

Abstract
Current adjuvant treatment regimens available for the treatment of glioblastoma are widely ineffective and offer a dismal prognosis. Advancements in conventional treatment strategies have only yielded modest improvements in overall survival. Immunotherapy remains a promising adjuvant in the treatment of GBM through eliciting tumor specific immune responses capable of producing sustained antitumor response while minimizing systemic toxicity. Heat Shock Proteins (HSP) function as intracellular chaperones and have been implicated in the activation of both innate and adaptive immune systems. Vaccines formulated from HSP-peptide complexes, derived from autologous tumor, have been applied to the field of immunotherapy for glioblastoma. The results from the phase I and II clinical trials have been promising. Here we review the role of HSP in cellular function and immunity, and its application in the treatment of glioblastoma.
Keywords: vaccine, heat shock protein, glioblastoma, glioma, immunotherapy, clinical trial
Introduction
A diagnosis of glioblastoma (GBM) portends a bleak prognosis due to the malignant properties intrinsic to the neoplasm and the limited therapeutic options available for treatment. The current standard-of-care, acknowledged as the “Stupp protocol”, remains relatively poor, resulting in a median overall survival (OS) of 14.6 months with adjuvant temozolomide-based chemoradiation after optimal resection[1]. Concurrent with the disheartening GBM statistics over the last decade were advances in immunotherapy treatments for metastatic systemic-based cancers. These advances, along with our increased knowledge of the altered genomic landscape of GBM in predicting potential antigens, have sparked and propelled great interest in not only harnessing the immune system to target glioblastoma, but also investigating how GBM modulates the immune system.
As opposed to lower grade glial neoplasms, GBM is highly antigenic and is enriched with lymphocytic infiltrates. A large contributor to the infiltrative cohort of lymphocytes phenotypically express CD4+ CD25+ FOXP3+ markers, thus identifying them as a regulatory T-cell (TReg) population which may serve to limit the immunogenic response of infiltrative cytotoxic lymphocytes (CTLs)[2]. Direct cellular inhibition of lymphocytes is also attributed to glioma cells as they acquire a higher expression of B7-H1 (PD-L1) with the loss of the PTEN tumor suppressor gene [3]. Additional immune-limiting barriers that patients face are attributed to systemic immunosuppression propagated by the neoplasm itself as well as the suppression imposed by the cytotoxic nature of the chemotherapeutic agent administered after resection[4]. Thus, current and future immunotherapy agents are tasked with the challenge of eliciting an immunogenic response towards GBM that can overcome both the local and systemic state of immunosuppression.
Immunotherapy can be stratified into either an active or passive form in terms of function. An example of a passive intervention would be monoclonal antibodies directed at aberrant neoplastic proteins or even immune checkpoint proteins which restrain immunogenic attack on a specific target. This method is considered passive because it does not directly stimulate the host immune system and relies on continuous exogenous introduction in order to gain benefit from the therapy. In contrast, the active arm of immunotherapy aims to educate the host immune system so it can autonomously train naïve immune cells against antigenic targets. An example of this would consist of vaccines which may be taken up and presented by resident antigen presenting cells (APCs) to lymphocytes.
There are a number of vaccines which have been recruited for the treatment against glioblastoma[5]. Peptide vaccines introduce short protein sequences of known antigenic entities within glioblastoma (e.g. EGFRviii) in order to elicit an immune response against the neoplastic cells harboring the mutant proteins [6]. Autologous vaccines are based on retrieving a patient’s peripheral blood cells, modifying them (e.g. stimulating with known tumor antigens or altering the autologous tumor cells with viruses) and re-infusing the primed immune cells back into the host[7]. Dendritic-cell-based vaccines pulse dendritic cells, isolated from peripheral blood mononuclear cells, with glioma antigens retrieved from resected tumorso they can stimulate naïve lymphocytes when reintroduced into the host[8]. In this review, we will focus primarily on heat shock protein (HSP)-peptide based vaccines. This vaccination method deals with the isolation and purification of HSPs from resected GBM patients with subsequent reinfusion of the complex to allow the chaperone to interact with APCs, thus primingthe lymphocytes with a varied cohort of antigenic peptides.
Cellular function of heatshock proteins
Heat shock proteins (HSPs), also known as chaperone proteins, are abundant across mammalian cell types, in which they play a vital role in the stress response to cellular insults including hyperthermia, inflammation, hypoxia, oxidative stress, and radiation.[9] The assembly and transport of nascent proteins within the cell relies on the activity of HSPs, especially in adverse intracellular situations where HSPs function to stabilize proteins and prevent aggregation.[10] In addition, HSPs also act to resolve protein aggregates, reassemble salvageable misfolded proteins, and guide the degradation of unsalvageable misfolded proteins following the resolution of cellular insults.[11] As a result, it is believed that HSPs are transcriptionally upregulated in cancer where there exists increased translation of abnormal protein products.[12] Analyses based on molecular weight and phylogenetics have distinguished five major HSP families, however only HSP gp 96, HSP 90, HSP 70, HSP 110, and HSP 170 have demonstrated immunogenic interactions as membrane-bound and extracellular components.[13, 14]
Specifically, in GBM, elevated constitutive and inducible expression of HSP27, αB-crystallin, HSP72, HSP73, and HSP90 has been reported both in vitro and in vivo.[15, 16] Moreover, HSP27, HSP60, HSP70, and HSP90 have been shown to be present in GBM released exosomes.[16] Of particular interest to GBM, however, are HSP70 and HSP90. The HSP70 family functions to inhibit cell stress induced apoptotic pathways, facilitate protein folding, and guide protein transport across membranes.[11] Recently, increased transcription of HSP70 mRNA was shown to correlate with glioma grade.[17] Moreover, HSPs within the HSP70 family of chaperone proteins were the first HSPs shown to bind antigenic peptides.[18] The family of HSPs to which HSP90 belongs is largely responsible for protein folding, protein stabilization, and peptide loading onto MHC class I molecules. Importantly, HSP90 substrates (including EGFRvIII, FAK, AKT, hTERT, p53, cdk4, MAPK, and PI3K) are involved in key tumor initiation and proliferation signaling pathways.[11] Similar to HSP70, HSP90 is also associated with the binding of tumor antigens that can elicit a tumor rejection response.[19, 20] As a result, HSPs have been targeted as potential vehicles by which to present tumor specific antigens in glioblastoma to elicit an antitumor immune response.
Heatshock proteins in immunity
A strength possessed by the HSP-vaccine is the ability to stimulate both the innate and adaptive immune responses. Alone, neither the HSP or the isolated peptides are immunogenic; only when complexed they are able to elicit an MHC-class I CD8+ cytotoxic T-lymphocyte (CTL) response[24]. Classically, exogenous antigens have been known to primarily be presented via MHC class II on APCs which promotes the interaction with T-helper cells (CD4+). HSP complexes have the added benefit of being able to undergo the endogenous MHC class I pathway to induce a CD8+ response. The CD91 receptor allocated on APCs is responsiblefor the uptake of the HSP complex into the cell [25]. Upon internalization, the complex undergoes processing via proteasomes, gets transported into the ER, and is ultimately loaded onto to MHC-class I for presentation to CD8+ CTLs[26]. In addition to this cytosolic pathway, an endosomal method which is proteasome-independent is also possible for loading the peptides onto MHC class I[27]. A small portion of the internalized HSP complex can also enter an acidic compartment which leads to MHC-class II loading of the peptide for the stimulation of CD4+ cells[28]. There are also other potential receptors which the HSP complex may interact, leading to a non-CD91 presentation of chaperoned proteins[29].
HSP-peptide complexes (HSPPCs) have the ability to interact with a number of cell surface receptors on APC’s which induces downstream activation of the NF-κB pathway (Figure 1). Some of these receptors are believed to include: CD36/CD91/CD40/CD14/Toll-like receptor 2 (TLR2) /Toll-like receptor 4(TLR4). There are potentially other cell surface receptors that interact with the HSP complex that have yet to be elucidated. In macrophages in particular, HSP stimulates the secretion of proinflammatory cytokines such as tumor necrosis factor α (TNFα), granulocyte macrophage colony-stimulating factor (GM-CSF), IL-12, and IL-1β. IL-12 may serve to activate the cytotoxic activity of both lymphocytes and natural killer (NK) cells. Additionally, HSP complexes are able to augment the production and secretion of nitric oxide in both dendritic cells and macrophages[30]. Interestingly, the combination of these secretory products induced by the interaction of HSP with cell surface receptors on macrophages coincide with the proinflammatory phenotype of macrophages (M1). The HSPPC additionally induces immature dendritic cells to undergo maturation which is noted by the increased expressivity of MHC class II and CD86 as well as the increased secretion of IL-12 and TNFα which further potentiate a proinflammatory response[31].
Figure 1. HSP-peptide complex interaction with antigen presenting cells.
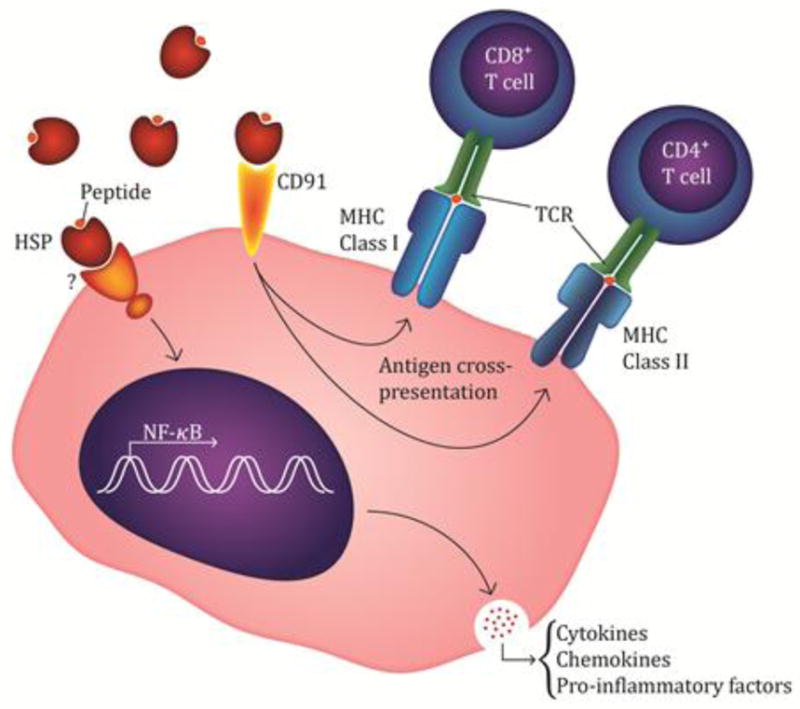
The proposed mechanism by which the HSP-peptide complex interacts with APC’s consists of cell surface receptor interaction. Primarily, CD91 has been shown to endocytose the complex and via either proteasome dependent or independent pathways lead to the presentation via MHC-class I receptor. In addition, a portion of the internalized complex enters an acidic compartment which leads to its loading onto MHC-class II receptors. Additional cell surface receptors, such as TLR2/TLR4, and others that have not been elucidated are also involved in eliciting a downstream effect which leads to the activation of the NF-κB pathway. Upon activation, proinflammatory cytokines and chemokines are generated and secreted in order to further augment a proinflammatory response.
One advantage possessed by HSP-based vaccinations is that they are not specific to one pre-defined antigen. While other vaccine modalities target one specific GBM antigen (e.g. EGFRviii), HSPPCs manage to present various types of potential antigenic proteins upon vaccination. This is a crucial facet to this vaccine methodology due to the intratumoral heterogeneity posed by glioblastoma. One of the hallmarks of cancer in general is that of immunoediting which selects for the non-immunogenic subset of cells within a tumor to survive and thrive. By vaccinating individuals with a patient-specific polyvalent HSPPC vaccine, it may provide an added advantage compared to vaccination against one specific antigen. An additional advantage of HSPPC vaccines is that they manifest their immunogenic benefit via multiple mechanisms which augment cytotoxic effects via other cell types in addition CTLs. A potential drawback to blindly vaccinating against unknown antigenic variants is that the immune system may be trained to target antigens which are not ultimately essential and expressed only in a minority of neoplastic cells. Although, this limitation may be hampered via epitope spreading whereby immune cells originally primed for a specific antigenic epitope can detect different unrelated epitopes, allowing for the detection of new antigens on the peptide[32]. This may theoretically further increase the antigenic repertoire of the induced immune response, thus allowing the immune system to target neoplastic cells with distinct antigenic epitopes differing from the initial epitope used for lymphocytic priming.
Heatshock protein vaccines
Following positive results acquired in the preclinical setting, phase I, II, and III clinical trials were conducted to investigate the safety and efficacy of vaccination with autologous tumor-derived HSP-peptide complex based antitumor vaccines in various tumor types.. The majority of HSP vaccine trials have utilized heat shock protein-peptide complex-96 (HSPPC-96), comprising autologous antigenic peptides chaperoned by HSP glycoprotein-96 (HSP gp-96) While other HSP families have share similar theoretical advantages for clinical translation, early pilot studies have demonstrated HSPPC-96 to be safe with minimal toxicity, and feasible with regards to purification and production as a clinical grade product. HSPPC-96 vaccines, developed from resected tumor specimens that are frozen and delivered to the vaccine manufacturer. Utilizing liquid chromatography, HSPs are isolated and further enriched by subsequent denaturing gel electrophoresis and anti-gp 96 western blot[33, 34]. Including sterility and endotoxin quality control screening, the manufacturing process takes three to four weeks from the date of tumor resection to vaccine release[35]. Typical vaccination schedules utilizing HSPPC-96 require weekly vaccination for the initial four weeks of therapy, followed by biweekly administration until supply depletion.
Non-glioma malignancies
To date, the technique has been utilized in several cancer types in an attempt to exploit the distinctive, patient-specific immunogenic potential offered by HSPPC-96 vaccination with varying degrees of success. Janetzki et al was the first to investigate the application of autologous HSPPC-96 in human malignancies. [36] To establish the safety profile of HSPPC-96 and evaluate immune responses in this pilot study, patients with a variety of cancers refractory to standard therapies received HSPPC-96 vaccines prepared from resected tumor tissue. Results from the study demonstrated feasibility of vaccine production and lack of toxicity. While the limited number of patients and study design precluded a clear evaluation of clinical efficacy, robust immune responses following immunization were noted in a majority of patients characterized by increased levels of NK cells and expansion of tumor specific T cells, consistent with observations in preclinical murine studies. [37, 38] Rivoltini et al. similarly demonstrated that treatment with tumor derived HSPPC-96 in 10 patients with either melanoma or colon carcinoma led to activation and expansion of tumor antigen specific CD8+ T cells in vitro and in vivo. [39] Subsequently, a number of phase I/II studies further demonstrated the feasibility of vaccine production, lack of toxicity, and signs of clinical activity in a range of tumors including colorectal cancer [40], non-small cell lung cancer (NSCLC) [41], pancreatic adenocarcinoma [42], and melanoma [35, 43, 44]. These studies demonstrated feasibility and safety, while also noting evidence of post vaccination tumor specific immune responses.
Two phase III clinical trials followed these studies, comprising the largest studies on HSPPC-96 tumor vaccines to date. The first randomized, multicenter trial enrolled 322 patients with metastatic melanoma and compared autologous HSPPC-96 vaccine to controls who received physician’s choice of therapy, comprised of Dacarbazine, Temozolomide (TMZ), IL-2, complete tumor resection either alone or in combination.[45] OS, via intention to treat analysis, did not differ between the vaccine and control arms. Consistent with limitations cited in phase I/II trials, vaccine production was constrained by the availability of adequate resected tumor tissue and technical challenges. Only 61.8% of patients assigned to the vaccine arm received one or more doses. For those who received vaccinations, the number of doses was also highly variable. Controlling for the bias in which patients living longer would be able to receive more vaccine doses through landmark analysis, subset analysis revealed that patients harboring M1a and M1b disease substages who received a greater number of immunizations survived longer than those who received fewer vaccinations (1+ vs 10+ doses). Thus, clinical efficacy was most evident in those with earlier stages of disease receiving higher number of doses. [45] The second a multicenter randomized phase III trial investigated efficacy of HSPPC-96 vaccine versus observation following nephrectomy in 728 patients with locally advanced renal cell carcinoma.[46] On median follow up of 1.9 years, there were no differences in recurrence (37.7% vs. 39.8%) or survival (19.4% vs. 19.6%) between the treatment and observation groups, respectively., Post-hoc subgroup analysis demonstrated a PFS benefit in intermediate risk patients as defined by ECOG risk stratification. [47] Rate of recurrence was lower in the treatment versus observation group in intermediate risk patients (15.2% vs 26.4%, p=0.026). However, there were no difference in overall survival. While this suggested increased vaccine efficacy in patients with less advanced disease, this should be interpreted with caution given the limitations of post-hoc analysis [48] No clinical benefit was seen in patients who were at high risk. These large phase III trials highlighted a number of factors concerning HSPPC-94 vaccines. First, increased number of vaccine doses was correlated with improved clinical response. Second, vaccine was most effective in patients with less advanced disease, possibly secondary to increasing numbers of mechanisms by which more advanced staged malignancies evade an immune mediated antitumor response.
Glioblastoma
In a phase I dose escalation trial, Crane et al. investigated of the role of HSPPC-96 in the vaccination of patients with recurrent high grade glioma.[49] Twelve patients met postsurgical study criteria (table 1). Patients either received 25μg HSPPC-96 every 2 weeks totaling 4 vaccinations or 25μg HSPPC-96 weekly for a total of 4 vaccinations. After the first 4 vaccination treatments, all patients were placed on a biweekly dosing schedule. Overall, this vaccine strategy appeared safe and tolerable with no significant toxicities encountered. A tumor-specific peripheral immune response to vaccine administration was present in 11 of the 12 patients. Restimulation of peripheral blood leukocytes with autologous HSP ex vivo demonstrated increased T cell proliferation and significant increase in IFN-γ production. These peripheral immune assays correlated with the proinflammatory immunogenic response induced by the vaccine. This was demonstrated in the 7 patients that underwent subsequent tumor biopsies after receiving the vaccine; their tumors harbored IFN-γ positive NK and T-cells which demonstrated that immune effector cells were localizing to the tumor site. Immune response was associated with clinical outcome, with a median OS of 47 weeks in immune responders compared to 16 weeks in nonresponders. [49]
Table 1.
Inclusion/Exclusion criteria for the phase I trial of autologous HSPPC-96 in the recurrent setting of glioblastoma [49].
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
|
In a subsequent open label phase II multicenter clinical trial, 68 adult patients with recurrent GBM were enrolled and underwent gross total resection. Only 41 patients met pre- and postoperative criteria (Table 2).[50] All patients received 25μg HSPPC-96 weekly for 4 weeks, followed by a biweekly dosing schedule. Only 3 patients failed to receive the protocol minimum of 4 doses. There were 17 vaccine attributable grade 3–4 adverse effects. Median and 6 month PFS were 19.1 weeks and 29.3%, respectively. Median and 6 months OS were 42.6 weeks and 29.3%, respectively. Evaluation of the prognostic impact of immunological status through subgroup analysis based on absolute lymphocyte count (ALC) demonstrated that an ALC above the median of the cohort was associated with improved survival on univariate (49.1 vs 37.1 weeks, p = 0.39) and multivariate analysis (HR 4.0, CI 1.4–11.8; p = 0.012). Results are promising in comparison to historical controls within similarly surgically focused trials for recurrent GBM. Examples of these include the PRECISE phase III Trial. Treatment in this study consisted of convection-enhanced delivery of a chimeric cytotoxin comprising human interleukin-13 fused to a truncated form of pseudomonas exotoxin (Cintredekin Besudotox) which was compared to implanted Gliadel wafers following resection in the management of recurrent GBM. Median OS was 36.4 weeks in patients receiving the chimeric cytotoxin and 35.3 weeks for the group receiving Gliadel Wafers. [51]
Table 2.
Inclusion/Exclusion criteria for the phase II trial of autologous HSPPC-96 in the recurrent setting of glioblastoma [50].
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
Postoperative criteria:
|
For recurrent GBM, the HSPPC-96 vaccination trial uniquely demonstrated both a peripheral and tumoral immune response which correlated with clinical outcome. A strong association between pre-vaccination lymphopenia and significantly worse outcomes further elaborates on the role of GBM mediated immunosuppression and possible benefit of addressing a patient’s immune status prior to vaccination. One of the methods in which GBM exerts a state of immunosuppression is by inducing B7-H1 expression in both circulating and tumor-infiltrating macrophages. Patients that demonstrated monocytes with high expression of B7-H1 had significantly worse median PFS when compared to patients with low B7-H1 expressing monocytes (10 vs 17 months respectively).[52] Since vaccine efficacy is dependent on a viable immunological response, addressing these immunologic deterrents may yield promising results.
Additionally, there is a completed multicenter trial with data pending publication. This phase II single arm study investigated the application of autologous HSPPC-96 vaccine in newly diagnosed adult patients with GBM undergoing standard of therapy (NCT00905060). Patients received weekly intradermal injections of vaccine for 4 consecutive weeks following tumor resection and adjuvant radiation therapy and temozolomide.
Ongoing clinical trials
After the encouraging results demonstrated by the previous phase II trial of HSPPC-96 on recurrent glioblastomas, a subsequent multi-institutional trial sponsored by the Alliance for Clinical Trials in Oncology (ALLIANCE) is currently recruiting (NCT01814813). This trial will help provide evidence as to whether if HSPPC-96 can prolong overall survival in cases of recurrent GBM as an adjuvant therapeutic agent. The study will consist of three different arms which include: HSPPC-96 with concomitant bevacizumab, HSPPC-96 with administration of bevacizumab at tumor progression, and bevacizumab alone. In addition to the primary measure of OS, secondary outcomes evaluated will include PFS, in addition to the safety and tolerability of the combinatorial therapy. Samples collected throughout the trial will be utilized to correlate immune responders to HSPPC-96 with survival outcome as well as investigating whether lymphocytic infiltrates at tumor baseline correlate with the response to the vaccine. There is also a phase I trial in Beijing, China studying the safety and efficacy of autologous HSP gp96 in newly diagnosed supratentorial gliomas (NCT02122822).
Limitations
Application of HSPPC-96 vaccine has demonstrated promise but is not without limitations. Acquisition of adequate tissue for vaccine production has been challenging in previous clinical trials. In the HSPPC-96 phase II trial for vaccination against GBM, the authors note that while vaccines were unable to be produced in 13 out of 63 patients, modifications and improvement in technique with patients enrolled later in the study led to improved rates of vaccine yield.[56] The inclusion criteria requiring near-complete tumor resection limiting patient eligibility may limit the generalizability of HSPPC-96 vaccine from the phase II trial.[56, 57] Progression free survival in the phase II study was not significantly improved compared to conventional salvage therapy for recurrent GBM. However distinguishing tumor recurrence from pseudoprogression due to treatment-related changes on postoperative imaging can be difficult. The trial demonstrated promising median overall survival, which provides a more definitive measure of clinical efficacy.
Conclusion
Immunotherapy remains a promising adjuvant in the treatment of GBM through eliciting tumor specific immune responses capable of producing sustained antitumor response while minimizing systemic toxicity. Theoretically, HSP vaccines provides a number of advantages including direct interaction APCs for antigen internalization and presentation, stimulation of both innate and adaptive immune responses. HSP vaccines allow for the delivery of a patient specific polyvalent vaccine that does not require identification of specific immunogenic GBM antigens. Multiple antigens are used to minimize the risk of immune evasion, which may occur with vaccines that utilize a single antigen. Within clinical trials, HSPPC-96 was safe with minimal adverse effects in the treatment of a variety of cancer types. Efficacy has been variable and improvements in outcomes were not seen in a variety of cancer types. However, HSPPC-96 has been most promising in phase I and II trials with recurrent GBM, a devastating disease with limited treatment options, as demonstrated by superior outcomes compared to historical controls. Challenges include acquisition of sufficient tumor for vaccine production and requirement of gross total resection, which may not always be achievable. However, with increasing experience through past and ongoing trials, issues with vaccine yield, patient selection, and screening will be optimized. HSPPC-96 is a promising immunotherapeutic adjuvant for treatment of GBM, and pending results from completed and ongoing trials will help further elaborate on the role of HSPPC-96 in the treatment of this devastating disease with limited treatment options.
Reference List
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organisation for R, Treatment of Cancer Brain T Radiotherapy G, National Cancer Institute of Canada Clinical Trials G. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 4.Waziri A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurgery clinics of North America. 2010;21:31–42. doi: 10.1016/j.nec.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Ampie L, Woolf EC, Dardis C. Immunotherapeutic advancements for glioblastoma. Frontiers in oncology. 2015;5:12. doi: 10.3389/fonc.2015.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, 2nd, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ, Bigner DD. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schirrmacher V, Haas C, Bonifer R, Ahlert T, Gerhards R, Ertel C. Human tumor cell modification by virus infection: an efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene therapy. 1999;6:63–73. doi: 10.1038/sj.gt.3300787. [DOI] [PubMed] [Google Scholar]
- 8.Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nature reviews Molecular cell biology. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 10.Craig E, Weissman JS. Heat shock proteins and molecular chaperones: Mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–372. doi: 10.1016/0092-8674(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 11.Graner MW, Bigner DD. Chaperone proteins and brain tumors: Potential targets and possible therapeutics. Neuro-Oncology. 2005;7:260–278. doi: 10.1215/s1152851704001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implicaitons. Cell Stress & Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. Journal of leukocyte biology. 2007;81:15–27. doi: 10.1189/jlb.0306167. [DOI] [PubMed] [Google Scholar]
- 14.Binder RJ. Heat shock protein vaccines: from bench to bedside. Int Rev Immunol. 2006;25:353–375. doi: 10.1080/08830180600992480. [DOI] [PubMed] [Google Scholar]
- 15.Hermisson M, Strik H, Rieger J, Dichgans J, Meyermann R, Weller M. Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology. 2000;54:1357–1365. doi: 10.1212/wnl.54.6.1357. [DOI] [PubMed] [Google Scholar]
- 16.Graner MW, Cumming RI, Bigner DD. The heat shock response and chaperones/heat shock proteins in brain tumors: surface expression, release, and possible immune consequences. The Journal of Neuroscience. 2007;27:11214–11227. doi: 10.1523/JNEUROSCI.3588-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaman GM, Dennison SR, Chatfield LK, Phoenix DA. Reliability of HSP70 (HSPA) expression as a prognostic marker in glioma. Molecular and cellular biochemistry. 2014;393:301–307. doi: 10.1007/s11010-014-2074-7. [DOI] [PubMed] [Google Scholar]
- 18.Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245:385–390. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proceedings of the National Academy of Sciences. 1986;83:3407–3411. doi: 10.1073/pnas.83.10.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ullrich SJ, Robinson EA, Law LW, Willingham M, Appella E. A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proceedings of the National Academy of Sciences. 1986;83:3121–3125. doi: 10.1073/pnas.83.10.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:3407–3411. doi: 10.1073/pnas.83.10.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ullrich SJ, Robinson EA, Law LW, Willingham M, Appella E. A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:3121–3125. doi: 10.1073/pnas.83.10.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava PK, Maki RG. Stress-induced proteins in immune response to cancer. Current topics in microbiology and immunology. 1991;167:109–123. doi: 10.1007/978-3-642-75875-1_7. [DOI] [PubMed] [Google Scholar]
- 24.Blachere NE, Li Z, Chandawarkar RY, Suto R, Jaikaria NS, Basu S, Udono H, Srivastava PK. Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. The Journal of experimental medicine. 1997;186:1315–1322. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Binder RJ, Srivastava PK. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6128–6133. doi: 10.1073/pnas.0308180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 27.Castellino F, Boucher PE, Eichelberg K, Mayhew M, Rothman JE, Houghton AN, Germain RN. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. The Journal of experimental medicine. 2000;191:1957–1964. doi: 10.1084/jem.191.11.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsutake T, Sawamura T, Srivastava PK. High efficiency CD91- and LOX-1-mediated representation of gp96-chaperoned peptides by MHC II molecules. Cancer immunity. 2010;10:7. [PMC free article] [PubMed] [Google Scholar]
- 29.Berwin B, Hart JP, Pizzo SV, Nicchitta CV. Cutting edge: CD91-independent cross-presentation of GRP94(gp96)-associated peptides. Journal of immunology. 2002;168:4282–4286. doi: 10.4049/jimmunol.168.9.4282. [DOI] [PubMed] [Google Scholar]
- 30.Panjwani NN, Popova L, Srivastava PK. Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. Journal of immunology. 2002;168:2997–3003. doi: 10.4049/jimmunol.168.6.2997. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nature reviews Immunology. 2002;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- 32.Vanderlugt CL, Begolka WS, Neville KL, Katz-Levy Y, Howard LM, Eagar TN, Bluestone JA, Miller SD. The functional significance of epitope spreading and its regulation by co-stimulatory molecules. Immunological reviews. 1998;164:63–72. doi: 10.1111/j.1600-065x.1998.tb01208.x. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava PK. Purification of heat shock protein-peptide complexes for use in vaccination against cancers and intracellular pathogens. Methods. 1997:165–171. doi: 10.1006/meth.1997.0464. [DOI] [PubMed] [Google Scholar]
- 34.See AP, Pradilla G, Yang I, Han S, Parsa AT, Lim M. Heat shock protein-peptide complex in the treatment of glioblastoma. Expert review of vaccines. 2011;10:721–731. doi: 10.1586/erv.11.49. [DOI] [PubMed] [Google Scholar]
- 35.Belli F, Testori A, Rivoltini L, Maio M, Andreola G, Sertoli MR, Gallino G, Piris A. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: Clinical and immunologic findings. J Clin Oncol. 2002;20:4169–4180. doi: 10.1200/JCO.2002.09.134. [DOI] [PubMed] [Google Scholar]
- 36.Janetzki S, Palla D, Rosenhauer V, Lochs H, Lewis JJ, Srivastava PK. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: a pilot study. International journal of cancer Journal international du cancer. 2000;88:232–238. doi: 10.1002/1097-0215(20001015)88:2<232::aid-ijc14>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Hishii M, Andrews D, Boyle LA, Wong JT, Pandolfi F, van den Elsen PJ, Kurnick JT. In vivo accumulation of the same anti-melanoma T cell clone in two different metastatic sites. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1378–1383. doi: 10.1073/pnas.94.4.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jager E, Ringhoffer M, Dienes HP, Arand M, Karbach J, Jager D, Ilsemann C, Hagedorn M, Oesch F, Knuth A. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. International journal of cancer Journal international du cancer. 1996;67:54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 39.Rivoltini L, Castelli C, Carrabba M, Mazzaferro V, Pilla L, Huber V, Coppa J, Gallino G, Scheibenbogen C, Squarcina P, Cova A, Camerini R, Lewis JJ, Srivastava PK, Parmiani G. Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. Journal of immunology. 2003;171:3467–3474. doi: 10.4049/jimmunol.171.7.3467. [DOI] [PubMed] [Google Scholar]
- 40.Mazzaferro V, Coppa J, Carrabba MG, Rivoltini L, Schiavo M, Regalia E, Mariani L, Camerini T, Marchiano A, Andreola S, Camerini R, Corsi M, Lewis JJ, Srivastava PK, Parmiani G. Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:3235–3245. [PubMed] [Google Scholar]
- 41.Santis G, Senzer NN, Champagne P, Isakov L, Teofilovici F. Phase II feasibility study of autologous vaccine (HSPPC-96) in patients with resectable lung cancer. Journal of Clinical Oncology. 2008;26 [Google Scholar]
- 42.Maki RG, Livingston PO, Lewis JJ, Janetzki S, Klimstra D, Desantis D, Srivastava PK, Brennan MF. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Digestive diseases and sciences. 2007;52:1964–1972. doi: 10.1007/s10620-006-9205-2. [DOI] [PubMed] [Google Scholar]
- 43.Pilla L, Patuzzo R, Rivoltini L, Maio M, Pennacchioli E, Lamaj E, Maurichi A, Massarut S, Marchiano A, Santantonio C, Tosi D, Arienti F, Cova A, Sovena G, Piris A, Nonaka D, Bersani I, Di Florio A, Luigi M, Srivastava PK, Hoos A, Santinami M, Parmiani G. A phase II trial of vaccination with autologous, tumor-derived heat-shock protein peptide complexes Gp96, in combination with GM-CSF and interferon-alpha in metastatic melanoma patients. Cancer immunology, immunotherapy : CII. 2006;55:958–968. doi: 10.1007/s00262-005-0084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oki Y, McLaughlin P, Fayad LE, Pro B, Mansfield PF, Clayman GL, Medeiros LJ, Kwak LW, Srivastava PK, Younes A. Experience with heat shock protein-peptide complex 96 vaccine therapy in patients with indolent non-Hodgkin lymphoma. Cancer. 2007;109:77–83. doi: 10.1002/cncr.22389. [DOI] [PubMed] [Google Scholar]
- 45.Testori A, Richards J, Whitman E, Mann GB, Lutzky J, Camacho L, Parmiani G, Tosti G, Kirkwood JM, Hoos A, Yuh L, Gupta R, Srivastava PK, Group CS. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26:955–962. doi: 10.1200/JCO.2007.11.9941. [DOI] [PubMed] [Google Scholar]
- 46.Wood C, Srivastava P, Bukowski R, Lacombe L, Gorelov AI, Gorelov S, Mulders P, Zielinski H, Hoos A, Teofilovici F, Isakov L, Flanigan R, Figlin R, Gupta R, Escudier B, Group CRS. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. 2008;372:145–154. doi: 10.1016/S0140-6736(08)60697-2. [DOI] [PubMed] [Google Scholar]
- 47.Messing EM, Manola J, Wilding G, Propert K, Fleischmann J, Crawford ED, Pontes JE, Hahn R, Trump D Eastern Cooperative Oncology Group/Intergroup t . Phase III study of interferon alfa-NL as adjuvant treatment for resectable renal cell carcinoma: an Eastern Cooperative Oncology Group/Intergroup trial. J Clin Oncol. 2003;21:1214–1222. doi: 10.1200/JCO.2003.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Wood CG, Srivastava P, Lacombe L, Gorelov AI, Gorelov S, Mulders P, Zielinski H, Teofilovici F, Isakov L, Escudier B. Survival update from a multicenter, randomized, phase III trial of vitespen versus observation as adjuvant therapy for renal cell carcinoma in patients at high risk of recurrence. Journal of Clinical Oncology. 2009;27 [Google Scholar]
- 49.Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, Butowski N, Chang SM, Clarke J, Berger MS, McDermott MW, Prados MD, Parsa AT. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19:205–214. doi: 10.1158/1078-0432.CCR-11-3358. [DOI] [PubMed] [Google Scholar]
- 50.Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, Butowski NA, Chang SM, Clarke JL, McDermott MW, Prados MD, Sloan AE, Bruce JN, Parsa AT. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-oncology. 2014;16:274–279. doi: 10.1093/neuonc/not203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, Shaffrey M, Ram Z, Piepmeier J, Prados M, Croteau D, Pedain C, Leland P, Husain SR, Joshi BH, Puri RK, Group PS. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-oncology. 2010;12:871–881. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bloch O, Kaur R, Aghi M, McDermott M, Berger M, Parsa A. Glioma-Induced Immunosuppression Shortens Progression-Free Survival In A Trial of Immunotherapy For Glioblastoma. J Neurosurg. 2013;119:A565–A565. [Google Scholar]
- 53.Khasraw M, Ameratunga MS, Grant R, Wheeler H, Pavlakis N. Antiangiogenic therapy for high-grade glioma. The Cochrane database of systematic reviews. 2014;9:CD008218. doi: 10.1002/14651858.CD008218.pub3. [DOI] [PubMed] [Google Scholar]
- 54.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 55.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ, Jr, Mehta MP. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bloch O, Parsa AT. Heat shock protein peptide complex-96 (HSPPC-96) vaccination for recurrent glioblastoma: a phase II, single arm trial. Neuro-oncology. 2014;16:758–759. doi: 10.1093/neuonc/nou054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chamberlain MC. Is there a role for vaccine-based therapy in recurrent glioblastoma? Neuro-oncology. 2014;16:757. doi: 10.1093/neuonc/nou031. [DOI] [PMC free article] [PubMed] [Google Scholar]