

Abstract
4-[18F]fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG, [18F]1) is a new cardiac sympathetic nerve radiotracer with kinetic properties favorable for quantifying regional nerve density with PET and tracer kinetic analysis. An automated synthesis of [18F]1 was developed in which the intermediate 4-[18F]fluoro-m-tyramine ([18F]16) was prepared using a diaryliodonium salt precursor for nucleophilic aromatic [18F]fluorination. In PET imaging studies in rhesus macaque monkeys, [18F]1 demonstrated high quality cardiac images with low uptake in lungs and liver. Compartmental modeling of [18F]1 kinetics provided ‘net uptake rate’ constants Ki (mL/min/g wet) and Patlak graphical analysis of [18F]1 kinetics provided Patlak slopes Kp (mL/min/g). In pharmacological blocking studies with the norepinephrine transporter inhibitor desipramine (DMI), each of these quantitative measures declined in a dose-dependent manner with increasing DMI doses. These initial results strongly suggest that [18F]1 can provide quantitative measures of regional cardiac sympathetic nerve density in human hearts using PET.
Introduction
The autonomic nervous system plays an important role in governing cardiac performance. The sympathetic and parasympathetic branches of the autonomic system work in opposition to each other in the heart, modulating cardiac output to meet the oxygen demands of organs and tissues in the body. Stimulation of the sympathetic branch (which uses norepinephrine as the neurotransmitter) increases cardiac output by increasing heart rate and myocardial contractility. Stimulation of the parasympathetic branch (which uses acetylcholine as the neurotransmitter) slows heart rate and decreases contractility, decreasing cardiac output. Cardiac autonomic dysfunction is known to occur in many cardiac diseases, including sudden cardiac death,1 heart failure,2 diabetic autonomic neuropathy3 and cardiac arrhythmias.4 With the goal of increasing our understanding of the underlying mechanisms of autonomic dysfunction and its contribution to the manifestation and progression of cardiac diseases, several radiopharmaceuticals have previously been developed for noninvasive imaging of presynaptic sympathetic nerve terminals in the heart.5 Examples include [123I]m-iodobenzylguanidine ([123I]MIBG) for single-photon scintigraphic imaging using gamma cameras or SPECT systems and [11C]-(−)-m-hydroxyephedrine ([11C]HED) for PET (Figure 1).6
Figure 1.
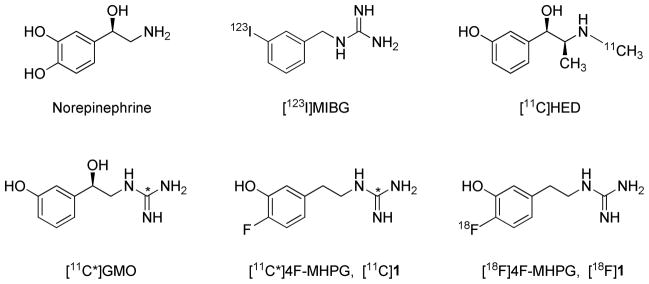
Structures of the endogenous neurotransmitter norepinephrine and radiotracers for imaging cardiac sympathetic innervation.
These tracers are actively transported into presynaptic sympathetic nerve terminals by the norepinephrine transporter (NET).6 Once inside neurons, they are taken up into norepinephrine storage vesicles by the second isoform of the vesicular monoamine transporter (VMAT2).7 The neuronal uptake rates of [123I]MIBG and [11C]HED are very rapid, which results in high quality heart images due to high uptake of radioactivity in the neurons. However, the rapid NET transport rates of [123I]MIBG and [11C]HED also causes them to be ‘flow-limited’ tracers.8 A flow-limited tracer exhibits very rapid uptake into tissue spaces after its initial extraction from plasma into extracellular spaces. Extraction from plasma is equal to E·F, where E is the ‘unidirectional extraction fraction’ of the tracer (often close to 1.0) and F is blood flow. Since delivery of the tracer into extracellular spaces by blood flow is the rate-limiting step in the net tissue uptake of a tracer with these kinetic properties, the term ‘flow-limited tracer’ is often used in this case.
The flow-limited neuronal uptake of [123I]MIBG and [11C]HED leads to limitations in the clinical utility of these tracers, in terms of their ability to provide accurate measures of regional cardiac sympathetic nerve density. First, their kinetics cannot be successfully analyzed in a straightforward manner using kinetic analysis techniques, such as compartmental modeling, to provide quantitative measures of regional nerve density. This forces the use of semi-quantitative measures of tracer retention as surrogate measures of nerve density, such as the heart-to-mediastinum ratio (HMR) for [123I]MIBG9 or the ‘retention index’ (RI) value for [11C]HED.6 However, the flow-limited neuronal uptake of these compounds causes their retention measures to be insensitive to substantial nerve losses in regions with mild to moderate levels of cardiac denervation.10 Only when regional nerve losses are relatively severe do measures of tracer retention begin to decline from control levels.
We hypothesized that these obstacles to accurate quantification of cardiac sympathetic nerve density could be overcome by designing a new tracer with specific kinetic properties to minimize flow-limitation effects.8 First, a slower transport rate into neurons by NET was needed, making this the rate-limiting step in the neuronal uptake of the tracer. Second, efficient uptake into norepinephrine storage vesicles by VMAT2 was desired, leading to prolonged neuronal retention times. The kinetics of a tracer with these properties can be analyzed using standard kinetic analysis methods to yield quantitative measures of regional sympathetic nerve density. These measures are much more sensitive to low levels of nerve losses that occur early in the progression of cardiac denervation than the tracer retention measures currently used for existing tracers.
We previously evaluated a series of radiolabeled phenethylguanidines, some of which were found to possess the desired kinetic properties described above.8 N-[11C]guanyl-(−)-m-octopamine ([11C]GMO, Figure 1) was one of the compounds identified to have a slower NET transport rate and a very long neuronal retention time in isolated rat hearts. Tracer kinetic analyses of myocardial [11C]GMO kinetics from PET studies in non-human primates proved to be very successful,11 supporting our hypothesis that tracers with slow NET transport rates and long neuronal retention times offer improved quantitative measures of cardiac sympathetic nerve density.
While our preclinical imaging studies with [11C]GMO have been very promising, using fluorine-18 as the positron-emitting radiolabel offers some advantages over carbon-11. Mainly, the longer half-life of fluorine-18 (T1/2 = 109.8 min vs. 20.4 min for carbon-11) makes it feasible to distribute large batches of the radiopharmaceutical to stand-alone PET centers that do not have a cyclotron and radiochemistry facility. It also allows PET imaging studies of several patients to be performed using a single batch of the radiopharmaceutical. In our studies with 11C-labeled phenethylguanidines, [11C]4-fluoro-m-hydroxyphenethylguanidine ([11C]4F-MHPG, [11C]1, Figure 1) was also found to exhibit a slower NET transport rate and very long neuronal retention times in isolated rat heart studies. MicroPET imaging studies with [11C]1 in non-human primates showed favorable imaging properties and in vivo kinetics. The encouraging results with [11C]1 led us to synthesize the fluorine-18 labeled analog 4-[18F]fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG, [18F]1, Figure 1). Recently, we reported on a multi-step radiosynthesis of [18F]1 which, while successful, only provided limited quantities of the final product.12 Initial bioevaluation studies of [18F]1 provided results similar to those previously seen with [11C]1, encouraging us to pursue first-in-human imaging studies with this new tracer. However, since the initial method of preparing [18F]1 was not practical for routine production, the goal of this study was to develop a new approach to the radiosynthesis of [18F]1.
We describe here a novel automated method for preparing [18F]1 at sufficiently high radiochemical yields and specific activities for clinical PET studies. We also provide results from initial bioevaluations of [18F]1, including isolated rat heart studies, microPET imaging studies in non-human primates and studies of tracer metabolism. Radiation absorbed dose estimates for human PET studies with [18F]1, based on biodistribution studies in rats, were also generated. The successful application of tracer kinetic analysis methodology to the myocardial kinetics of [18F]1 in non-human primates strongly suggests that this radiotracer can be used in humans to provide robust and sensitive quantitative measures of regional sympathetic nerve density using PET.
Results and Discussion
Chemistry
Our goal was the development of a radiosynthetic pathway consisting of an 18F-labeling step followed by one or two straightforward steps to yield [18F]1. Many methods have been developed for the incorporation of fluorine-18 into a deactivated electron-rich aromatic ring by a nucleophilic aromatic substitution reaction.13–16 Recently, the use of diaryliodonium salts has received considerable attention as a one-step method of introducing fluorine-18 into ring structures with high radiochemical yields in electron-rich aromatic systems.17–21 This approach provides specific activities that are sufficiently high for successful PET imaging studies of receptors in various organs.22–25 Although this method has been shown to be suitable for preparing small 18F-labeled molecules with relatively simple structures, difficulties are sometimes encountered when trying to label more complex molecular structures, as evidenced by a lack of consistency in radiochemical yields with more complex diaryliodonium salts precursors.16
We began with a systematic exploration of various diaryliodonium salt precursors to achieve synthesis of [18F]1 by direct no-carrier-added (NCA) nucleophilic aromatic [18F]fluorination in electron-rich aromatic systems. Several diaryliodonium salt precursors for [18F]1 were prepared by selecting from three main structural elements: (a) the choice of asymmetric diaryliodonium or arylheteroaryliodonium, (b) the choice of the protecting groups for the phenol and guanidine moieties, and (c) the choice of different counter anions to influence the 18F-labeling yield. It was previously reported that the electron-rich 2-thienyl group in arylheteroaryliodonium provides a regiospecific single radioactive product with high radiochemical yields (20–61%) due to the variation of the electron density, especially in diaryliodonium derivatives with an ortho-methoxy substituent.26 On the basis of these results, we prepared 2-benzyloxy-4-{2′-(N,N′-bis(tert-butyloxycarbonyl)-N″-guanidinyl}ethylphenyl(2-thienyl)iodonium tosylate (9) as a precursor for preparing [18F]1, as shown in Scheme 1. The starting material 3-hydroxy-4-iodobenzyl alcohol (2) was protected with a benzyl group, followed by bromination with phosphorous tribromide (PBr3) to afford the benzylbromide 4. 3-Benzyloxy-4-iodophenethylamine hydrochloride (6) was prepared by cyanation of 4 and reduction with 1.0 M BH3-THF complex followed by acidification with hydrogen chloride, sequentially. Condensation of 6 with 1,3-bis(tertbutoxycarbonyl)-2-methyl-2-thiopseudourea afforded the di-N,N′-Boc-protected iodophenethylguanidine 7. Reaction of the iodo-compound 7 with bis(trimethyl)tin in the presence of a catalytic amount of tetrakis(triphenylphosphine)palladium provided the trimethylstannane compound 8. [2-hydroxy(tosyloxy)iodo]thiophene,27 which was generated in situ by a mixture of 2-(diacetoxyiodo)thiophene28 with para-toluenesulfonic acid under nitrogen atmosphere, was reacted with trimethylstannane 8 to give (2-thienyl)iodonium tosylate 9 in 69% yield as a yellow solid. Different counter anions of phenyl(2-thienyl)iodonium salts were tested to determine their effect on 18F-radiofluorination.26 Among various counter anions, in some cases bromide has been found to be a preferred anion, increasing the radiochemical yield.29 Therefore, we converted di-N,N′-Boc protected (2-thienyl)iodonium tosylate 9 into a corresponding bromide 10 in 89% yield by a metathesis reaction using KBr in a mixed solution of MeCN and H2O. The structure of precursor 10 and its counter anion was confirmed by the absence of proton signals for tosylate in the 1H NMR spectra.
Scheme 1.

a) BnCl, K2CO3, DMF, 1 h, 130 °C, 92%; b) PBr3, CH2Cl2, rt, 16 h, 81%; c) NaCN, DMF, rt, 2 h, 85%; d) 1.0 M BH3-THF complex, THF, reflux, 2 h, 69%; e) 1,3-N,N′-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, DMF, 0 °C-rt, 24 h, 78%; f) Sn2Me6, Pd(PPh3)4, toluene, reflux, N2, 30 min, 46%; g) i) 2-(Diacetoxyiodo)thiophene, p-TsOH·H2O, MeCN, CH2Cl2, N2, rt, 1 h; ii) 8, MeCN, CH2Cl2, N2, rt, 20 h, 69%; h) KBr, MeCN, H2O, 60 °C-rt, 1 h, 89%.
Radiochemistry
We attempted 18F-radiofluorination into the di-N,N′-Boc-protected guanidine diaryliodonium salt precursors 9 and 10. Unfortunately, incorporation of [18F]fluoride ion was never observed under a variety of reaction conditions. For example, different sources of [18F]F− were investigated, including K[18F]F-Kryptofix® (K[18F]F-K222), tetrabutylammonium [18F]fluoride ([18F]TBAF) and Cs[18F]F. Conventional heating and microwave heating were tested. Varying the reaction temperature in different solvents such as DMF and MeCN, with or without H2O and/or a radical scavenger (2,2,6,6-tetramethylpiperidine-N-oxyl, TEMPO) did not produce any [18F]fluoride ion incorporation. Diaryliodonium salts under the quite harsh basic conditions of radiofluorination may decompose in many ways including photoinitiated free radical pathways.30 Also, [18F]fluorination can be extremely sensitive to the structure of the diaryliodonium salt, particularly in the case of electron-rich salts. Exploring possible reasons for this disappointing outcome, we hypothesized that the guanidinyl group might be causing a disturbance in the [18F]fluorination reaction. To test this hypothesis, we studied [18F]fluorination of some related iodonium salts, specifically the m-tyramines 13 and 14 (Scheme 2) in which an aminoethyl group replaced the guanidinylethyl group in the target compound.
Scheme 2.
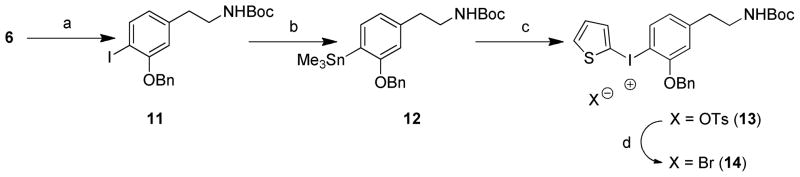
a) (Boc)2O, Et3N, DMF, 130 °C, 1 h, 78%; b) Sn2Me6, Pd(PPh3)4, toluene, reflux, N2, 30 min, 81%; c) i) 2-(Diacetoxyiodo)thiophene, p-TsOH·H2O, MeCN, CH2Cl2, N2, rt, 1 h; ii) 12, MeCN, CH2Cl2, N2, rt, 18 h, 94%; d) KBr, MeCN, H2O, 60 °C-rt, 1 h, 71%
We prepared N-Boc protected aminoethylphenyl(2-thienyl)iodonium salts 13 and 14 (Scheme 2) by adapting the procedure shown in Scheme 1. Radiofluorination of 14 was carried out under various conditions, including different [18F]fluoride ion sources and reaction solvents. The reaction was tested using both a manual synthesis and an automated production using a TRACERlab FXFN system (GE Medical Systems) to produce N-Boc-3-benzyloxy-4-[18F]fluoro-m-tyramine, [18F]15 (Scheme 3). The reaction of K[18F]F-K222 or [18F]TBAF with precursor 14 in anhydrous DMF or MeCN (exposed to ambient room light) afforded 18F-radiofluorinated product [18F]15 in low yields (2–6%). When it was reacted with Cs[18F]F under general anhydrous radiofluorination conditions, the yield of [18F]15 was again low (~4%). We guessed that these low radiochemical yields might be due to the low solubility of Cs[18F]F in the reaction solvent (DMF or MeCN). Addition of TEMPO (1.0 mg) and some water (10 μl) to the solvent (DMF, 500 μl) significantly increased the radiochemical yields to ~45% for manual syntheses.
Scheme 3.

This encouraging result led us to design the radiosynthesis of the target [18F]1 through [18F]15. We were able to achieve a fully automated radiosynthesis of [18F]1 using two GE TRACERlab FXFN radiosynthesis systems in adjacent hot-cells, linked in series. In the first FXFN system, the diaryliodonium salt precursor 14 was reacted with Cs[18F]F to prepare the product [18F]15 (Scheme 3). Removal of the N-Boc protecting group in [18F]15 was achieved by treatment with 1.0 N HCl at 100 °C for 10 min to yield the 4-[18F]fluoro-m-tyramine intermediate [18F]16. This reaction mixture was passed through a C-18 Sep-Pak, which was eluted with 1.0 mL of ethanol to transfer pure [18F]16 directly into the reaction vial of the second FXFN system. To convert the tyramine intermediate [18F]16 into the desired guanidine [18F]1 (Scheme 3), several guanylating reagents were evaluated, including cyanamide, S-alkylisothiouronium salts, aminoiminomethanesulfonic acid, 1H-pyrazole-1-carboxamidine hydrochloride and benzotriazole-1-carboxamidinium tosylate. N,N′-bis-(tert-butoxycarbonyl)-5-chloro-1H-benzotriazole-1-carboxamidine (17) proved to be most effective for the guanylation of [18F]16.31 The reaction of [18F]16 with 17 was carried out in the second FXFN system with DIEA in MeCN at 45 °C for 15 min. Radiochemical yields were >95% as assessed by radio-TLC. Simultaneous cleavage of both of the benzyl ether and N,N′-di-Boc protecting groups in the product [18F]18 with 1.0 N HBr at 120 °C for 15 min followed by HPLC purification provided the desired compound [18F]1. The structure of the product was confirmed by comparing its retention time against a cold standard of 1 using reverse-phase HPLC. The total synthesis time of the fully automated process is ~ 150 min. Final yields of 55–125 mCi of [18F]1 product were obtained from 1.4 Ci of [18F]fluoride ion at end of bombardment (EOB), with overall radiochemical yield averaging 7 ± 3% (decay-corrected based on starting activity) and >99% radiochemical purity. Specific activities (SA) were 1.2 ± 0.3 Ci/μmol at end of synthesis (EOS), significantly higher than those obtained with our initial radiolabeling approach.12 However, these specific activities are lower than levels typically achieved in nucleophilic fluorine-18 labeling, partly due to the relatively long synthesis time.
Radiofluorination of the diaryliodonium salt precursor 14 to produce [18F]16 (N-Boc-3-benzyloxy-4-[18F]fluoro-m-tyramine) gave ~20% yield using the automated radiofluorination system. Using a manual process to prepare the radiofluorinated tyramine 15 provided significantly higher yields (~45% yield). Thus, one area of future work will be to optimize conditions in the automated process to increase the incorporation of fluorine-18 into the diaryliodonium salt precursor. Also, we are currently working on establishing the entire synthesis on a dual-reactor GE TRACERlab FX N Pro system, which should improve radiochemical yields and reduce the overall synthesis time.
Kinetic Studies in Isolated Rat Heart
Kinetic studies of [18F]1 were performed in an isolated working rat heart model. The ‘extraneuronal uptake’ pathway into myocytes (‘uptake-2’) was blocked pharmacologically by adding 54 μM corticosterone to all heart perfusates.32 [18F]1 at a very low concentration (total [18F]1 + [19F]1 mass concentration < 10 nM) was infused into the heart for 10 min to measure its uptake rate into cardiac sympathetic neurons (Kup; mL/min/g wet). The heart was then switched to normal heart perfusate for 120 min to measure clearance rates from tissue. The kinetics of [18F]1 are almost identical to those of the carbon-11 analog [11C]1 in the isolated rat heart (Figure 2). For the studies shown, each tracer exhibited a neuronal uptake rate Kup = 0.77 mL/min/g wet, and the major clearance half-times from sympathetic neurons were very long (T1/2 > 24 h).
Figure 2.

Kinetics of [11C]1 and [18F]1 in isolated rat hearts. The radiotracer is infused at very low concentrations into the heart for 10 min to measure the tracer uptake rate into cardiac sympathetic neurons (Kup; mL/min/g wet), then the heart is switched to normal heart perfusate to measure tracer clearance rates over a 2 h period. Note the very long retention times of these tracers due to efficient trapping inside norepinephrine storage vesicles.
Cardiac Imaging Studies of [18F]1 in Monkeys
MicroPET imaging studies with [18F]1 in rhesus macaque monkeys were performed to assess cardiac image quality and tracer kinetics. In control studies (n = 4), the images showed relatively uniform uptake of [18F]1 throughout the left ventricle (Figure 3A). Uptake of the tracer into lung tissue was very low. Also, liver uptake of [18F]1 was relatively low in comparison with levels previously observed for [11C]-(−)-m-hydroxyephedrine and [11C]-(−)-epinephrine in this species, providing improved image contrast between heart and liver. Final heart-to-blood ratios averaged 3.0 ± 0.5 and heart-to-liver ratios were 2.5 ± 0.3. Faint uptake of fluorine-18 was observed in the spinal column on transaxial and sagittal images (data not shown) consistent with a small amount of in vivo defluorination of [18F]1.
Figure 3.

Coronal microPET images of [18F]1 in rhesus macaque monkeys, showing a control study (A) and four studies using desipramine (DMI) to induce varying degrees of pharmacological blockade of cardiac NET. The DMI doses used were 0.010 mg/kg (B), 0.0316 mg/kg (C), 0.10 mg/kg (D) and 1.0 mg/kg (E). For the highest DMI block condition (E), myocardial levels of [18F]1 at the end of the study (80–90 min) are lower than the activity levels in whole blood.
To verify that cardiac uptake of [18F]1 was specific for sympathetic neurons, a series of imaging studies with the potent NET inhibitor desipramine (DMI) were performed. Four doses of DMI were tested: 0.010 mg/kg, 0.0316 mg/kg, 0.10 mg/kg and 1.0 mg/kg (i.v.). Heart retention of [18F]1 progressively decreased with increasing dose levels of DMI (Figures 3B–E). At the highest DMI dose (1.0 mg/kg) there was little uptake of tracer in heart tissue (Figure 3E), demonstrating high selectivity of [18F]1 for presynaptic sympathetic nerve terminals.
Metabolism of [18F]1
During microPET imaging studies, six venous blood samples were drawn from the monkey to assess the metabolic breakdown of [18F]1. Plasma was separated from whole blood and processed for injection onto a reverse-phase HPLC system equipped with a radiation detector (Synergi 10μ Hydro-RP column, 4.6 × 250 mm, 60 mM sodium phosphate buffer, pH 5.4 with 10% ethanol, flow rate 1.0 mL/min). A representative HPLC/radiation detection curve for a blood sample taken 2 min after tracer administration is shown in Figure 4A. In this system, [18F]1 has retention time Rt ~ 11.4 min while the main radiometabolite formed has Rt ~ 8.2 min.
Figure 4.

Radiodetection data from reverse-phase HPLC analysis of [18F]1 and its radiolabeled metabolites. The top curve (A) is data for a plasma sample taken from a rhesus macaque monkey 2 min after administration of [18F]1, which shows a polar metabolite at retention time Rt ~ 8.2 min. The bottom curve (B) is data from an in vitro incubation of [18F]1 with a monkey liver cystosol fraction plus the cofactor for sulfur conjugation, 3′-phospho-adenosine-5′-phosphosulfate (PAPS), at 37 °C for 20 min. The retention time of the sulfur conjugate of [18F]1 is the same as that of the main metabolite observed in the monkey study (top curve), strongly suggesting that sulfur conjugation is the primary metabolic pathway for [18F]1 in rhesus macaques. The structure of the sulfur conjugate of [18F]1 is shown (C).
We believe that the major radiometabolite formed, which is more polar than the parent compound [18F]1, is the sulfur conjugate of [18F]1. We performed in vitro incubations of [18F]1 with a monkey liver cytosol fraction preparation and the sulfur conjugation cofactor 3′-phospho-adenosine-5′-phosphosulfate (PAPS) at 37 °C. After 1 min of incubation, ~70% of the [18F]1 added was sulfur conjugated, and after 20 min of incubation, 100% was in the sulfur conjugated form. The retention time of the sulfur conjugate of [18F]1 produced in vitro was the same as the main radiometabolite observed in monkey plasma (Figure 4B), strongly suggesting that sulfur-conjugated [18F]1 is the metabolite formed in this species (Figure 4C). The metabolism of [18F]1 in rhesus monkeys was biphasic, with most of the tracer metabolized fairly rapidly, followed by a slower phase of breakdown (Figure 5). The average time at which 50% of the tracer in plasma was in the form of metabolites (T1/2) was 2.5 ± 0.8 min (n = 7). The use of DMI to pharmacologically block cardiac NET had no significant effect on the observed metabolic breakdown rate.
Figure 5.

Metabolic breakdown of [18F]1 in the plasma of a rhesus macaque monkey. After correcting the HPLC-radiodetection data for fluorine-18 decay, the fraction of radioactivity associated with intact [18F]1 was determined for each plasma sample.
Samples of whole blood (2 mL) were ‘spiked’ with a small amount of [18F]1 and incubated at 37 °C for 65–75 min and were processed in the same manner as the venous blood samples. In all cases, < 7% of the total activity was in the form of the sulfur-conjugate of [18F]1, with the remainder intact [18F]1, indicating that metabolic breakdown of the tracer in blood is a very slow process.
Analysis of the venous blood samples to determine partitioning of [18F]1 between plasma and whole blood (Cp/Cwb) showed a consistent tendency of the tracer to stay in plasma. A typical mean ratio of Cp/Cwb for the 6 blood samples in one study was 1.24 ± 0.10. Across all 7 studies, the mean ratio averaged 1.25 (range 1.18–1.31). The global mean for all samples in all studies was 1.25 ± 0.09. The NET inhibitor DMI appeared to have no influence on the blood partitioning data.
Myocardial Kinetics of [18F]1
Region-of-interest (ROI) analysis was used to extract time-activity curves for [18F]1 in whole blood, Cwb(t), and for left ventricular tissue, Ct(t), from the dynamic PET scans. An example of the kinetics of [18F]1 in rhesus macaque monkey is shown in Figure 6. In whole blood, following the peak of the bolus injection, total activity levels in blood, Cwb(t), stayed roughly constant from around t = 10 min out to 85 min. Because of the rapid sulfur conjugation of [18F]1, after ~ 30 min most of the activity in blood is in the form of the sulfur conjugate of [18F]1. Myocardial activity climbed for 10 to 30 min after injection and then remained constant. The lack of any further accumulation of [18F]1 into cardiac nerve terminals after this time is indirect evidence that the sulfur conjugate of [18F]1 is inactive at the norepinephrine transporter (NET). Thus only intact [18F]1 molecules are taken up into presynaptic sympathetic nerve terminals.
Figure 6.

Kinetics of [18F]1 in whole blood (triangles) and myocardial tissue (circles) in a rhesus macaque monkey. The final concentrations of [18F]1 in tissue and blood, expressed as ‘standardized uptake values’ (SUV), were 3.6 and 1.2, respectively.
Analyses of Tracer Kinetics
As described in the Experimental section, data from the analyses of the blood samples for metabolites and of the partitioning of radioactivity between plasma and whole blood were used to convert the image-derived whole-blood time activity curve Cwb(t) to an estimate of the plasma time-activity curve Cp(t). The kinetics of [18F]1 in plasma, Cp(t), and in heart tissue, Ct(t) were analyzed quantitatively using two well-established approaches, compartmental modeling and Patlak graphical analysis. Compartmental modeling of the kinetics of [18F]1 using a compartment model structure with irreversible uptake into neurons (Figure 7) was robust for all experimental conditions, converging quickly in each case to a single global minimum. The rate constants K1 (mL/min/g), k2 (min−1) and k3 (min−1) and a ‘blood volume fraction’ BV (dimensionless) were estimated for each study. In addition, a ‘net uptake rate’ constant Ki (mL/min/g), which reflects the net rate of tracer accumulation into tissue compartments, was calculated for each study as: Ki = (K1k3)/(k2 + k3). Estimated values of k3 did not decline with increasing doses of DMI. This was not completely unexpected as similar results have been observed with other radiopharmaceuticals, such as 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG).33 However the ‘net uptake rate’ constant Ki (mL/min/g), calculated from the parameter estimates, did decline in a dose-dependent manner with increasing DMI dose. For control studies, the average estimated ‘net uptake rate’ constant was Ki = 0.341 ± 0.041 mL/min/g. Patlak graphical analysis uses a mathematical transformation of the kinetic data Cp(t) and Ct(t) to generate a ‘Patlak plot’, which has a characteristic linear phase.34 The slope of the linear portion of a Patlak plot, Kp (mL/min/g), provides an alternative estimate of the ‘net uptake rate’ constant Ki (mL/min/g). Thus for the model structure used (Figure 7), the slope of the Patlak plot is given by Kp ≅ (K1k3)/(k2 + k3). For the control studies, the Patlak plots were highly linear (r2 > 0.99) with an average measured Patlak slope Kp = 0.302 ± 0.031 mL/min/g. For the four DMI block studies, again highly linear Patlak plots were seen, and the measured Patlak slopes declined in a DMI-dose dependent manner (Figure 8). The decline in the estimated Ki or Kp values with increasing doses of DMI each followed a sigmoidal dose-response curve (Figure 9). For the Ki data, a half-maximal inhibitory concentration (IC50) of 0.051 mg/kg DMI was estimated, with a Hill slope nH = −0.79. For the Kp data, IC50 = 0.025 mg/kg DMI and nH = − 0.68. These results suggest that estimates of the ‘net uptake rate’ constant Ki, obtained from either compartmental modeling or Patlak analysis of [18F]1 kinetics, can serve as sensitive and robust measures of regional cardiac sympathetic nerve density.
Figure 7.

Compartmental model structure used for tracer kinetic analysis of the myocardial kinetics of [18F]1 in rhesus macaque monkeys.
Figure 8.

Patlak graphical analysis of the myocardial kinetics of [18F]1 kinetics in rhesus macaque monkeys under control conditions (n = 4) and following DMI block of cardiac NET at four different DMI dose levels. Patlak plots were highly linear in all cases (r2 > 0.99) and the Patlak slopes declined in a dose-dependent manner with increasing DMI doses.
Figure 9.

Dose response curves of ‘net uptake constants’ Ki (mL/min/g) derived from either compartmental modeling (circles) or Patlak graphical analysis (triangles) of [18F]1 kinetics in rhesus macaque monkeys.
Oncology applications: Uptake of [18F]1 into adrenal glands
In addition to using [18F]1 to quantify regional cardiac sympathetic nerve density, it may also be possible to use this radiotracer for localizing neuroendocrine tumors, such as neuroblastoma and pheochromocytoma.35 To assess the ability of [18F]1 to localize in these types of tumors, we examined its uptake into the adrenal glands of rhesus macaque monkeys. In control studies, the adrenal glands were clearly visualized, especially the adrenal on the right side of the coronal PET images, which was better separated from the kidney than the contralateral adrenal gland (Figure 10, top row). There was also significant uptake of [18F]1 in the cortex of the kidney. It is not known if this is nonspecific uptake into, for example, the proximal tubules in the renal cortex, or if it is specific to sympathetic innervation in the kidneys. The kinetics of [18F]1 in the adrenal gland are very similar to those seen in heart tissue (data not shown). In the 1.0 mg/kg DMI block study described previously, the adrenal glands were scanned for 10 min following the 90 min cardiac PET study. No uptake of [18F]1 was seen in the adrenal glands after DMI block, demonstrating that [18F]1 uptake into the adrenal gland is primarily mediated by NET transport (Figure 10, bottom row). Together, these data suggest that [18F]1 could potentially find oncology applications in the nuclear medicine clinic.
Figure 10.

Uptake of [18F]1 into the adrenal glands of a rhesus macaque monkey. Coronal and sagittal slice PET images are shown for control conditions (top row) and following DMI block (1 mg/kg) of NET uptake (bottom row). In controls, the images show there is very high uptake into the renal cortex as well as high uptake into the one adrenal gland contained in these image slices (arrows, top row). Under DMI block conditions, [18F]1 uptake into the same adrenal gland is completely absent (bottom row), indicating that uptake of [18F]1 into the adrenal gland is specific to NET transport.
Human Radiation Dosimetry Estimates
The biodistribution of [18F]1 into the organs of rats at four time points (Table 1) were used to derive human radiation absorbed dose estimates using the OLINDA/EXM 1.0 software package.36 From the acquired data, the maximum percentage of the injected dose observed in the gastrointestinal tract (12.6%) was assumed to enter the small intestine using the ICRP 30 GI Tract model incorporated into the OLINDA/EXM program. Also, in separate microPET imaging studies in rats, 19.9 ± 0.2% of the injected dose was found to be excreted through the bladder with a biological half-time of 0.20 ± 0.02 h. These values were entered into the Dynamic Bladder model of OLINDA/EXM, assuming a 4.0 h void interval. Absorbed doses estimates for the reference adult male organ model of OLINDA/EXM are shown in Table 2. Organs with the highest absorbed dose estimates included the urinary bladder wall (0.666 rad/mCi), upper lower intestine (0.221 rad/mCi), small intestine (0.201 rad/mCi) and the heart wall (0.201 rad/mCi). An ‘effective dose’ of 0.091 rem/mCi was estimated for [18F]1. Under US federal regulations governing research with a new radiopharmaceutical (21CFR361.1), the maximum allowable radiation absorbed dose to an individual organ (other than the gonads) is 5 rad. Considering the maximum estimated absorbed dose (urinary bladder wall; 0.666 rad/mCi) initial PET studies in human subjects could use a maximum of 7.5 mCi of [18F]1. This would correspond to an ‘effective dose’ of 0.68 rem.
Table 1.
Biodistribution of [18F]1 into major organs in rats (%ID·kg/g).
| Organ | 5 min | 30 min | 60 min | 120 min |
|---|---|---|---|---|
| Brain | 0.004 ± 0.001 | 0.003 ± 0.000 | 0.002 ± 0.000 | 0.001 ± 0.001 |
| Eyeballs | 0.039 ± 0.006 | 0.040 ± 0.004 | 0.041 ± 0.006 | 0.026 ± 0.006 |
| Heart | 0.746 ± 0.067 | 0.602 ± 0.111 | 0.526 ± 0.116 | 0.347 ± 0.055 |
| Lung | 0.267 ± 0.045 | 0.136 ± 0.032 | 0.091 ± 0.015 | 0.036 ± 0.006 |
| Liver | 0.477 ± 0.110 | 0.406 ± 0.071 | 0.350 ± 0.077 | 0.233 ± 0.049 |
| Pancreas | 0.137 ± 0.024 | 0.145 ± 0.021 | 0.172 ± 0.042 | 0.121 ± 0.016 |
| Spleen | 0.268 ± 0.044 | 0.262 ± 0.055 | 0.253 ± 0.054 | 0.235 ± 0.066 |
| Adrenal | 0.203 ± 0.083 | 0.181 ± 0.069 | 0.193 ± 0.053 | 0.133 ± 0.021 |
| Kidney | 1.398 ± 0.530 | 0.183 ± 0.055 | 0.186 ± 0.090 | 0.078 ± 0.024 |
| Stomach | 0.116 ± 0.021 | 0.110 ± 0.004 | 0.115 ± 0.029 | 0.089 ± 0.019 |
| Ovary* | 0.194 ± 0.048 | 0.171 ± 0.020 | 0.142 ± 0.052 | 0.129 ± 0.029 |
| Uterus* | 0.268 ± 0.044 | 0.155 ± 0.001 | 0.112 ± 0.046 | 0.081 ± 0.006 |
| Testes* | 0.018 ± 0.002 | 0.010 ± 0.001 | 0.008 ± 0.003 | 0.004 ± 0.001 |
| Muscle† | 0.019 ± 0.005 | 0.025 ± 0.011 | 0.016 ± 0.002 | 0.015 ± 0.002 |
| Bone† | 0.060 ± 0.015 | 0.039 ± 0.015 | 0.027 ± 0.009 | 0.016 ± 0.005 |
| Blood† | 0.062 ± 0.004 | 0.022 ± 0.003 | 0.016 ± 0.002 | 0.009 ± 0.001 |
(n = 2) animals used for these values.
Values expressed as %ID/g.
Table 2.
Radiation absorbed dose estimates (rem/mCi) for [18F]1 for the reference adult male model of OLINDA/EXM 1.0.
| Target organ | Total dose (rad/mCi) | Dose for 10 mCi (rad) |
|---|---|---|
| Adrenals | 0.059 | 0.59 |
| Brain | 0.025 | 0.25 |
| Breasts | 0.026 | 0.26 |
| Gallbladder wall | 0.061 | 0.61 |
| LLI wall | 0.107 | 1.07 |
| Small intestine | 0.201 | 2.01 |
| Stomach wall | 0.048 | 0.48 |
| ULI wall | 0.221 | 2.21 |
| Heart wall | 0.109 | 1.09 |
| Kidneys | 0.078 | 0.78 |
| Liver | 0.096 | 0.96 |
| Lungs | 0.035 | 0.35 |
| Muscle | 0.037 | 0.37 |
| Ovaries | 0.088 | 0.88 |
| Pancreas | 0.063 | 0.63 |
| Red marrow | 0.037 | 0.37 |
| Osteogenic cells | 0.049 | 0.49 |
| Skin | 0.026 | 0.26 |
| Spleen | 0.079 | 0.79 |
| Testes | 0.041 | 0.41 |
| Thymus | 0.033 | 0.33 |
| Thyroid | 0.029 | 0.29 |
| Urinary bladder wall | 0.666 | 6.66 |
| Uterus | 0.103 | 1.03 |
| Total body | 0.041 | 0.41 |
|
| ||
| (rem/mCi) | (rem) | |
|
| ||
| Effective dose | 0.091 | 0.91 |
Conclusion
An automated radiosynthesis of [18F]1 capable of preparing the compound at moderate radiochemical yields and specific activities was successfully developed. Initial biological evaluations of [18F]1 in rhesus macaque monkeys were highly encouraging and strongly suggest that this agent is capable of providing accurate and robust quantitative measures of regional cardiac sympathetic nerve density in human subjects. If this proves true, [18F]1 could be used clinically to accurately assess the regional distribution of functional sympathetic nerve terminals in diseases associated with cardiac sympathetic dysfunction, including sudden death, heart failure, cardiac arrhythmias and diabetic autonomic neuropathy. Also, [18F]1 may find an additional clinical role as a diagnostic imaging agent for localizing adrenergic tumors, such as neuroblastoma and pheochromocytoma, using PET.
EXPERIMENTAL SECTION
Chemistry
Experimental procedures for the synthetic preparation of selected compounds (3–10, 15 and 18), as well as detailed methods for the isolated rat heart studies are included in the Supporting Information File.
3-Benzyloxy-4-iodophenethylamine tert-butylcarbamate (11)
To a solution of 3-benzyloxy-4-iodophenethylamine hydrochloride 6 (1.1 g, 2.8 mmol) and triethylamine (1.6 mL, 11.3 mmol) in DMF (10 mL) was added (Boc)2O (650 mg, 3.0 mmol) at room temperature and the resulting solution was heated at 120 °C for 1 h. The reaction mixture was treated with saturated NH4Cl (50 mL) and extracted with ethyl acetate (25 mL × 2). The combined organic layers were washed by brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 20% ethyl acetate in hexane) to afford the product (1.13 g, 88%) as a white solid; mp 105–107 °C; 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 7.8 Hz, 1H), 7.52-7.50 (m, 2H), 7.41-7.38 (m, 2H), 7.36-7.31 (m, 1H), 6.71 (s, 1H), 6.58 (d, J = 7.6 Hz, 1H), 5.13 (s, 2H), 4.53 (bs, 1H, NH), 3.34 (q, J = 6.1 Hz, 2H), 2.74 (t, J = 6.1 Hz, 2H), 1.44 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 157.2, 140.9, 139.4, 136.4, 128.5, 127.9, 127.0, 123.3, 113.3, 84.1, 70.8, 41.5, 36.0, 28.4; MS (ESI) m/z 476 (M+Na)+. Anal. Calcd. For C20H24INO3: C, 52.99; H, 5.34; N, 3.09. Found: C, 52.77; H, 5.39, N, 2.96.
3-Benzyloxy-4-trimethylstannylphenethylamine tert-butylcarbamate (12)
Hexamethylditin (0.84 mL, 4.04 mmol) was added to a solution of compound 11 (916 mg, 2.02 mmol) and a tetrakis(triphenylphosphine)-palladium (117 mg, 0.1 mmol) in anhydrous toluene (6.0 mL) at room temperature under nitrogen. The reaction mixture was heated to 120–130 °C for 30 min under N2, cooled down to room temperature and filtered through a Celite pad. Celite pad was washed with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (silica gel, 5% to 10% ethyl acetate in hexane) to afford the product (800 mg, 81%) as a white solid; mp 90–92 °C; 1H NMR (500 MHz, CDCl3) δ 7.42-7.40 (m, 4H), 7.39-7.32 (m, 2H), 6.84 (d, J = 7.1 Hz, 1H), 6.73-6.72 (m, 1H), 5.03 (s, 2H), 4.57 (bs, 1H, NH), 3.39 (q, J = 6.3 Hz, 2H), 2.79 (t, J = 6.7 Hz, 2H), 1.45 (s, 9H), 0.15 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 163.2, 155.8, 141.3, 137.0, 136.6, 128.4, 128.1, 127.8, 127.5, 121.6, 110.7, 69.9, 67.3, 41.7, 36.4, 28.4, −9.2; MS (ESI) m/z 492 (M+H)+. Anal. Calcd. For C23H33NO3Sn: C, 56.35; H, 6.79; N, 2.86. Found: C, 56.53; H, 6.92, N, 2.96.
2-Benzyloxy-4-(2′-t-butoxycarbonylaminoethyl)phenyl(2-thiophenyl)iodonium tosylate (13)
A solution of 2-(diacetoxy)iodothiophene (164 mg, 0.5 mmol) in CH2Cl2 (1.5 mL) was added to a solution of p-toluenesulfonic acid hydrate (96 mg, 0.5 mmol) in MeCN (1.5 mL) at room temperature under nitrogen atmosphere. The white precipitate was immediately generated and the mixture was stirred for 1 h. A solution of compound 12 (246 mg, 0.5 mmol) in CH2Cl2 (1.5 mL) and MeCN (1.5 mL) was added slowly to the reaction mixture. After the white precipitate was disappeared, the mixture was stirred at room temperature for 18 h under nitrogen atmosphere. The solvent was removed under reduced pressure and the crude product was sonicated with diethyl ether (50 mL). The precipitate was filtered, washed with diethyl ether and dried in vacuo to give the product (332 mg, 94%) as a white solid. mp 157–160 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.24 (d, J = 9.1 Hz, 1H), 7.91 (d, J = 4.9 Hz, 1H), 7.78 (d, J = 2.9 Hz, 1H), 7.53-7.40 (m, 7H), 7.27 (bs, 1H), 7.11-7.10 (m, 3H), 6.94-6.92 (m, 2H), 5.33 (s, 2H), 3.17 (q, J = 6.4 Hz, 2H), 2.74 (t, J = 6.9 Hz, 2H), 2.28 (s, 3H), 1.33 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ 156.2, 155.6, 148.3, 146.0, 140.4, 138.5, 136.3, 130.0, 129.1, 128.7, 126.1, 124.7, 114.9, 107.4, 100.6, 78.3, 71.5, 41.3, 36.1, 28.8, 21.4; MS (ESI) m/z 536 (M−OTs)+. HRMS (EI) calcd for C24H27ION3S 536.0751, found 536.0749.
2-Benzyloxy-4-(2′-t-butoxycarbonylaminoethyl)phenyl(2-thiophenyl)iodonium bromide (14)
A solution of KBr (144 mg, 1.2 mmol) in H2O (1.0 mL) was added slowly to a solution of compound 13 (200 mg, 0.28 mmol) in MeCN (1.0 mL) at 60 °C. The reaction mixture was stirred at room temperature for 1 h. The precipitate was washed with ice H2O (10 mL), filtered off, washed further with Et2O by several times and dried in vacuo to give the product (123 mg, 71%) as a white solid. mp 160–163 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.19 (d, J = 8.3 Hz, 1H), 7.84 (dd, J = 5.2, 1.0 Hz, 1H), 7.68 (d, J = 3.2 Hz, 1H), 7.52-7.39 (m, 5H), 7.22 (bs, 1H), 7.07-7.05 (m, 1H), 6.94 (t, J = 5.4 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 5.31 (s, 2H), 3.16 (q, J = 6.5 Hz, 2H), 2.73 (t, J = 7.1 Hz, 2H), 1.33 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ 156.1, 155.5, 147.5, 139.2, 137.2, 136.4, 136.2, 129.6, 129.2, 128.9, 128.4, 124.4, 114.7, 109.5, 104.3, 78.2, 71.3, 41.2, 36.0, 28.8; MS (ESI) m/z 536 (M−Br)+. Anal. Calcd. For C24H27BrION3S: C, 46.77; H, 4.42; N, 2.27. Found: C, 46.72; H, 4.46, N, 2.31.
4-Fluoro-3-hydroxyphenethylguanidine hydrochloride (4F-MHPG, 1)
1.0 N HCl (5.5 mL, in Et2O) was added to a solution of N,N′-bis(tert-butoxycarbonyl)-N″-4-fluoro-3-hydroxyphenethylguanidine12 (130 mg, 0.32 mmol) in MeOH (1.0 mL) and CH2Cl2 (0.1 mL) at room temperature. The reaction mixture was stirred for 24 h and concentrated under reduced pressure. The generated precipitate was washed three times with diethyl ether and dried in vacuo to give the desired product (66 mg, 87%) as a white solid. mp 178–181 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.84 (bs, 1H), 7.73 (t, J = 5.5 Hz, 1H), 7.48 (bs, 2H), 7.06-7.02 (m, 1H), 6.87 (dd, J = 8.5, 2.0 Hz, 1H), 6.69-6.60 (m, 1H), 3.47 (bs, 1H), 3.30 (q, J = 6.8 Hz, 2H), 2.66 (t, J = 7.3 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 157.4, 150.4 (J = 237.9 Hz), 145.1 (J = 11.9 Hz), 135.3, 120.0 (J = 6.8 Hz), 118.7, 116.3 (J = 18.1 Hz), 42.5, 34.4; MS (EI) m/z 198 (M+). HRMS (EI) calcd for C9H13FN3O of the free base 198.1037, found 198.1040.8, 12
Radiochemistry
Two GE TRACERlab FXFN modules were used in sequence for a fully automated synthesis of [18F]1. The first module was used for production of 3-benzyloxy-4-[18F]fluoro-m-tyramine [18F]16, while the second module was used to convert [18F]16 into the final product [18F]1, as described in Scheme 3.
Radiosynthesis of 3-benzyloxy-4-[18F]fluoro-m-tyramine ([18F]16)
The reaction reagents were placed in vials of the GE TRACERLab FXFN module as follows: Cs2CO3 (0.5 mL, 0.5 M in H2O) in vial 1; MeCN (1.0 mL) in vial 2; diaryliodonium salt precursor 14 (5.5-6.0 mg) in anhydrous DMF (0.5 mL) and TEMPO (1.0 mg) in H2O (20 μL) in vial 3; HCl solution (1.0 mL, 1.0 N in H2O) in vial 4; anhydrous EtOH (1.0 mL) in vial 7; H2O (5.0 mL) in vial 9; NaOH solution (10 mL, 0.2 M in H2O) in dilution flask.
[18F]F− was prepared by the 18O(p,n)18F reaction using H2 18O as the target material in a GE PETrace cyclotron. It was isolated from the enriched water by trapping on Chromafix-HCO3 (Sep-Pak® Light QMA) cartridge (pre-activated with 10 mL of ethanol and 10 mL of H2O) and eluted from the cartridge into the reactor vial with a solution of 0.5 mL Cs2CO3 (0.5 M). 1.0 mL of MeCN was added to the reactor vessel and then water/acetonitrile was evaporated at 80 °C under vacuum with a nitrogen stream to yield a dried Cs[18F]F. After cooling to 60 °C, a mixture solution of 0.5 mL of DMF, 20 μL of H2O, containing 5.5-6.0 mg of the diaryliodonium salt precursor 14 and 1.0 mg of TEMPO was added from vial 3 to the glassy carbon reactor vessel containing Cs[18F]F. The sealed reaction mixture was heated at 150 °C for 25 min to form [18F]15. After cooling to 100 °C, 1.0 mL of 1.0 N HCl solution was added to the reaction mixture. The reaction solution was heated at 100 °C for 10 min and then directly transferred to the dilution flask containing 10 mL of 0.2 M NaOH solution. The diluted solution was stirred for 3 min and passed through a C-18 Sep-Pak® cartridge which was pre-activated with 10 mL of ethanol and 10 mL of water. The cartridge was washed with 5.0 mL of H2O and purged for 2 min with nitrogen to remove most of the moisture. The trapped intermediate [18F]16 was eluted from the cartridge with 1.0 mL of anhydrous EtOH. This eluted solution was transferred into the reaction vial of the second GE TRACERlab FXFN module. In general, ~1.4 Ci of [18F]F− was used and 170–250 mCi of [18F]16 was obtained in 15 ± 3 % radiochemical yield and > 95% radiochemical purity.
Radiosynthesis of 4-[18F]fluoro-m-hydroxyphenethylguanidine ([18F]1)
The reaction reagents were placed in vials of the second GE TracerLab FXFN module as follows: A solution of N′,N″-bis-(tert-butoxycarbonyl)-5-chloro-1H-benzotriazole-1-carboxamidine 17 (5.0 mg) and DIEA (0.1 mL) in MeCN (0.4 mL) in vial 1; HBr solution [0.35 mL, 1.0 N solution prepared by a mixed solution of 48% HBr (0.25 mL) with MeCN (1.0 mL)] in vial 2; NaOH solution (0.7 mL, 1.0 N in H2O) in vial 3; buffer solution (0.9 mL, 5% EtOH in 60 mM NaH2PO4) in vial 4.
The transferred EtOH solution of [18F]16 (170–250 mCi) was evaporated at 80 °C under a stream of nitrogen and then cooled to 40 °C. A solution of 5.0 mg of N′,N″-bis-(tert-butoxycarbonyl)-5-chloro-1H-benzotriazole-1-carboxamidine 17 in a mixed solution of 0.1 mL of DIEA and 0.4 mL of MeCN was added to the reactor vessel containing [18F]16 and the resulting mixture was heated at 45 °C for 15 min. 0.35 mL of 1.0 N HBr solution was added from vial 2 to the reactor vessel and the reaction mixture was heated at 120 °C for 15 min. After cooling to 100 °C, 0.7 mL of 1.0 N NaOH solution and 0.9 mL of buffer solution were added from vial 4 to the reaction mixture. The crude product was injected onto a reverse-phase HPLC column (Phenomenex Synergi 10 micron Hydro-RP 80A, 250 × 10 mm, 5% EtOH in 60 mM NaH2PO4 buffer, flow rate 4.0 mL/min., λ224 nm) and [18F]1 was collected at 28–30 min. In general, 55–125 mCi of [18F]1 was obtained in 7 ± 3% overall radiochemical yield (decay-corrected based on starting activity) and >99% radiochemical purity. Specific activity (SA) was determined by injecting a sample of [18F]1 with known activity (μCi) onto the HPLC system. The area under the UV absorbance peak associated with the [18F]1 radioactivity peak was compared against a predetermined standard curve to estimate the total mass ([18F]1 + [19F]1) in μg. The ratio of 18F-activity to total mass (converted from μg to μmol using the molecular weight of [19F]1) gave the specific activity, which averaged 1.2 ± 0.3 Ci/μmol. Total synthesis time from end of bombardment (EOB) was 150 min.
Animal Care
The care of all animals used in this study was done in accordance with the Animal Welfare Act and the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.37 Animal protocols were approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan.
Isolated rat heart studies
Kinetic studies of [18F]1 in an isolated working rat heart system were performed to measure its neuronal uptake rate (Kup; mL/min/g wet) and neuronal clearance rate (T1/2; h) using previously reported methods.8 Details are provided in the Supporting Information File.
Biodistribution Studies for Human Radiation Dosimetry Estimates
Sprague-Dawley rats (280–330 g) were purchased from Charles River Laboratories, Inc., Wilmington, MA. Four time points were studied: 5 min, 30 min, 1 h and 2 h. For each time point, 2 male rats and 2 female rats under light isoflurane anesthesia received bolus tail-vein injections of 20–120 μCi of [18F]1 in 0.05–0.10 mL of isotonic 60 mM sodium phosphate buffer, pH 5.4. The animals were then allowed to recover and resume normal activity. Groups of animals were killed by decapitation while under isoflurane anesthesia. Organs were quickly removed and sectioned into smaller pieces (< 1 g each) for gamma counting. Organ sections, blood samples and urine samples were placed in previously weighed counting tubes. Tubes were reweighed to determine tissue masses and then counted in a gamma-counter (MINAXI Auto-Gamma 5500; Perkin-Elmer, Wellesley, MA). The radioactivity remaining in each carcass after removal of all samples was measured in a dose calibrator (CRC712M, Capintec, Ramsey, NJ). For each organ, the decay-corrected counts in all organ sections, expressed in disintegrations per minute (dpm), were summed together and divided by the total mass of the sections to determine radiotracer tissue concentration (dpm in organ/g). The activity in the injected dose of [18F]1 was converted from μCi into dpm. The measured tissue activity concentrations were then normalized to the total injected dose (dpm) and the animal’s body weight (kg) as: Animal organ concentration (%ID·kg/g) = (dpm in organ/g)/[(dpm injected)/(kg body weight)].38 For the major organs the tracer organ concentrations in %ID·kg/g were scaled to estimates of human organ uptake values (%ID/organ) by multiplying each value by standardized human organ weights and dividing by the total body mass of 73 kg, as defined for the reference adult male model of the OLINDA/EXM software package.39
To better define the kinetics of elimination of [18F]1 through urinary excretion, microPET imaging studies were performed in two rats. Rats were anesthetized with isoflurane and a transmission scan was acquired using a rotating 68Ge/68Ga rod source for attenuation corrections. Dynamic PET data were acquired in list-mode for 90 min after injection (i.v.) of 0.5 mCi of [18F]1. List-mode emission data were rebinned into a 17-frame dynamic sequence (17×300 s). Rebinned emission data were corrected for attenuation and scatter, and transaxial images reconstructed using OSEM/MAP reconstruction. A region-of-interest was drawn on the bladder and used to extract a time-activity curve for the appearance of activity in urine. Nonlinear regression analysis was used to fit the urine time-activity data to a one phase exponential association model to determine the fraction of the injected dose excreted in the urine (Prism v.3.03, GraphPad Software).
The OLINDA/EXM 1.0 software package36 was used to generate human radiation absorbed dose estimates. The estimates of %ID/organ for human organs vs. time were entered into the Kinetics Input Form of OLINDA/EXM to generate ‘normalized cumulated activity’ values Ni for each organ. Rat biodistribution data for excretion through the gastrointestinal (GI) tract were modeled using the ICRP 30 GI Tract model feature of OLINDA/EXM. Kinetic results from the urinary excretion microPET studies in rats (% excreted, urinary clearance biological half-time) were entered into the Voiding Bladder model feature of OLINDA/EXM, using a 4.0 h bladder voiding interval. A normalized cumulated activity value for the total body NTB was estimated from the urinary and GI tract excretion kinetics.39 An estimate of the normalized cumulated activity for the ‘remainder’ of the body, Nrem, exclusive of the major organs, was estimated from the N values for total body and the major organs as: Nrem = NTB − ΣNi. This value was entered into the Kinetics Input Form of OLINDA/EXM. Radiation absorbed dose estimates were then calculated for the reference man organ model of OLINDA/EXM.
Cardiac PET Imaging Studies
Cardiac PET studies were performed in rhesus macaque monkeys using a microPET P4 primate scanner (Siemens/CTI Concorde Microsystems, Knoxville, TN). After the monkey was anesthetized, a percutaneous angiocather was placed in the saphenous vein of each leg (one for tracer injection, one for blood sampling). Heart rate (bpm), blood oxygen saturation levels (SpO2) and body temperature were monitored continuously (SurgiVet V3404P). A transmission scan was acquired using a rotating 68Ge/68Ga rod source for attenuation corrections. Dynamic PET data were acquired in list-mode for 90 min after intravenous injection of 5–6 mCi of [18F]1. List-mode emission data were rebinned into a 27-frame dynamic sequence (12×10 s, 2×30 s, 2×60 s, 2×150 s, 2×300 s, 7×600 s). Rebinned emission data were corrected for attenuation and scatter, and transaxial images reconstructed using maximum a posteriori (MAP) reconstruction40, an iterative method that accounts for the detector point spread function in the model of the system.
Radiometabolites in Monkey Plasma
Before the imaging study, a blood sample (1.5–2.0 mL) was drawn and 0.1 mCi of [18F]1 added and incubated at 37 °C for 60–70 min to determine the tracer’s stability in blood. During the PET scan six venous blood samples (1.5–2.0 mL) were drawn (typically, t = 1, 2, 4, 10, 30 and 60 min) to assess radiometabolites in plasma and the partitioning of [18F]1 between plasma and red blood cells (RBCs). Blood samples were collected into heparinized blood tubes, transferred to a 2.0 mL Eppendorf vial, and centrifuged for 1 min at 12000 × g to separate plasma and RBCs. Plasma was deproteinized by adding perchloric acid (HClO4), final concentration 0.4N, and centrifuged for 5 min at 12000 × g. The supernatant was removed, neutralized with KOH (pH 7.0–7.5) and filtered twice through 0.22 μm filters (Millipore Millex/GS). A portion of this solution was analyzed by a reverse-phase HPLC (Phenomenex Synergi 10 micron Hydro-RP 80A, 250 × 4.6 mm, 10% EtOH in 60 mM NaH2PO4 buffer, pH 5.4, flow rate 1.0 mL/min., λ224 nm) and radiation detection (Ortec Model 905-4 NaI(Tl) detector). The blood sample ‘spiked’ with [18F]1 was processed in the same manner. Aliquots (0.1 mL) of whole blood, plasma, final supernatant and pellets were counted in a gamma counter. Count data (corrected for decay) were used to determine the relative concentrations of [18F]1 in plasma and whole blood (Cp/Cwb) for each sample. HPLC/radiation detection data were decay corrected and processed for peak analysis (ACD/ChromProcessor v.10; ACD Inc., Toronto, Canada) to determine the percentage of total activity associated with intact [18F]1 (fintact) for each plasma sample. A mathematical function describing the time course of the metabolic breakdown of [18F]1 in plasma, fintact(t), was obtained by nonlinear regression analysis (Prism 3.0, GraphPad Software, San Diego, CA).
In Vitro Sulfoconjugation Assay
A 20 μL aliquot of monkey liver cytosol (#452461, BD Biosciences, San Jose, CA) was thawed on ice. A 10 μL aliquot of 10 mM sulfotranferase cofactor PAPS (adenosine-3′-phosphate-5′-phosphosulfate lithium salt hydrate; #A1651, Sigma-Aldrich, Milwaukee, WI) was added to 50 μL of 1.0 mM Tris-HCl buffer (pH 7.4) and 170 μL of ultrapure water (18 M·cm MilliQ water system, Millipore, Billerica, MA) and incubated at 37 °C for 5 min.41 Approximately 20 μCi of [18F]1 in 250 μL of ultrapure water was added to the reaction mixture (final volume 500 μL) and incubated at 37 °C for either 1 min or 20 min. The reaction was terminated by centrifugation at 16,000 × g for 5 min at 4 °C. The supernatant was filtered (Millex-GS 0.22 μm, Millipore, Billerica, MA) and analyzed using HPLC with radiation detection as described above for the rhesus macaque plasma samples.
Tracer Kinetic Analyses
Summed images of the final four transaxial PET frames were used to draw regions-of-interest (ROIs) on the myocardial wall and on the blood pool in the basal portion of the left ventricular chamber to extract time-activity curves for myocardial tissue Ct(t) and whole blood Cwb(t). The whole blood time-activity curve was used to estimate the plasma concentration of intact [18F]1 vs. time, Cp(t), by multiplying Cwb(t) by the function describing the tracer’s metabolic breakdown, fintact(t) and the average measured relative activity concentrations in plasma and whole blood, Cp/Cwb. Thus, Cp(t) = Cwb(t)·fintact(t)·[Cp/Cwb]. The plasma time-activity curve Cp(t) was used with the tissue time-activity curve Ct(t) for compartmental modeling. The simplified compartmental model (Figure 7) was used to obtain estimates of the rate constants K1 (mL/min/g), k2 (min−1) and k3 (min−1) and a blood volume fraction BV (dimensionless). The estimated rate constants for each study were used to calculate a ‘net uptake rate’ constant Ki = (K1k3)/(k2 + k3), with units mL/min/g, which reflects the rate of [18F]1 accumulation into sympathetic nerve terminals. The kinetics of [18F]1 in heart tissue and plasma were also analyzed using Patlak graphical analysis34. After construction of a Patlak plot from the kinetic data, Cp(t) and Ct(t), the linear portion of the plot was analyzed with linear regression to determine the Patlak slope, Kp (mL/min/g). Under ideal conditions, the Patlak slope Kp is a direct measure of the ‘net uptake rate’ constant Ki, and thus for the compartmental model structure used is also approximately equal to (K1k3)/(k2 + k3).
Pharmacological Blocking Studies
In addition to microPET imaging studies under control conditions (n = 4), a series of pharmacological blocking studies were performed with the potent NET inhibitor desipramine (DMI). The goal of these studies was to assess the ability of the quantitative parameters from compartmental modeling and Patlak analysis to track progressively lower cardiac NET densities induced pharmacologically by increasing amounts of administered DMI. All studies were performed in the same monkey to minimize biological variation between studies. The DMI dose (dissolved into 2.0 mL of saline) was infused intravenously over 20 min using a Harvard infusion pump. [18F]1 was injected 10 min after the end of the DMI infusion. DMI doses (n = 1 each) were: 0.010 mg/kg, 0.0316 mg/kg, 0.10 mg/kg and 1.0 mg/kg. The measured kinetic parameters Ki from compartmental modeling and Kp values from Patlak analysis, as a function of DMI dose, were fit to a sigmoidal dose-response model with variable slope using nonlinear regression (Prism 3.0, GraphPad Software, San Diego, CA).
PET Imaging of [18F]1 Uptake into Adrenal Glands
These studies were performed using the same imaging methods and protocols described above for the cardiac PET imaging studies, except that the animal was positioned in the scanner gantry to focus on the adrenal glands and kidneys (rather than the heart), and no blood samples were drawn.
Supplementary Material
Acknowledgments
We thank the staff of the University of Michigan Cyclotron Facility for their many contributions. This work was supported by PHS grant R01-HL079540 from the National Heart Lung and Blood Institute, National Institutes of Health, Bethesda, MD USA.
ABBREVIATIONS USED
- DMI
desipramine
- [11C]HED
[11C]-(−)-m-hydroxyephedrine
- [123I]MIBG
[123I]metaiodobenzylguanidine
- NET
norepinephrine transporter
- TEMPO
2,2,6,6-tetramethylpiperidine-N-oxyl
- VMAT2
vesicular monoamine transporter, isoform 2
Footnotes
Experimental procedures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vaseghi M, Shivkumar K. The role of the autonomic nervous system in sudden cardiac death. Prog Cardiovasc Dis. 2008;50:404–419. doi: 10.1016/j.pcad.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parati G, Esler M. The human sympathetic nervous system: its relevance to hypertension and heart failure. Eur Heart J. 2012;33:1058–1066. doi: 10.1093/eurheartj/ehs041. [DOI] [PubMed] [Google Scholar]
- 3.Pop-Busui R. Cardiac autonomic neuropathy in diabeties: a clinical perspective. Diabetes Care. 2010;33:434–441. doi: 10.2337/dc09-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zipes DP, Rubart M. Neural modulation of cardiac arrhythmias and sudden cardiac death. Heart Rhythm. 2006;3:108–113. doi: 10.1016/j.hrthm.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thackeray JT, Bengel FM. Assessment of cardiac autonomic function using PET imaging. J Nucl Cardiol. 2013;20:150–165. doi: 10.1007/s12350-012-9644-4. [DOI] [PubMed] [Google Scholar]
- 6.Raffel DM, Wieland DM. Assessment of cardiac sympathetic nerve integrity with positron emission tomography. Nucl Med Biol. 2001;28:541–559. doi: 10.1016/s0969-8051(01)00210-4. [DOI] [PubMed] [Google Scholar]
- 7.Wieland DM, Brown LE, Rogers WL, Worthington KC, Wu JL, Clinthorne NH, Otto CA, Swanson DP, Beierwaltes WH. Myocardial imaging with a radioiodinated norepinephrine storage analog. J Nucl Med. 1981;22:22–31. [PubMed] [Google Scholar]
- 8.Raffel DM, Jung YW, Gildersleeve DL, Sherman PS, Moskwa JJ, Tluczek LJ, Chen W. Radiolabeled phenethylguanidines: novel imaging agents for cardiac sympathetic neurons and adrenergic tumors. J Med Chem. 2007;50:2078–2088. doi: 10.1021/jm061398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson AF, Matsuoka DT. Influence of myocardial region of interest definition on quantitative analysis of planar 123I-mIBG images. Eur J Mol Imaging. 2013;40:558–564. doi: 10.1007/s00259-012-2336-0. [DOI] [PubMed] [Google Scholar]
- 10.Raffel DM. Targeting norepinephrine transporters in cardiac sympathetic nerve terminals. In: Welch MJ, Eckelman WC, editors. Targeted Molecular Imaging. CRC Press; Boca Raton: 2012. pp. 305–320. [Google Scholar]
- 11.Raffel DM, Koeppe RA, Jung YW, Gu G, Jang KS, Sherman PS, Quesada CA. Quantification of cardiac sympathetic nerve density of N-11C-guanyl-meta-octopamine and tracer kinetic analysis. J Nucl Med. 2013;54 doi: 10.2967/jnumed.113.20659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang KS, Jung YW, Sherman PS, Quesada CA, Gu G, Raffel DM. Synthesis and bioevaluation of [18F]4-fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG): a novel radiotracer for quantitative PET studies of cardiac sympathetic innervation. Bioorg Med Chem Lett. 2013;23:1612–1616. doi: 10.1016/j.bmcl.2013.01.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pages T, Langlois BR. Fluorination of aromatic compounds from 1-aryl-3,3-dimethyltriazenes and fluoride anions in acidic medium: 1. A model for 18F labelling. J Fluorine Chem. 2001;107:321–327. [Google Scholar]
- 14.Pages T, Langlois BR, Le Bars D, Landais P. Fluorination of aromatic compounds from 1-aryl-3,3-dimethyltriazenes and fluoride anions in acidic medium: 2. Synthesis of (S)-[18F]-3-fluoro-α-methylphenylalanine. J Fluorine Chem. 2001;107:329–336. [Google Scholar]
- 15.Chu CK, Kim JH, Kim DW, Chung KH, Katzenellenbogen JA, Chi DY. Aromatic fluorination by decomposition of triazenes in ionic liquids. Bull Korean Chem Soc. 2005;26:599–602. [Google Scholar]
- 16.Hostetler ED, Jonson SD, Welch MJ, Katzenellenbogen JA. Synthesis of 2-[18F]fluoroestradiol, a potential diagnostic imaging agent for breast cancer: strategies to achieve nucleophilic substitution of an electron-rich aromatic ring with [18F]F−. J Org Chem. 1999;64:178–185. doi: 10.1021/jo981619a. [DOI] [PubMed] [Google Scholar]
- 17.Pike VW, Aigbirhio FI. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts - a novel single step route to no-carrier-added [18F]fluoroarenes. J Chem Soc Chem Commun. 1995;21:2215–2216. [Google Scholar]
- 18.Cai L, Lu S, Pike VW. Chemistry with [18F]fluoride ion. Eur J Org Chem. 2008;2008:2853–2873. [Google Scholar]
- 19.Wang B, Qin L, Neumann KD, Uppaluri S, Cerny RL, DiMagno SG. Improved arene fluorination for I(III) salts. Org Lett. 2010;12:3352–3355. doi: 10.1021/ol101154h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun JH, Lu S, Pike VW. Rapid and efficient radiosyntheses of meta-substituted [18F]fluoroarenes from [18F]fluoride ion and diaryliodonium tosylates within a microreactor. Eur J Org Chem. 2011;2011:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tredwell M, Gouverneur V. 18F labeling of arenes. Angew Chem Int Ed. 2012;51:11426–11437. doi: 10.1002/anie.201204687. [DOI] [PubMed] [Google Scholar]
- 22.Lee BC, Lee KC, Lee H, Mach RH, Katzenellenbogen JA. Strategies for the labeling of halogen-substituted peroxisome proliferator-activated receptor γ ligands: potential positron emission tomography and single photon emission computed tomography imaging agents. Bioconjugate Chem. 2007;18:514–523. doi: 10.1021/bc060191g. [DOI] [PubMed] [Google Scholar]
- 23.Lee BC, Dence CS, Zhou H, Parent EE, Welch MJ, Katzenellenbogen JA. Fluorine-18 labeling and biodistribution studies on peroxisome proliferator-activated receptor γ ligands: potential positron emission tomography imaging agents. Nucl Med Biol. 2009;36:147–153. doi: 10.1016/j.nucmedbio.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Telu S, Chun JH, Simeon FG, Lu S, Pike VW. Syntheses of mGluR5 PET radioligands through the radiofluorination of diaryliodonium tosylates. Org Biomol Chem. 2011;9:6629–6638. doi: 10.1039/c1ob05555k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon BS, Kil HS, Park JH, Kim JS, Park J, Chi DY, Lee BC, Kim SE. Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with evaluation of their stability and selectivity. Org Biomol Chem. 2011;9:8346–8355. doi: 10.1039/c1ob06277h. [DOI] [PubMed] [Google Scholar]
- 26.Ross TL, Ermert J, Hocke C, Coenen HH. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J Am Chem Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 27.Kazmierczak P, Skulski L, Kraszkiewicz L. Synthesis of (diacetoxyiodo)arenes or iodylarenes from iodoarenes, with sodium periodate as the oxidant. Molecules. 2001;6:881–891. [Google Scholar]
- 28.Chun JH, Pike VW. Regiospecific syntheses of functionalized diaryliodonium tosylates via [hydroxy(tosyloxy)iodo]arenes generated in site from (diacetoxyiodo)arenes. J Org Chem. 2012;77:1931–1938. doi: 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chun JH, Lu S, Lee YS, Pike VW. Fast and high-yield microreactor syntheses of ortho-substituted [18F]fluoroarenes from reactions of [18F]fluoride ion with diaryliodonium salts. J Org Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carroll MA, Nairne J, Smith G, Widdowson DA. Radical scavengers: a practical solution to the reproducibility issue in the fluoridation of diaryliodonium salts. J Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 31.Musiol HJ, Moroder L. N-N′-Di-tert-butoxycarbonyl-1H-benzotriazole-1-carboxamidine derivatives are highly guanidinylating reagents. Org Lett. 2001;3:3859–3861. doi: 10.1021/ol010191q. [DOI] [PubMed] [Google Scholar]
- 32.Salt PJ. Inhibition of noradrenaline uptake2 in the isolated rat heart by steroids, clonidine and methoxylated phenylethylamines. Eur J Pharmacol. 1972;20:329–340. doi: 10.1016/0014-2999(72)90194-x. [DOI] [PubMed] [Google Scholar]
- 33.Ratib O, Phelps ME, Huang SC, Henze E, Selin CE, Schelbert HR. Positron tomography with deoxyglucose for estimating local myocardial glucose metabolism. J Nucl Med. 1982;23:577–586. [PubMed] [Google Scholar]
- 34.Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow. 1985;5:584–590. doi: 10.1038/jcbfm.1985.87. [DOI] [PubMed] [Google Scholar]
- 35.Low G, Dhliwayo H, Lomas DJ. Adrenal neoplasms. Clin Radiol. 2012;67:988–1000. doi: 10.1016/j.crad.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine. J Nucl Med. 2005;46:1023–1027. [PubMed] [Google Scholar]
- 37.National Research Council. Guide for the Care and Use of Laboratory Animals. U.S. Department of Health and Human Services, National Institutes of Health; Bethesda, MD: 1985. [Google Scholar]
- 38.Kirschner AS, Ice RD, Beierwaltes WH. Radiation dosimetry of 131I-19-iodocholesterol: the pitfalls of using tissue concentration data, the author’s reply. J Nucl Med. 1975;16:248–249. [Google Scholar]
- 39.Stabin MG. Fundamentals of Nuclear Medicine Dosimetry. Springer; New York: 2008. p. 237. [Google Scholar]
- 40.Qi J, Leahy RM. Resolution and noise properties of MAP reconstruction for fully 3DPET. IEEE Trans Med Imaging. 2000;19:493–506. doi: 10.1109/42.870259. [DOI] [PubMed] [Google Scholar]
- 41.Narimatsu S, Kobayashi N, Asaoka K, Masubuchi Y, Horie T, Hosokawa M, Ishikawa T, Ohmori S, Kitada M, Miyano J, Kataoka H, Yamamoto S. Highperformance liquid chromatographic analysis of the sulfation of 4-hydroxypropranolol enantiomers by monkey liver cytosol. Chirality. 2001;13:140–147. doi: 10.1002/1520-636X(2001)13:3<140::AID-CHIR1011>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.