

Abstract
Afferent (Af-Art) and efferent arterioles resistance regulate glomerular capillary pressure. The nephron regulates Af-Art resistance via: 1) vasoconstrictor tubuloglomerular feedback (TGF), initiated in the macula densa via Na-K-2Cl cotransporters (NKCC2) and 2) vasodilator connecting tubuloglomerular feedback (CTGF), initiated in connecting tubules via epithelial Na channels (ENaC). Furosemide inhibits NKCC2 and TGF. Benzamil inhibits ENaC and CTGF. In vitro, CTGF dilates preconstricted Af-Arts. In vivo, benzamil decreases stop-flow pressure (PSF), suggesting that CTGF antagonizes TGF; however, even when TGF is blocked, CTGF does not increase PSF, suggesting there is another mechanism antagonizing CTGF. We hypothesize that in addition to NKCC2, activation of Na/H exchanger (NHE) antagonizes CTGF, and when both are blocked CTGF dilates Af-Arts and this effect is blocked by a CTGF inhibitor benzamil. Using micropuncture, we studied the effects of transport inhibitors on TGF responses by measuring PSF while increasing nephron perfusion from 0 to 40 nl/min. Control TGF response (−7.9 ± 0.2 mmHg) was blocked by furosemide (−0.4 ± 0.2 mmHg; P < 0.001). Benzamil restored TGF in the presence of furosemide (furosemide: −0.2 ± 0.1 vs. furosemide+benzamil: −4.3 ± 0.3 mmHg; P < 0.001). With furosemide and NHE inhibitor, dimethylamiloride (DMA), increase in tubular flow increased PSF (furosemide+DMA: 2.7 ± 0.5 mmHg, n = 6), and benzamil blocked this (furosemide+DMA+benzamil: −1.1 ± 0.2 mmHg; P < 0.01, n = 6). We conclude that NHE in the nephron decreases PSF (Af-Art constriction) when NKCC2 and ENaC are inhibited, suggesting that in the absence of NKCC2, NHE causes a TGF response and that CTGF dilates the Af-Art when TGF is blocked with NKCC2 and NHE inhibitors.
Keywords: afferent arteriole, connecting tubuloglomerular feedback, tubuloglomerular feedback, stop-flow pressure, benzamil
since glomerular capillary pressure (GCP) plays the main role in controlling glomerular filtration rate (GFR), identifying the regulators of GCP is of extreme importance. GCP is determined by the resistance of afferent (Af-Art) and efferent arterioles (Ef-Art). Regulation of Af-Art resistance is similar to those of other arterioles. However, Af-Art resistance is also modified by two feedback mechanisms initiated in the nephron: 1) the vasoconstrictor tubuloglomerular feedback (TGF), initiated in the macula densa via the Na-K-2Cl cotransporters (NKCC2) (1, 20), and 2) vasodilator connecting tubuleoglomerular feedback (CTGF) initiated in the connecting tubule (CT) via the epithelial Na channels (ENaC) (2, 3, 9). TGF also regulates Ef-Arts, but instead of vasoconstriction it induces Ef-Art vasodilatation (19). Ef-Art resistance is also regulated by eicosanoids released from the glomerulus (17).
In vivo, when CTGF is inhibited with benzamil, TGF is potentiated causing a greater decrease in stop-flow pressure (PSF), suggesting that CTGF normally antagonizes TGF (24). Also, CTGF participates in TGF resetting (23). However, in vivo, even when TGF is blocked by the NKCC2 inhibitor furosemide, CTGF does not cause an increase in PSF (vasodilation), suggesting that there is another mechanism(s) that could antagonize CTGF. The vasodilator effect of CTGF in vivo had only been proven indirectly by observing that when CTGF is inhibited, it causes a potentiation of TGF as measured by a further decrease in PSF. Therefore, it is possible that other processes initiated by Na transport along the distal nephron may regulate Af-Art resistance and antagonize CTGF.
Recent studies have shown that in addition to expressing NKCC2, macula densa cells functionally and immunologically express Na/H exchanger (NHE) (6, 15). The distal convoluted tubule reabsorbs NaCl via thiazide-sensitive Na-Cl cotransporters (NCC) (13, 16). Thus, NCC may also be involved in the regulation of Af-Art resistance.
We hypothesize that in addition to NKCC2, under some circumstances NHE can also mediate a vasoconstrictor mechanism that antagonizes CTGF. Thus, when both NKCC2 and NHE are blocked, CTGF increases PSF due to Af-Art dilation (18). To test this hypothesis, we used the nephron micropuncture technique in vivo. We measured two consecutive TGF responses by increasing the perfusion of the nephron from 0 to 40 nl/min, while adding the drugs to the tubular perfusate that blocks transporters and measuring PSF as an index of GCP. The limitation of this technique is that a decrease in PSF or GCP could be due to an Af-Art constriction and/or Ef-Art dilation and vice versa, an increase in PSF could be due to an Af-Art dilation and/or Ef-Art constriction. Thus, the data presented here were interpreted taking into consideration these dual effects on TGF.
METHODS
Male Sprague-Dawley rats weighing 314.3 ± 3.6 g were used in this study. All experiments were approved by the Henry Ford Health System Institutional Animal Care and Use Committee (IACUC) and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were fed standard rat chow and given tap water ad libitum. Micropuncture studies were performed as previously described (12, 20). The rats were anesthetized; the left kidney was exposed, placed in a Lucite cup, and immobilized by surrounding it loosely with saline-soaked cotton. After 30–45 min of equilibration, grease was injected into an early segment of a proximal tubule, and two pipettes were inserted. One pipette for perfusion was inserted downstream from the grease block and attached to the nanoliter infusion pump. A second pipette (for measuring PSF), was inserted upstream from the grease block and attached to a micropressure system (model 900A; World Precision Instruments, Sarasota, FL). To generate a PSF curve, the late proximal perfusion rate was incrementally increased from 0 to 10, 20, 30, and 40 nl/min while PSF was measured. Each perfusion rate was maintained for 1–5 min, as required, to observe a stable PSF. We performed two consecutive TGF responses while adding drugs that inhibit the transporters to the tubular perfusate. The following drugs were used: NKCC2 inhibitor furosemide, ENaC inhibitor benzamil, NCC blocker hydrochlorothiazide (HCTZ), and NHE inhibitor dimethylamiloride (DMA).
Statistics.
Data are expressed as means ± SE. As the data were normally distributed, we used both Student's two-sample t-tests and Student's paired t-tests on repeated-data measurements. Hochberg's step-up procedure was used to adjust the P values for multiple comparisons to control for the family-wise type 1 error rate, predefined as 0.05.
RESULTS
Time control TGF responses and effect of inhibiting TGF with NKCC2 blocker furosemide on PSF.
To study whether TGF responses varied with time, late proximal tubule perfusion was increased twice from 0 to 40 nl/min while PSF was measured. We found that increasing the tubule perfusion decreased PSF, reflecting Af-Art constriction and/or Ef-Art dilation. There was no difference between the first and second curves, indicating that this response was reproducible over time (Fig. 1A). As expected, this response was blocked by the addition of the NKCC2 inhibitor furosemide (10−4 M; Fig. 1B). These data indicate that NKCC2 in the macula densa is a major component of the TGF response.
Fig. 1.
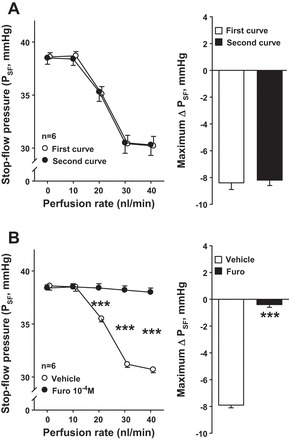
A: time control experiments. Left: increasing the perfusion rates in the late proximal tubule 2 consecutive times decreased stop-flow pressure (PSF) in a reproducible manner (○, first curve; ●, second curve). Right: maximum PSF responses in the first and second curves. B: effect of inhibiting tubuloglomerular feedback (TGF) with Na-K-2Cl cotransporter (NKCC2) blocker furosemide (furo) on PSF. Left: increasing the perfusion rates in the late proximal tubule decreased PSF (○, vehicle). This response was completely blocked when the NKCC2 inhibitor furosemide was added to the perfusate (●, furosemide). Right: maximum PSF responses in the vehicle and furosemide curves. ***P < 0.001.
Effect of inhibiting TGF and CTGF simultaneously on a TGF-like response.
To test whether the effect of furosemide on TGF is reproducible, we generated two consecutive PSF response curves in the presence of furosemide. There was no difference between the first and second curves, and the maximum PSF response in both curves was not significantly different from zero (Fig. 2A). However, in the presence of furosemide, when CTGF was inhibited by the ENaC blocker benzamil (10−6 M), we observed a decrease in PSF in response to increasing the nephron perfusion (Fig. 2B). This was blocked by addition of the NHE inhibitor DMA (10−4 M; Fig. 2C). These data indicate that in the presence of furosemide, there is an additional constrictor component, a TGF-like response, mediated by NHE and initiated by increasing luminal perfusion of the nephron. This TGF-like response was only evident when both TGF and CTGF were inhibited.
Fig. 2.
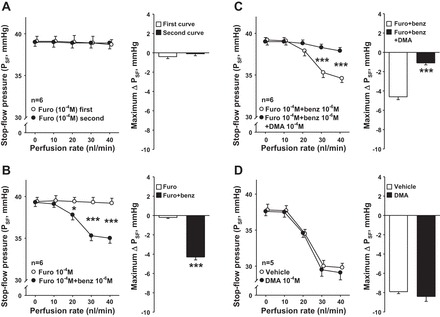
A: time control experiments with furosemide treatment. Left: in the presence of the NKCC2 blocker furosemide, PSF remained unchanged in a reproducible manner while increasing the perfusion rates 2 consecutive times in the late proximal tubule (○, first curve; ●, second curve). Right: maximum PSF responses in the first and second curves. B: effect of inhibiting TGF and connecting tubuloglomerular feedback (CTGF) simultaneously on a TGF-like response. Left: in the presence of furosemide (○), adding epithelial Na channel (ENaC) blocker benzamil (benz; ●) caused PSF to decrease in response to increasing the nephron perfusion, suggesting that when NKCC2 and CTGF are both blocked there is an additional constrictor phenomenon initiated in the nephron. Right: maximum PSF responses in the furosemide and furosemide+benzamil curves. *P < 0.05, ***P < 0.001. C: effect of Na/H exchanger (NHE) inhibition on PSF in the absence of TGF and CTGF. Left: in the presence of furosemide and benzamil (○), adding NHE blocker dimethylamiloride (DMA; ●) prevented the decrease in PSF in response to increasing the nephron perfusion, suggesting that when TGF and CTGF are blocked, NHE can induce a TGF-like response. Right: maximum PSF responses in the furosemide+benzamil and furosemide+benzamil+DMA curves. ***P < 0.001. D: effect of NHE blocker DMA on PSF. Left: increasing the perfusion rates in the late proximal tubule decreased PSF (○, vehicle). Addition of NHE blocker DMA (●) did not affect the PSF response induced by increasing the perfusion rates. Right: maximum PSF responses in vehicle and DMA curves.
We also tested the effect of DMA alone. We found that increasing the tubule perfusion rate decreased PSF similarly in the vehicle- and DMA-treated groups (Fig. 2D). These data suggest that NHE by itself has no effect on TGF or CTGF.
Effect of simultaneously inhibiting NKCC2 and NHE on CTGF response.
In the presence of furosemide, increasing tubular perfusion caused no change in PSF. However, inhibition of both NKCC2 and NHE via addition of furosemide and DMA caused PSF to increase in response to increasing the nephron perfusion (Fig. 3A). These data suggest that the vasodilator effect of CTGF can be observed as an increase in PSF when both NKCC2 and NHE are inhibited. The increase in PSF could be mainly due to a decrease in the resistance of Af-Art and/or an increase in the resistance of Ef-Art during the CTGF. The increase in PSF was due to CTGF, since it was blocked by the addition of benzamil to the perfusate (Fig. 3B).
Fig. 3.
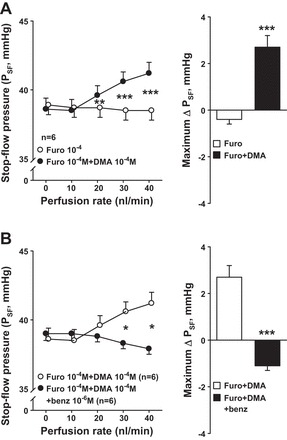
A: effect of simultaneously inhibiting NKCC2 and NHE on CTGF response. Left: in the presence of furosemide (○), adding NHE blocker DMA (●) caused PSF to increase in response to increasing nephron perfusion, suggesting that when NKCC2 and NHE are both blocked, the afferent arteriole (Af-Art) vasodilator effect induced by CTGF can be revealed with increasing nephron perfusion. Right: maximum PSF responses in the furosemide and furosemide+DMA curves. **P < 0.01, ***P < 0.001. B: effect of inhibiting CTGF with ENaC blocker benzamil on PSF when both NKCC2 and NHE are blocked. Left: in the presence of furosemide and DMA (○), increasing the nephron perfusion caused an increase in PSF; addition of benzamil to the perfusate blocked this effect (●). Right: maximum PSF responses in the furosemide+DMA curve and furosemide+DMA+benzamil curve. Benzamil prevented the Af-Art vasodilation observed when both NKCC2 and NHE are blocked, indicating that CTGF causes Af-Art vasodilation that can be revealed when both NKCC2 and NHE are completely blocked. *P < 0.05, ***P < 0.001.
Effect of the NCC blocker HCTZ on PSF when NKCC2 and CTGF are inhibited.
In the presence of furosemide and benzamil, PSF decreased in response to increasing the nephron perfusion. Addition of the NCC blocker HCTZ (10−3 M) did not affect the decrease in PSF (Fig. 4). These data suggest that nephron NCC does not participate in the control of Af-Art tone.
Fig. 4.
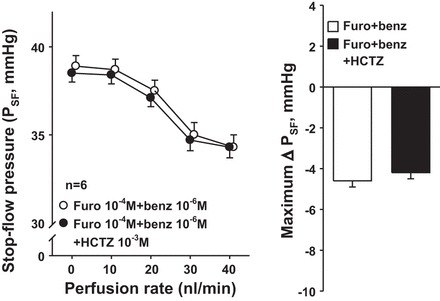
Effect of the Na-Cl cotransporter (NCC) blocker hydrochlorothiazide (HCTZ) on PSF when NKCC2 and CTGF are inhibited. Left: in the presence of furosemide and benzamil (○), adding the NCC inhibitor HCTZ (●) did not affect PSF, suggesting that the distal convoluted tubule does not participate in the regulation of Af-Art tone. Right: maximum PSF responses in furosemide+benzamil and furosemide+benzamil+HCTZ curves.
DISCUSSION
We hypothesize that in addition to NKCC2, under some circumstances NHE can mediate a vasoconstrictor mechanism that antagonizes CTGF. Thus, when both NKCC2 and NHE are blocked, CTGF increases PSF due to Af-Art dilation. As expected, we found that the NKCC2 inhibitor furosemide completely blocked TGF. However, when we perfused the nephron with furosemide plus the ENaC inhibitor benzamil to block CTGF, we observed a decrease in PSF. These data suggest a novel Af-Art constrictor and/or Ef-Art dilator mechanism initiated by the nephron. We showed that this vasoconstrictor mechanism can be blocked by inhibiting NHE, but not NCC, and when both NKCC2- and NHE-mediated mechanisms are blocked, CTGF causes an increase in PSF due to Af-Art dilatation.
TGF is a constrictor mechanism initiated by apical NKCC2 in the macula densa (2, 3, 9). Loop diuretics, such as furosemide, added to the tubular perfusate can reduce renal vascular resistance by blocking the constrictor effect of TGF (4, 7, 8, 25). When we inhibited TGF by adding furosemide to the tubule perfusate, the reduction in PSF caused by increasing nephron perfusion was completely blocked as expected.
In contrast to TGF, CTGF is a vasodilator mechanism initiated in the CT by the ENaC, by an increase in NaCl (18). In vitro CTGF dilates Af-Arts while in vivo it antagonizes the decrease in PSF caused by TGF (18, 24). If TGF and CTGF were the only two mechanisms that control PSF, one would expect that blocking TGF with furosemide would reveal CTGF-induced increase in PSF in response to increasing the nephron perfusion. However, here we show that when TGF was blocked with furosemide, increasing the tubular perfusion did not increase PSF. This observation led us to hypothesize that when NKCC2 is blocked with furosemide, there is another constrictor mechanism that opposes CTGF.
Recent studies have shown that in addition to expressing NKCC2, macula densa cells functionally and immunologically express Na/H exchanger 2 (NHE2) at the apical membrane and NHE4 at the basolateral membrane. These two isoforms likely participate in Na transport, pHi, and cell volume regulation, and may be involved in the regulation of TGF (6, 15). Thus, we tested whether NHE mediated the decrease in PSF caused by increasing tubular perfusion when both furosemide and benzamil were present to inhibit NKCC2 and ENaC. Here, we report for the first time the existence of the Af-Art constrictor phenomenon that is initiated in the nephron by increasing the luminal perfusion, and that it can be blocked by the NHE inhibitor DMA.
Using micropuncture, one cannot definitively define the segment or the exact mechanism by which the NHE-mediated decrease in PSF occurs. However, the literature suggests reasonable explanation. Bell and colleagues (14) postulated that Na-K-ATPase does not play a significant role in pumping Na out of the macula densa due to low amounts of active Na-K-ATPase in the basolateral membrane of the macula densa. One may hypothesize that NHE mediates the removal of Na from the cell interior in exchange for H. Thus, intracellular pH will become relatively acidic at some point after the luminal NaCl is raised. We reported that NHE activity is necessary for an increase in luminal Na to activate nitric oxide production in the macula densa, and this blunts TGF (11), and that in vitro, in the absence of furosemide, blockade of NHE with DMA potentiates TGF (21). Now we show data that during the TGF blockade with furosemide, by inhibiting NKCC2, inhibition of NHE causes vasodilation rather than the vasoconstriction. How can one explain such data? In short, when luminal NaCl increases and NKCC2 is active, it mediates Na entry, intracellular Na increases, and NHE restores intracellular Na, as put forth by Bell and colleagues (6). NHE is driven in reverse mode (Na out and protons in) by the elevated intracellular Na. However, when NKCC2 is inhibited, it cannot cause an increase in intracellular Na and there is a large gradient for Na entry. This gradient drives Na entry by the NHE. NHE-mediated Na entry causes an increase in intracellular Na, just as NKCC2 would have if it was not inhibited, and the increase in intracellular Na initiates TGF just as it would have if NKCC2 had mediated its entry. Thus, the vasoconstriction caused by increasing luminal NaCl that is mediated by NHE can only be seen when NKCC2 is inhibited and it can operate in a mode that is the opposite of what happens when NKCC2 is not blocked by furosemide.
Alternatively, NHE-induced reduction in PSF may be initiated in a different nephron segment such as CT. Since the CT is in close proximity to the Af-Art and since the intercalated cells of the CT abundantly express NHE in the luminal membrane (5), it is possible that CT intercalated cells mediate this constrictor phenomenon.
We also perfused the nephron with HCTZ, to test whether HCTZ-sensitive NCC plays a role in the regulation of Af-Art constriction, but we found no effect. Recently, another Na transporter has been described in the nephron, the Na-driven Cl/HCO3 exchanger (NDCBE), which can mediate electroneutral Na reabsorption by acting jointly with pendrin (10). Since this transport is also sensitive to HCTZ, our findings also ruled out the role for NDCBE in controlling Af-Art tone. Since this is the first report on NHE-induced Af-Art constrictor mechanism, many questions still remain to be answered, in particular defining the exact segment of the distal nephron that initiates this novel mechanism.
Given our finding that both NKCC2 and NHE mediate a decrease in PSF, we tested whether we could measure a CTGF-dependent increase in PSF. To do this, we studied the effect of furosemide and DMA on changes in PSF induced by increasing tubular perfusion. Here, we report for the first time that in vivo CTGF increased PSF in absolute terms when NHE and NKCC2 were inhibited. This increase in PSF could be due to either dilation of the Af-Art or constriction of Ef-Art during the CTGF. However, CTGF is initiated by the ENaC in the CT that has contact with the Af-Art but not with the Ef-Art, thus it is unlike that the increase in PSF is mediated by Ef-Art constriction. These data support the hypothesis that when NKCC2 and CTGF are blocked, activation of NHE in the nephron constricts the Af-Art and antagonizes CTGF in response to the increase in tubular perfusion. Furthermore, it also supports the hypothesis that when in vivo both NKCC2 and NHE are blocked, CTGF causes an increase in PSF or Af-Art dilation.
During the treatment with furosemide and benzamil, the NHE-induced decrease in PSF may participate in nephron autoregulation of glomerular filtration. It is also possible that it participates in antagonizing CTGF. An increase in CTGF may explain the higher glomerular pressure and renal damage in salt-sensitive hypertensive individuals (22), such as African-Americans, the elderly, and the diabetic. ENaC-blocking drugs (potassium-sparing diuretics), by blocking CTGF and decreasing glomerular perfusion pressure, could be useful in preventing hypertensive nephrosclerosis. Also, CTGF is a novel regulatory mechanism of the renal microcirculation that may explain the Af-Art dilatation and increased GFR observed during high-salt intake, perhaps by antagonizing or resetting TGF. During high-salt intake, O2 consumption by the nephron is higher because of increased Na+ reabsorption; thus, CTGF could help protect the kidney from ischemia by increasing renal blood flow. On the other hand, CTGF may be detrimental in certain situations, such as in diabetes with osmotic diuresis, where Af-Art dilation might increase intraglomerular pressure and glomerular damage.
In summary, our studies provide the first evidence for the existence of a constrictor phenomenon initiated in the nephron that controls Af-Art tone and that is apparent when both NKCC2 and CTGF are inhibited. We showed that inhibiting both NKCC2 and CTGF causes constriction of the Af-Art in response to increases in nephron tubular flow rate and that is mediated by NHE, rather than by NCC. We also showed that when NHE and NKCC2 are both blocked, CTGF causes in vivo vasodilation of the Af-Art, seen as an absolute increase in PSF.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.W., J.L.G., E.L.P., and O.A.C. conception and design of research; H.W., Y.R., S.R.M., P.L., and K.K. performed experiments; H.W., Y.R., E.L.P., and O.A.C. analyzed data; H.W., M.A.D., S.R.M., P.L., K.K., J.L.G., B.J., and O.A.C. interpreted results of experiments; H.W., M.A.D., Y.R., B.J., and O.A.C. prepared figures; H.W., M.A.D., and B.J. drafted manuscript; H.W., M.A.D., Y.R., S.R.M., P.L., K.K., J.L.G., B.J., E.L.P., and O.A.C. approved final version of manuscript; J.L.G., B.J., and O.A.C. edited and revised manuscript.
REFERENCES
- 1.Arima S, Ito S. Role of renal eicosanoids in the control of intraglomerular and systemic blood pressure during development of hypertension. Contrib Nephrol 143: 65–76, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Bell PD, Lapointe JY. Characteristics of membrane transport processes of macula densa cells. Clin Exp Pharmacol Physiol 24: 541–547, 1997. [DOI] [PubMed] [Google Scholar]
- 3.Bell PD, Lapointe JY, Cardinal J, Chang YS. Transport pathways in macula densa cells. Kidney Int Suppl 32: S59–S64, 1991. [PubMed] [Google Scholar]
- 4.Brunkhorst R, Muller-Ott K, Gutsche HU, Niedermayer W. Effect of furosemide, bumetanide and piretanide on the sensor of the tubuloglomerular feedback mechanism. Proc Eur Dial Transplant Assoc 15: 613–616, 1978. [PubMed] [Google Scholar]
- 5.Chambrey R, Warnock DG, Podevin RA, Bruneval P, Mandet C, Blair MF, Barity J, Paillard M. Immunolocalization of the Na+/H+ exchanger isoform NHE2 in rat kidney. Am J Physiol Renal Physiol 275: F379–F386, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Fowler BC, Chang YS, Laamarti A, Higdon M, Lapointe JY, Bell PD. Evidence for apical sodium proton exchange in macula densa cells. Kidney Int 47: 746–751, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Gutsche HU, Brunkhorst R, Muller-Ott K, Franke H, Niedermayer W. Effect of diuretics on the tubuloglomerular feedback response. Can J Physiol Pharmacol 62: 412–417, 1984. [DOI] [PubMed] [Google Scholar]
- 8.Johnston PA, Kau ST. The effect of loop of Henle diuretics on the tubuloglomerular feedback mechanism. Methods Find Exp Clin Pharmacol 14: 523–529, 1992. [PubMed] [Google Scholar]
- 9.Lapointe JY, Bell PD, Cardinal J. Direct evidence for apical Na+:2Cl−:K+ cotransport in macula densa cells. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1466–F1469, 1990. [DOI] [PubMed] [Google Scholar]
- 10.Leviel F, Hubner CA, Houillier P, Morla L, El MS, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu R, Carretero OA, Ren Y, Garvin JL. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int 67: 1837–1843, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz JN. Micropuncture of the kidney: a primer on techniques. In: Comprehensive Physiology. John Wiley & Sons, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Peti-Peterdi J, Bebok Z, Lapointe JY, Bell PD. Novel regulation of cell [Na+] in macula densa cells: apical Na+ recycling by H-K-ATPase. Am J Physiol Renal Physiol 282: F324–F329, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Peti-Peterdi J, Chambrey R, Bebok Z, Biemesderfer D, St. John PL, Abrahamson DR, Warnock DG, Bell PD. Macula densa Na+/H+ exchange activities mediated by apical NHE2 and basolateral NHE4 isoforms. Am J Physiol Renal Physiol 278: F452–F463, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Reilly RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev 80: 277–313, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Ren Y, Garvin JL, Falck JR, Renduchintala KV, Carretero OA. Glomerular autacoids stimulated by bradykinin regulate efferent arteriole tone. Kidney Int 63: 987–993, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int 71: 1116–1121, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Ren Y, Garvin JL, Liu R, Carretero OA. Possible mechanism of efferent arteriole (Ef-Art) tubuloglomerular feedback. Kidney Int 71: 861–866, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Schnermann J, Traynor T, Yang T, Arend L, Huang YG, Smart A, Briggs JP. Tubuloglomerular feedback: new concepts and developments. Kidney Int 54, Suppl 67: S40–S45, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Carretero OA, Garvin JL. Inhibition of apical Na+/H+ exchangers on the macula densa cells augments tubuloglomerular feedback. Hypertension 41: 688–691, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, D'Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback in hypertension. Hypertension 62: 738–745, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, D'Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback mediates acute tubuloglomerular feedback resetting. Am J Physiol Renal Physiol 302: F1300–F1304, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Garvin JL, D'Ambrosio MA, Ren Y, Carretero OA. Connecting tubule glomerular feedback antagonizes tubuloglomerular feedback in vivo. Am J Physiol Renal Physiol 299: F1374–F1378, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright FS, Schnermann J. Interference with feedback control of glomerular filtration rate by furosemide, triflocin, and cyanide. J Clin Invest 53: 1695–1708, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]