

Abstract
The use of 4-acetoxy-2,2-dimethylbutanoyl protecting group for the C2-hydroxyl allows the selective formation of β-glycosides without producing α-glycosides. This very bulky protecting group can be removed under mild conditions.
Keywords: β-Glycoside formation, Carbohydrate protecting groups, β-Glucan
Methodology for the synthesis of complex oligosaccharides has progressed remarkably during the past decade primarily due to the discovery of new anomeric activation procedures and coupling protocols.1 In spite of this progress, the successful linking of two saccharide units can still be thwarted by unexpected difficulties. An example of this is the recent disclosure by Kong and co-workers that, in certain cases, α-linked products are unexpectedly obtained in glycosylations using trichloroacetimidate activated donors with a C2 ester capable of neighboring group participation.2
The stereoselective preparation of β-glycosidic linkages are most reliably formed using a 2-carboxyprotected (aka ‘disarmed’3) glycosyl donor (A).4 Neighboring group participation of the 2-carboxy group assists the departure of the leaving group (to give B) and nucleophilic attack of the glycosyl acceptor affords the disaccharide (C). A problem sometimes encountered during the preparation of β-glycosides using this approach is the production of orthoesters (D) instead of the desired glycoside (Scheme 1).5 The orthoesters complication has not been a major concern since there are numerous reports that the orthoesters can easily be converted into the β-glycoside.6 The reliability of the stereochemical outcome from coupling a disarmed glycosyl donor with a glycosyl acceptor has been called into question by Kong and co-workers work.2 Although the exact mechanism of α-glycoside formation is unknown, Kong and co-workers proposed that the orthoesters is an intermediate in the formation of the α-glycoside. While Kong and co-workers showed the utility of this process for the preparation of 1,3-glucans with mixed α- and β-link-ages,2 the unexpected formation of the α–linkage is a major impediment to the preparation of β-1,3-glucans, an important class of immune stimulators.7
Scheme 1.

Recently, during the preparation of a small library of β-1,3-polyglycosides (β-1,3-glucans), we encountered a similar problem with a lack of stereoselectivity in glycosylations under conditions where β-selectivity was expected. As shown in Table 1, the reaction of various donors (1a–f) with 1,2,4,6-tetra-O-acetyl-α-d-glycopyranose 2 or the β-linked disaccharide 3 afforded disaccharide or trisaccharide products whose newly formed glycosidic linkage ranged from β-only to α-only.
Table 1.
Glycosylation using glycosyl donors with various ester-protecting groups at C2
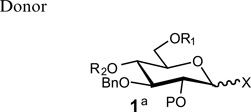 |
Acceptors | |
|---|---|---|
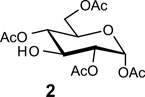 |
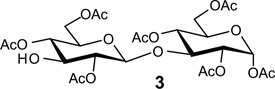 |
|
| 1a P=Ac X= O-trichloroacetimidateb | 84% (α:β 1:1) | NA |
| 1b P=Bz X= O-trichloroacetimidateb | NA | 86% (α only) |
| 1c P = ClCH2CO X = O-trichloroacetimidateb | NA | 85% (α only) |
| 1d P=CH3OCO X = O-trichloroacetimidateb | 85% (α:β 1:1) | 83% (α only) |
| 1e P = (CH3)3CCO X = O-trichloroacetimidateb | 80% (α only) | 81% (α:β 2:3) |
| 1f P = (CH3)3CCO X = SPhc | NA | 82% (α:β 1:3) |
Compound 1a–e, R1 = Ac, R2 = Ac; compound 1f, R1:R2 = CHPh.
Activation with TMSOTf.
Activation with NIS/AgOTf.
As shown in Table 1, formation of the unexpected α-linkage occurs both for trichloroacetimidate activation and for thioglycoside mediated couplings. The use of pivaloate esters at C2 is clearly beneficial to the formation of the β-glycoside but does not completely inhibit formation of the α-glycoside. In spite of the stereochemical benefits of the C2-Piv group we were concerned that the harsher conditions required for the deprotection of multiple pivaloate esters (at sterically hindered secondary centers) would be a limitation in the production of 1,3-β-glucans. Therefore we set out to prepare a protecting group with the steric advantages of the bulky pivaloyl group but which could be removed under mild conditions.
There were two reports in the literature where a similar problem has been faced and overcome. Crimmins et al.8 had utilized the 2,2-dimethyl-4-pentenoyl protecting group which could be removed by hydroboration, oxidation, and mild base treatment and Trost and Hembre9 had used the 4-tert-butyldimethylsilyloxy-2,2-dimethylbutanoyl protecting group which could be removed on treatment with fluoride anion. The additional steps required in the Crimmins approach made it less attractive and since we wanted to reserve the use of TBDMS for the C-3 hydroxyl group protection, Trost’s approach could not be used. The 4-acetoxy-2,2-dimethylbutanoyl group appeared to be a likely candidate. It was anticipated that base catalyzed hydrolysis of the 4-acetoxy group would initiate an intramolecular lactonization and loss of dimethylbutyrolactone. The preparation of 4-acetoxy-2,2-dimethylbutanoic acid and the corresponding acid chloride has been described10 but its potential utility as a protecting group has not been explored.
The 4-acetoxy-2,2-dimethylbutanoyl (ADMB) esters are easily prepared under the same conditions used for pivaloate esters and shows similar selectivity in carbohydrate acylations. Jiang and Chan has reported the selective pivaloylation of a number of mono- and disaccharides using pivaloyl chloride.11 Similarly, under the same conditions, we have found that treatment of methyl 4,6-benzylidene-α-d-glucoside and ethyl 4,6-benzylidene-1-thio-α-d-glucoside with 4-acet oxy-2,2-dimethylbutanoyl chloride permits selective acylation of the C2 hydroxyl group in 95% and 94% yield, respectively. The ease of removal of the ADMB ester group on treatment with catalytic quantity of diazabicycloundecane (DBU) in methanol at room temperature is demonstrated in Table 2.12
Table 2.
Comparison of hydrolysis of pivaloyl and 4-acetoxy-2,2-dimethylbutanoyl esters (ADMB)
| Substrate | Equiv of DBU | Time (h) | ROH yield (%) | ||
|---|---|---|---|---|---|
| 4 | 4a P = Piv | 0.5 | 24 | 0 | |
| 4b P = ADMB | 0.5 | 0.5 | 100 | ||
| 4b P = ADMB | 0.1 | 2 | 100 | ||
| 5 | 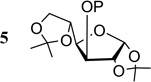 |
5a P = Piv | 0.5 | 10 | 12 |
| 5b P = ADMB | 0.5 | 1 | 100 | ||
| 5b P = ADMB | 0.1 | 3 | 100 | ||
| 6 | 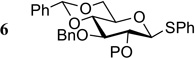 |
6a P = Piv | 0.5 | 24 | 0 |
| 6b P = ADMB | 0.1 | 3 | 98 | ||
| 7 | 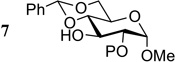 |
7a P = Piv | 0.5 | 2 | 30 |
| 7a P = Piv | 0.5 | 12 | 97 | ||
| 7b P = ADMB | 0.5 | 1 | 99 | ||
We had anticipated that the facile removal of the C2-ADMB group would be the only difference between it and the pivaloyl group. However, we were gratified to find that the problem of α-glycoside formation was eliminated when using the C2-ADMB group in carbohydrate coupling reactions. The couplings afford good yields (80–92%) of β-glycosides. For example, the coupling of glycoside 8, with disaccharide 3 gave an 83% yield of the all β-linked trisaccharide 9 as the only product (Table 3, entry 1) (comparing with the last entry in Table 1).
Table 3.
Glycosylation using glycosyl donors with C2 4-acetoxy-2,2-dimethylbutanoate protecting group
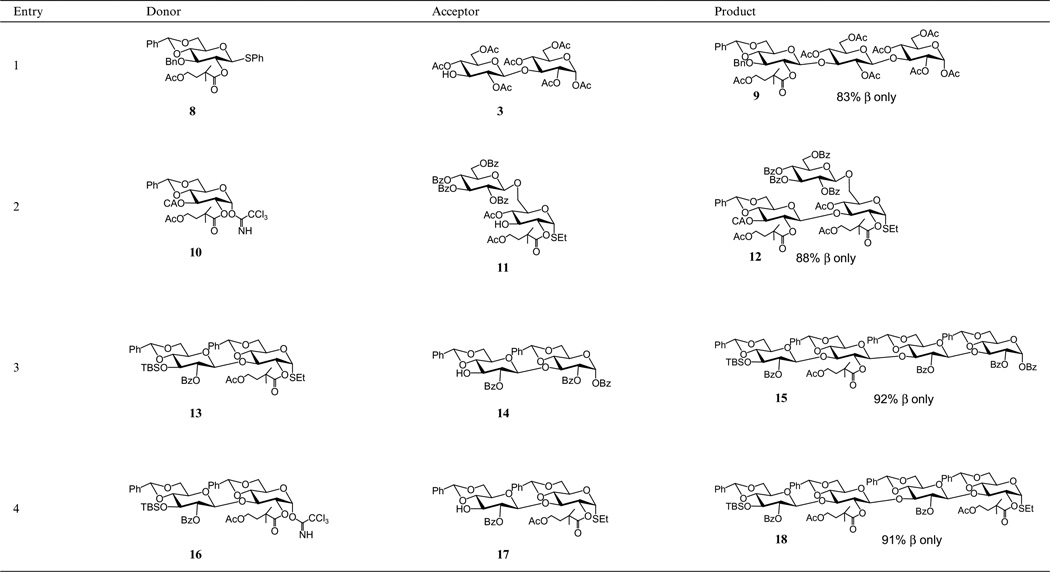 |
Other examples showing the glycosyl trichloroacetimidate and thioglycoside mediated couplings are given in Table 3. High yields of β-glycosides are obtained in all of the cases we have examined. The structures of the oligosaccharides are evident from their NMR spectra. The anomeric protons of oligosaccharide differ significantly with the α- and β-anomers showing proton coupling constants of ~3 and ~8 Hz, respectively. 13C NMR spectra were recorded giving the C-1 α in the δ 90–93 ppm range and the C-1 β in the δ 96–104 ppm range.13
Although it is unclear why the ADMB ester is beneficial to β-glycoside formation, if, as Kong and co-workers proposed,2 the orthoesters is an intermediate in the formation of the α-glycoside, the presence of the ADMB group could stabilize the dioxolenium ion and sterically prevent orthoesters formation (Scheme 2). Irrespective of the mechanism-of-action, the use of the ADMB group makes possible the selective formation of pure (1→3)-β glucan polymers. This may lead to the development of synthetic glucan ligands for biomedical applications.
Scheme 2.

References and notes
- 1.(a) Ernst B, Hart GW, Sinay P, editors. Carbohydrates in Chemistry and Biology. Vol. 1. Weinheim, Germany: Wiley-VCH; 2000. [Google Scholar]; (b) Hanessian S, editor. Preparative Carbohydrate Chemistry. New York: Marcel Dekker; 1997. [Google Scholar]
- 2.(a) Zeng Y, Ning J, Kong F. Tetrahedron Lett. 2000;43:3729. [Google Scholar]; (b) Zeng Y, Ning J, Kong F. Carbohydr. Res. 2003;338:307. doi: 10.1016/s0008-6215(02)00455-x. [DOI] [PubMed] [Google Scholar]
- 3.Paulsen H, Richter A, Sinnwell V, Stenzell W. Carbohydr. Res. 1978;64:339–362. [Google Scholar]; (b) Fraser Reid B, Udodong UE, Wu Z, Ottesson H, Merritt JR, Rao CS, Roberts C, Madsen R. Synlett. 1992:927–942. [Google Scholar]
- 4.Schmidt RR, Kinzy W. Adv. Carbohydr. Chem. Biochem. 1994;50:21–123. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Seeberger PH, Eckhardt M, Gutteridge CE, Danishefsky SJ. J. Am. Chem. Soc. 1997;119:10064–10072. [Google Scholar]; (b) Khan SH, O’Neil RA, editors. Modern Methods in Carbohydrate Synthesis. United States: Harwood Academic; 1996. p. 125. [Google Scholar]
- 6.(a) Wang W, Kong F. J. Org. Chem. 1998;63:5744. doi: 10.1021/jo981135e. [DOI] [PubMed] [Google Scholar]; (b) Sznaidman ML, Johnson SC, Crasto C, Hecht SM. J. Org. Chem. 1995;60:3942. [Google Scholar]; (c) Gass J, Strobl M, Kosma P. Carbohydr. Res. 1993;244:69. doi: 10.1016/0008-6215(93)80005-y. [DOI] [PubMed] [Google Scholar]
- 7.Brown GD, Gordon S. Nature. 2001;413:36. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 8.Crimmins MT, Carroll CA, Wells AJ. Tetrahedron Lett. 1998;39:7005–7008. [Google Scholar]
- 9.Trost BM, Hembre EJ. Tetrahedron Lett. 1999;40:219–222. [Google Scholar]
- 10. Hoffman WF. Monocyclic lactone derivatives of mevinolin and compactin as hydroxymethylglutaryl coenzyme A reductase inhibitors. 4665091. U.S. Patent. 1987 May 12;:96514. Briefly, 2-methylbutyrolactone is treated with LDA and then CH3I to afford 2,2-dimethylbutyrolactone. After distillation, the lactone is treated with 1 equiv of 1 N NaOH in methanol and stirred overnight at rt. The solution is concentrated on a rotary evaporator and dried azeotropically using toluene (temp <50 °C). The dried sodium salt is added to pyridine containing a catalytic amount of DMAP, cooled to 0 °C and 2 equiv of acetic anhydride was added dropwise. After stirring overnight and extraction a mixture of recyclized 2,2-dimethylbutyrolactone and 4-acetoxy-2,2-dimethylbutyric acid is obtained. The acid is separated by base extraction followed by careful acidification. Treatment with oxalyl chloride in benzene affords 4-acetoxy-2,2-dimethylbutanoyl chloride.
- 11.Jiang L, Chan TH. J. Org. Chem. 1998;63:6035–6038. doi: 10.1021/jo980294v. [DOI] [PubMed] [Google Scholar]
- 12.Typical procedure for the deprotection of 4-acetoxy-2,2-dimethylbutanoyl protecting group. To a solution of 6b (250 mg, 0.41 mmol) in CH2Cl2–MeOH (6 mL, 1:1) at room temperature was added 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) (7 µL, 0.04 mmol). After being stirred for 3 h, the solution was concentrated to a residue, which was purified by a silica gel column chromatography to give the alcohol (183 mg, 98%).
- 13.1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data for compounds 12, 15, and 18. Compound 12: 1H NMR δ 7.94–7.19 (m, 25H), 5.80 (t, J = 9.6 Hz, 1H, H-3″), 5.60 (t, J = 9.6 Hz, 1H, H-4″), 5.44 (dd, J = 9.6 Hz and 8.0 Hz, 1H, H-2′), 5.42 (s, 1H), 5.36 (d, J = 5.6 Hz, 1H, H-1), 5.20 (t, J = 9.2 Hz, 1H, H-2″), 4.82–4.74 (m, 4H, H-1, H-1′, H-2, H-3′), 4.65 (t, J = 10.0 Hz, 1H), 4.57 (dd, J = 12.4 Hz and 3.2 Hz, 1H), 4.42–4.29 (m, 3H), 4.11–3.90 (m, 9H), 3.68–3.44 (m, 4H), 2.20–2.09 (m, 2H), 1.92–1.83 (m, 2H), 1.96 (s, 3H), 1.93 (s, 3H), 1.89 (s, 3H), 1.75–1.69 (m, 2H), 1.21 (s, 3H), 1.18 (s, 3H), 1.05 (s, 6H), 0.89 (t, J = 7.6 Hz, 3H); 13C NMR δ 175.9, 175.7, 171.2, 171.1, 169.6, 166.8, 166.3, 160.0, 165.4, 165.3, 136.6, 133.7, 133.6, 133.5, 133.4, 130.1, 130.0, 129.7, 129.6, 129.4, 128.9, 128.8, 128.7, 128.6, 128.6, 128.5, 128.5, 126.3, 101.9, 101.3, 100.1, 79.7, 78.5, 75.1, 74.1, 73.5, 73.1, 72.5, 72.3, 71.9, 69.7, 69.1, 68.7, 68.5, 66.6, 63.2, 61.5, 61.1, 41.4, 41.0, 40.8, 38.3, 38.1, 25.7, 25.6, 25.3, 24.9, 23.2, 21.2, 21.1, 21.0, 14.3.