3,4-thiadiazole (entry 12, 65% yield). Importantly, heteroaromatic substrates that include a second halide substituent (F, Cl, Br) at a non-electrophilic site remain viable in this protocol (entries 4, 5, and 9), an important consideration with respect to (i) predictable regiocontrol of the arylation coupling site and (ii) the compatibility of this protocol with complementary coupling technologies in a synthetic sequence.
Table 2.
Coupling of five-membered heteroarenes: arene scopea
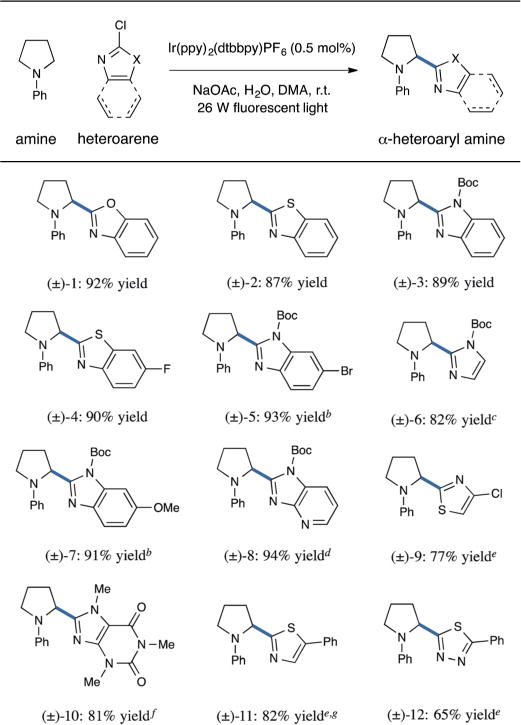
|
Conditions as in Table 1, entry 10 unless noted.
Chloroarene starting material and product are a 1 : 1 mixture of regioisomers with respect to position of the Boc group.
Performed using 3.0 equiv. arene to 1.0 equiv. amine and 30 equiv. H2O.
Performed at 0 °C.
Performed using 1.5 equiv. arene to 1.0 equiv. amine.
Performed with 40 equiv. H2O.
Performed without H2O.
Having successfully demonstrated α-heteroarylation with a variety of five-membered heteroarenes, we next anticipated that electrophilic six-membered heterocycles might also engage in this new α-amino coupling protocol (Table 3). In general, pyrimidine substrates were found to be viable, with a second electron-withdrawing substituent typically providing superior yields. Thus, 2-chloropyrimidines bearing ester, amide, or trifluoromethyl functionality at the 5-position undergo coupling with excellent efficiency (entries 1–3, 73–84% yield).27 Pyrimidines that incorporate a second aromatic ring (entry 4, 73% yield) or a halide substituent (entry 5, 74% yield) may also be successfully employed. Performing these reactions at subambient temperatures (0 °C) with added water is critical for suppressing arene decomposition and achieving high levels of reaction efficiency (see ESI†). Pyrimidines bearing a chloride leaving group at the 4-position also undergo coupling (entries 6–8, 72–92% yield), and ortho-substitution on the pyrimidine ring is tolerated, albeit with diminished yield (entry 9, 64% yield). Again, the compatibility of a second chloride substituent on the pyrimidine substrate allows for subsequent product functionalization via a range of transformations. Finally, this photoredox protocol may be used to install a variety of other 6-membered heteroarenes including purines (entry 10, 73% yield) and 1,3,5-triazines (entries 11 and 12, 88 and 84% yield).
Table 3.
Coupling of six-membered heteroarenes: arene scopea
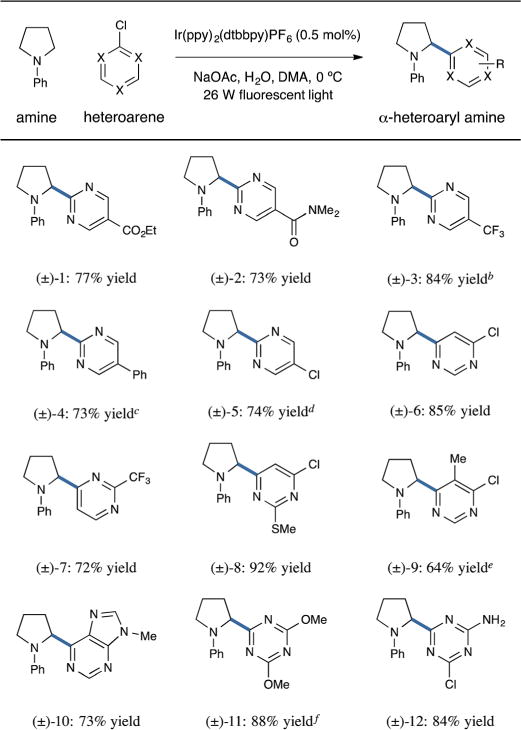
|
Conditions employ 25 equiv. H2O at 0 °C, unless otherwise noted.
Performed using 5 equiv. H2O, 1.0 equiv. amine, and 1.5 equiv. arene.
Performed at 10 °C.
Performed using 40 equiv. H2O, 1.0 equiv. amine, and 3.0 equiv. arene; product obtained as a single regioisomer.
Performed using 1.0 equiv. amine and 1.5 equiv. arene.
Performed using 50 equiv. H2O.
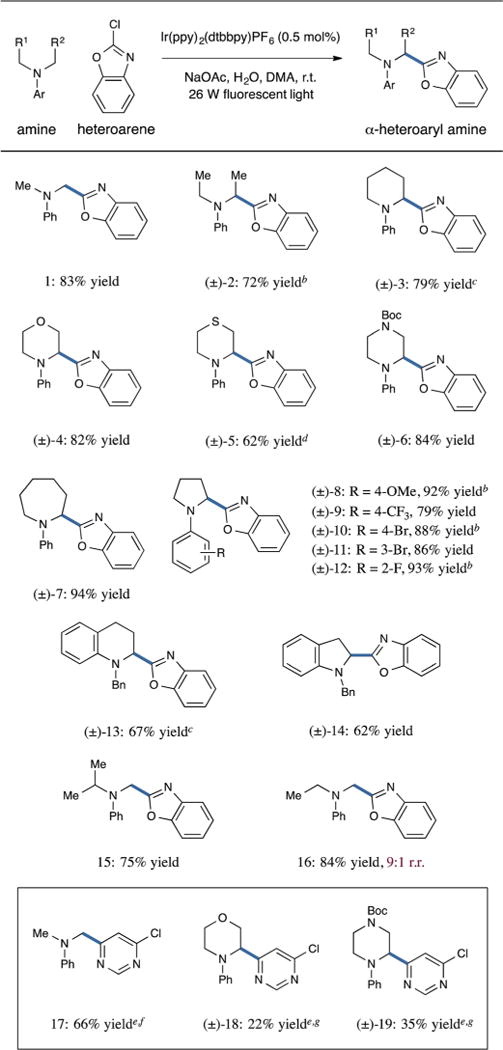
With regard to the scope of the amine coupling partner, we anticipated that a range of readily oxidized tertiary anilines would serve as competent substrates. Indeed, using 2-chlorobenzoxazole as a representative heteroarene, we were pleased to find that both acyclic (Table 4, entries 1 and 2, 83 and 72% yield) and cyclic dialkylanilines (entries 3 and 7, 79 and 94% yield) undergo α-arylation with excellent efficiency. Moreover, saturated heterocyclic ring systems such as morpholine, thiomorpholine, and piperazine were found to be amenable to this photoredox heteroarylation (entries 4–6, 62–84% yield). Electron-donating as well as withdrawing substituents on the N-aryl ring are tolerated, and may be incorporated at the para, meta, or ortho positions (entries 8–12, 79–93% yield). Medicinally relevant tetrahydroquinoline and indoline architectures can also be successfully employed, with arylation proceeding regioselectively at the α-amino ring position in lieu of the exocyclic benzylic site (entries 13 and 14, 67 and 62% yield). Unsymmetrical, acyclic dialkylanilines interestingly undergo functionalization at methyl α-substituents in preference to methine (entry 15, single regioisomer) and methylene (entry 16, 9 :1 r.r.) α-positions.28 Finally, we provide examples of heteroaryl chloride-amine combinations that undergo coupling with moderate levels of reaction efficiency. As shown, the use of N,N-dimethylaniline, N-phenylmorpholine, and N-phenylpiperazine with 4,6-dichloropyrimidine resulted in diminished yields (entries 17–19, 22–66% yield). While these transformations remain of value to medicinal chemists, we are focused upon overcoming these limitations in efficiency.29 To demonstrate the scalability of the arylation reaction, we additionally performed the coupling of 2-chlorobenzoxazole and tert-butyl-4-phenylpiperazine-1-carboxylate on a 4.0 mmol scale, providing the α-arylated product in 80% yield (1.22 g product, cf. Table 4, entry 6, 84% yield).
Table 4.
Photoredox amine α-heteroarylation: amine scopea
|
Conditions employ 10 equiv. H2O unless otherwise noted.
Performed with 5 equiv. H2O.
Performed at 0 °C.
Performed with 15 equiv. H2O.
Performed with 1.0 equiv. amine and 1.5 equiv. arene at 0 °C.
Performed with 40 equiv. H2O.
Performed with 20 equiv. H2O.
Mechanism
We have sought to obtain evidence for the proposed homolytic aromatic substitution mechanism as outlined in Scheme 1. Stern–Volmer fluorescence quenching studies using the preferred Ir(ppy)2(dtbbpy)PF6 photocatalyst revealed significant emission quenching by N-phenylpyrrolidine, yet no change in the emission was observed using a variety of chloroheteroarene substrates. This finding indicates that a reductive quenching cycle is operative (wherein the excited state of the catalyst first performs amine oxidation). To determine if the neutral α-amino radical 1 undergoes addition to the ground state heteroarene 3 (as proposed in Scheme 1) or if the chloroarene undergoes reduction via electron transfer with the transient IrII(ppy)2(dtbbpy) species (as observed in our cyanoarene studies),14 the reduction potentials of several chloroheteroarene substrates were measured by cyclic voltammetry. These potentials were found to range from −1.70 V to −2.19 V vs. SCE (see ESI†). More specifically, 2-chlorobenzoxazole was found to undergo reduction at −2.19 V vs. SCE, indicating that electron transfer from the IrII(ppy)2(dtbbpy) species is highly endothermic and suggesting that a radical anion-neutral radical coupling mechanism is not operative.30,31
Conclusions
In conclusion, we have leveraged photoredox catalysis to achieve the α-heteroarylation of tertiary amines with a diverse array of five-and six-membered heterocycles via a putative homolytic aromatic substitution pathway. Given the prevalence of heterocycles in medicinal agents, as well as the commercial availability of a large number of chloroheteroarenes, we expect that this reaction will find immediate application in the synthesis of biologically active compounds.
Supplementary Material
Acknowledgments
Financial support was provided by the NIHGMS (R01 01 GM093213-01), Merck and Amgen. C. K. P. is grateful for a Bristol-Myers Squibb graduate fellowship. We thank Yuan Hu for generous assistance with cyclic voltammetry.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, and spectral data for all new compounds are provided (97 pages) (PDF). See DOI: 10.1039/c4sc02155j
Notes and references
- 1.Ritchie TJ, Macdonald SJF, Peace S, Pickett SD, Luscombe CN. Med Chem Comm. 2012;3:1062. [Google Scholar]
- 2.McGrath NA, Brichacek M, Njardarson JT. J Chem Educ. 2010;87:1348. [Google Scholar]
- 3.(a) Smith MB, March J. Advanced Organic Chemistry. 5. Wiley-Interscience; New York: 2001. p. 850. [Google Scholar]; (b) Joule JA, Mills K. Heterocyclic Chemistry. 5. Wiley; Chichester: 2010. [Google Scholar]; (c) Bunnet JF, Zahler RE. Chem Rev. 1951;49:273. [Google Scholar]; (d) Bunnett JF. J Chem Educ. 1974;51:312. [Google Scholar]; (e) Walsh K, Sneddon HF, Moody CJ. Chem Sus Chem. 2013;6:1455. doi: 10.1002/cssc.201300239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805. [Google Scholar]; (b) Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hartwig JF. Angew Chem, Int Ed. 1998;37:2046. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (d) Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]
- 5.(a) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; (b) Carey JS, Laffan D, Thomson C, Williams MT. Org Biomol Chem. 2006;4:2337. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- 6.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, Frost DJ. Clin Cancer Res. 2007;13:2728. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 7.Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W. J Med Chem. 2002;45:1757. doi: 10.1021/jm0109513. [DOI] [PubMed] [Google Scholar]
- 8.Reuveni M. Eur J Plant Pathol. 2003;109:243. [Google Scholar]
- 9.(a) Dieter RK, Li S. Tetrahedron Lett. 1995;36:3613. [Google Scholar]; (b) Dieter RK, Li S. J Org Chem. 1997;62:7726. [Google Scholar]
- 10.(a) Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C. J Am Chem Soc. 2006;128:3538. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]; (b) Klapars A, Campos KR, Waldman JH, Zewge D, Dormer PG, Chen C. J Org Chem. 2008;73:4986. doi: 10.1021/jo8006804. [DOI] [PubMed] [Google Scholar]; (c) Barker G, O’Brien P, Campos KR. Org Lett. 2010;12:4176. doi: 10.1021/ol1017799. [DOI] [PubMed] [Google Scholar]; (d) Barker G, McGrath JL, Klapars A, Stead D, Zhou G, Campos KR, O’Brien P. J Org Chem. 2011;76:5936. doi: 10.1021/jo2011347. [DOI] [PubMed] [Google Scholar]
- 11.Yoshikai N, Mieczkowski A, Matsumoto A, Ilies L, Nakamura E. J Am Chem Soc. 2010;132:5568. doi: 10.1021/ja100651t. [DOI] [PubMed] [Google Scholar]
- 12.For reviews on amine α-functionalization:; (a) Campos KR. Chem Soc Rev. 2007;36:1069. doi: 10.1039/b607547a. [DOI] [PubMed] [Google Scholar]; (b) Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW. Chem–Eur J. 2012;18:10092. doi: 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]
- 13.(a) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (d) Teplý F. Collect Czech Chem Commun. 2011;76:859. [Google Scholar]
- 14.(a) McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]; A subsequent variant was reported using trialkylamines and aryl halides:; (b) Singh A, Arora A, Weaver JD. Org Lett. 2013;15:5390. doi: 10.1021/ol402751j. [DOI] [PubMed] [Google Scholar]
- 15.Renaud P, Giraud L. Synthesis. 1996:913. [Google Scholar]
- 16.(a) Minisci F. Synthesis. 1973:1. [Google Scholar]; (b) Minisci F. Top Curr Chem. 1976;62:1. doi: 10.1007/BFb0046046. [DOI] [PubMed] [Google Scholar]; (c) Minisci F, Vismara E, Fontana F. Heterocycles. 1989;28:489. [Google Scholar]; (d) Minisci F, Fontana F, Vismara E. J Heterocycl Chem. 1990;27:79. [Google Scholar]; (e) Duncton MAJ. MedChemComm. 2011;2:1135. [Google Scholar]
- 17.(a) Vaillard SE, Schulte B, Studer A. In: Radical-Based Arylation Methods in Modern Arylation Methods. Ackermann L, editor. Wiley-VCH; Weinheim: 2009. pp. 475–511. [Google Scholar]; (b) Studer A, Bossart M. In: Homolytic Aromatic Substitutions in Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 62–80. [Google Scholar]; (c) Bowman WR, Storey JMD. Chem Soc Rev. 2007;36:1803. doi: 10.1039/b605183a. [DOI] [PubMed] [Google Scholar]; (d) Traynham JG. Chem Rev. 1979;79:323. [Google Scholar]
- 18.For examples of homolytic aromatic substitution reactions employing photoredox catalysis:; (a) Cano-Yelo H, Deronzier A. J Chem Soc, Perkin Trans. 1984;2:1093. [Google Scholar]; (b) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]; (c) Nagib DA, MacMillan DWC. Nature. 2011;480:224. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hari DP, Schroll P, König B. J Am Chem Soc. 2012;134:2958. doi: 10.1021/ja212099r. [DOI] [PubMed] [Google Scholar]
- 19.For other relevant examples of homolytic aromatic substitution:; (a) Fiorentino M, Testaferrri L, Tiecco M, Troisi L. J Chem Soc, Chem Commun. 1977:316. [Google Scholar]; (b) Cowden CJ. Org Lett. 2003;5:4497. doi: 10.1021/ol035814+. [DOI] [PubMed] [Google Scholar]; (c) Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herlé B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Slinker JD, Gorodetsky AA, Lowry MS, Wang J, Parker S, Rohl R, Bernhard S, Malliaras GG. J Am Chem Soc. 2004;126:2763. doi: 10.1021/ja0345221. [DOI] [PubMed] [Google Scholar]; (b) Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712. [Google Scholar]
- 21.Seo ET, Nelson RF, Fritsch JM, Marcoux LS, Leedy DW, Adams RN. J Am Chem Soc. 1966;88:3498. [Google Scholar]
- 22.A pKa of 18.8 has been calculated for the radical cation of N,N-dimethylaniline in acetonitrile:; Dombrowski GW, Dinnocenzo JP, Zielinski PA, Farid S, Wosinska ZM, Gould IR. J Org Chem. 2005;70:3791. doi: 10.1021/jo047813g. [DOI] [PubMed] [Google Scholar]; The acetate base employed in this work may perform this deprotonation in organic solvents; the pKa of acetic acid in acetonitrile has been determined to be 23.5:; Kütt A, Leito I, Kaljurand I, Sooväli L, Vlasov VM, Yagupolskii LM, Koppel IA. J Org Chem. 2006;71:2829. doi: 10.1021/jo060031y. [DOI] [PubMed] [Google Scholar]
- 23.(a) Lilie J, Beck G, Henglein A. Ber Bunsenges Physik Chem. 1971;75:458. [Google Scholar]; (b) Grätzel M, Henglein A, Lilie J, Scheffler M. Ber Bunsenges Physik Chem. 1972;76:67. [Google Scholar]
- 24.While a cyclohexadienyl radical is not a perfect approximation for the heterocyclic dienyl radical 4, we anticipate that the heterocyclic radical will undergo reduction even more readily.
- 25.For more data on the effect of water, see ESI.† For another example of the beneficial role of water in single-electron transfer chemistry, see:; Hoshikawa T, Inoue M. Chem Sci. 2013;4:3118. [Google Scholar]
- 26.Reactivity in the absence of photocatalyst may involve electron transfer from excited electron donor-acceptor complexes, see:; (a) Foster R. J Phys Chem. 1980;84:2135. [Google Scholar]; (b) Arceo E, Jurberg ID, Álvarez-Fernández A, Melchiorre P. Nat Chem. 2013;5:750. doi: 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]
- 27.While we have previously demonstrated the installation of pyridine rings using the corresponding cyanoarenes (ref. 14), our efforts to couple more electron-deficient heteroaryl nitriles (such as cyanopyrimidines)have thus far been unsuccessful.
- 28.See ESI† for a proposed model for the observed regioselectivity.
- 29.We postulate that the superior reactivity of pyrrolidines arises from the ability of this ring system to accommodate conjugation between the α-amino SOMO and the nitrogen lone pair without incurring destabilizing eclipsing interactions. See:; (a) Wayner DDM, Clark KB, Rauk A, Yu D, Armstrong DA. J Am Chem Soc. 1997;119:8925. [Google Scholar]; (b) Griller D, Howard JA, Marriott PR, Scaiano JC. J Am Chem Soc. 1981;103:619. [Google Scholar]
- 30.Stern-Volmer quenching experiments were also performed using fac-Ir(ppy)3, a complex which in its photoexcited state ( vs. SCE)is slightly more reducing than IrII(ppy)2(dtbbpy)( vs. SCE). Minimal quenching (kqτ0 < 2 M−1)was observed in the presence of pyrimidine and purine substrates, and no quenching was detected in the presence of 2-chlorobenzoxazole (see ESI†).
- 31.It has furthermore been demonstrated that the reduction of chloroheteroarenes to the corresponding radical anions typically results in halide fragmentation to generate aryl radicals. For examples of this reactivity, see:; Carver DR, Komin AP, Hubbard JS, Wolfe JF. J Org Chem. 1981;46:294. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.