

Abstract
The speciation of pathogens can be driven by divergent host specialization. Specialization to a new host is possible via the acquisition of advantageous mutations fixed by positive selection. Comparative genome analyses of closely related species allows for the identification of such key substitutions via inference of genome-wide signatures of positive selection. We previously used a comparative genomics framework to identify genes that have evolved under positive selection during speciation of the prominent wheat pathogen Zymoseptoria tritici (synonym Mycosphaerella graminicola). In this study, we conducted functional analyses of four genes exhibiting strong signatures of positive selection in Z. tritici. We deleted the four genes in Z. tritici and confirm a virulence-related role of three of the four genes ΔZt80707, ΔZt89160 and ΔZt103264. The two mutants ΔZt80707 and ΔZt103264 show a significant reduction in virulence during infection of wheat; the ΔZt89160 mutant causes a hypervirulent phenotype in wheat. Mutant phenotypes of ΔZt80707, ΔZt89160 and ΔZt103264 can be restored by insertion of the wild-type genes. However, the insertion of the Zt80707 and Zt89160 orthologs from Z. pseudotritici and Z. ardabiliae do not restore wild-type levels of virulence, suggesting that positively selected substitutions in Z. tritici may relate to divergent host specialization. Interestingly, the gene Zt80707 encodes also a secretion signal that targets the protein for cell secretion. This secretion signal is however only transcribed in Z. tritici, suggesting that Z. tritici-specific substitutions relate to a new function of the protein in the extracellular space of the wheat-Z. tritici interaction. Together, the results presented here highlight that Zt80707, Zt103264 and Zt89160 represent key genes involved in virulence and host-specific disease development of Z. tritici. Our findings illustrate that evolutionary predictions provide a powerful tool for the identification of novel traits crucial for host adaptation and pathogen evolution.
Author Summary
Zymoseptoria spp provides a unique model system to study the underlying genetics of host specialization of plant pathogens. Closely related Zymoseptoria species, including the prominent wheat pathogen Z. tritici, have recently specialized to distinct grass hosts. Positively selected substitutions have played a central role in the acquisition of new host specificities. We have identified a small set of genes showing signatures of positive selection. We demonstrate that three of these four candidate genes play an important role during host infection. Two mutants of Z. tritici were impaired in virulence; a third mutant showed a hypervirulent phenotype. New protein specificities not only include changes at the amino acid sequence level but also at the level of the protein structure. We conducted a gene replacement experiment to test if mutant phenotypes in Z. tritici could be complemented by the insertion of orthologous genes from the two closely related species Z. pseudotritici and Z. ardabiliae. For two genes, we confirm that the species-specific protein changes are essential for proper protein functioning in Z. tritici; key traits involved in the evolution of virulence and host specificity of this prominent pathogen can be characterized via a combination of evolutionary predictions and functional analyses.
Introduction
Host specialization of pathogens can be a strong driver of diversification and speciation [1]. Host-driven speciation implies the specialization of traits involved in host interactions from the initial infection and the defeat of host defenses to within-host nutrient uptake, multiplication and reproduction. Even closely related host species may differ in their repertoire of defense-related genes, as well as their biochemical and physical properties. As a result, distinct selection pressures are imposed on infecting pathogens. Genes affected by divergent selection during host specialization can be recognized in pathogen genomes as outlier loci with an excess of genetic divergence [2]. Positive selection and adaptive changes at the amino acid level are in particular reflected by an accumulation of non-synonymous divergence at the nucleotide level.
In plant pathogens, divergent selection has been documented in several studies showing specialization of genes related to different functions, e.g., genes encoding effector proteins [3], secondary metabolites [4], toxins [5] and genes encoding cell wall-degrading enzymes [6]. The functional implications of divergent selection are, however, poorly understood. Dong and co-workers elegantly demonstrated that one amino acid change in the oomycete effector protein EpiC1 determines target specificity [3]. EpiC1 encodes a protease inhibitor that has evolved under strong positive selection during the divergence of the two plant pathogenic Phytophthora species P. infestans and P. mirabilis infecting different Solanum plants and Mirabilis jalapa, respectively. The adaptive changes in EpiC1 directly reflect differences in the protease targets of EpiC1 of P. infestans and P. mirabilis, and the study illustrates the direct functional effect of positive selection.
For the fungal wheat pathogen Zymoseptoria tritici (Mycosphaerella graminicola), the underlying genetics of host-pathogen interaction is poorly understood. Infection of Zymoseptoria spp. involves an initial biotrophic phase where infectious hyphae take up readily accessible sugars in the apoplast. Host cell death is induced after approximately two weeks and involves a strong proliferation of hyphae in substomatal cavities and the formation of asexual fruiting bodies, so-called pycnidia [7]. Specialization to wheat likely has involved the acquisition and fixation of adaptive substitutions in key genes playing a role in the molecular interaction between host and pathogen. Several non-characterized genes, including genes encoding both secreted and non-secreted proteins, were found to exhibit signatures of positive selection during divergence from the closest relatives Z. pseudotritici and Z. ardabiliae [7–8]. Z. pseudotritici and Z. ardabiliae were isolated from wild grasses in the Middle East and are unable to infect the host of Z. tritici (bread wheat). Divergence of these three species occurred very recently and involved only a few adaptive changes at the genome level [8]. It is likely that these few changes have been instrumental during specialization to distinct hosts. We hypothesize that genes subjected to positive selection during speciation of Z. tritici likely have played a role in the specialization to wheat. Using genome-wide analyses of non-synonymous to synonymous divergence, we previously identified a set of 27 positively selected genes in Z. tritici [8]. In this study, we aim to elucidate the underlying role of four selected genes showing increased ratios of non-synonymous to synonymous substitutions. We adopt a reverse genetic approach and show that three of the genes have a strong impact on virulence and reproduction of Z. tritici during infection of wheat. Besides amino acid changes, we describe structural variation in transcript lengths including the addition of a signal peptide in one Z. tritici-encoded protein.
Results
Selection of candidate genes
We previously conducted a comparative genome study in which we assessed pairwise K a/K s ratios for more than 9000 aligned genes in Z. tritici, Z. pseudotritici, Z. ardabiliae and Z. passerinii with the aim of identifying positively selected genes [8]. In the present study, we address the functional relevance of a subset of these positively selected genes in Z. tritici. We selected the four genes Zt80707, Zt89160, Zt103264 and Zt110804 for which we previously identified signatures of positive selection using two different approaches [8]. Zt80707 and Zt103264 were selected from a list of 27 candidates with K a > K s (computed according to Nei and Gojobori [9], FDR-adjusted p-value < 0.05; Z-test). Both genes show an increased accumulation of non-synonymous substitutions in pairwise comparisons between Z. tritici-Z. pseudotritici and Z. tritici-Z. ardabiliae. The two genes Zt89160 and Zt110804 were selected from the output of a maximum likelihood analysis of gene-wise branch specific d N/d S ratios in the three Zymoseptoria species. Both genes show increased d N/d S ratios exclusively in the Z. tritici branch. These four genes are all located on the core chromosomes in regions with a well-conserved synteny among the three Zymoseptoria species (Table 1). None of the genes have previously been characterized functionally in Z. tritici; however, the gene Zt89160 is predicted to encode a Regulator of Chromosome Condensation (RCC1) domain [10] and Zt110804 encodes a hypothetical protein containing a proline-rich region predicted to be involved in binding with other proteins [11]. The genes Zt80707 and Zt103264 show no homology to known proteins.
Table 1. Candidate genes and genomic coordinates in Z. tritici.
| Gene ID | Location | Length [aa] | K a/K s Z.tritici vs. Z.pseudotritici | Predicted function |
|---|---|---|---|---|
| 80707 | Chr_5:0657996–0658613 | 125 | 5.825 | Unknown / secreted |
| 89160 | Chr_1:2089079–2090359 | 394 | 5.392 | RCC1 domain |
| 103264 | Chr_2:1992714–1993521 | 173 | 2.303 | Unknown |
| 110804 | Chr_9:1317146–1318829 | 317 | 3.533 | Kinase |
Gene alignments showed that the structure of the reading frames of Zt89160 and Zt110804 are conserved between the three Zymoseptoria species with identical start and stop codon positions. However, we verified the open-reading frames of Zt80707 and Zt103264 by Rapid Amplification of cDNA Ends (RACE-PCR) in Z. tritici, Z. pseudotritici and Z. ardabiliae as the orthologous sequences for these two genes had different start and stop codon positions. Based on sequencing of transcripts, we revealed significant differences in transcript lengths for those two genes among the three Zymoseptoria species (Figs 1 and S1). According to these structural differences, we asked whether the genes are located next to transposons or in regions with frequent re-arrangements. To this end, we analyzed the four loci in Z. tritici (isolate IPO323) and compared them with the orthologous loci in the Z. pseudotritici isolate STIR04_2.2.1 (here termed Zp13) and the Z. ardabiliae isolate STIR04_1.1.1 (here termed Za17) [8,12]. We found transposable and repetitive elements in the vicinity of the neighboring genes (<30kbp) but none directly associated (<2kbp) with the candidate genes (S2 Fig). We next re-analyzed the extent of positive selection in the gene models of Zt80707 and Zt103264 corrected by RACE-PCR in Zymoseptoria spp using a maximum likelihood approach. We applied a branch model to obtain maximum likelihood estimates of branch-specific ω values (d N/d S ratios) [13]. This approach allowed us to compare the branch-specific accumulation of non-synonymous to synonymous divergence in the individual Zymoseptoria lineages for each gene. An ω value above 1 indicates that a gene has been subjected to positive selection in a particular branch of a phylogenetic tree during the divergence of lineages. Our analyses confirm previous results using the Nej and Gojobori K a/K s estimates [8] and show that all four genes have evolved under positive selection during divergence of Z. tritici (Zt89160 and Zt110804) or during divergence of all three Zymoseptoria lineages (Zt80707 and Zt103264) according to branch specific dN/dS ratios (S3 Fig).
Fig 1. Variation at the nucleotide and protein levels between orthologs of Zt80707.

A) Alignment of the protein Zt80707 of Z. tritici (Zt) and orthologs in the sister species Z. pseudotritici (Zp) and Z. ardabiliae (Za). The alignment demonstrates the difference in length of the orthologous proteins. The additional 25 amino acids at the N-terminal end of the Z. tritici ortholog encodes a weak signal peptide (red line). B) Nucleotide alignment of the genomic region encoding the Zt80707 protein including the nucleotide sequence encoding the signal peptide in Z. tritici and the corresponding up-stream sequences in Z. pseudotritici and Z. ardabiliae. Different start codon positions in Z. tritici, Z. pseudotritici and Z. ardabiliae identified by RACE-PCR are indicated with red lines.
Zt80707 encodes a signal peptide solely translated in Z. tritici
Remarkably, the gene Zt80707 showed different transcript lengths among the three species, Z. tritici, Z. pseudotritici and Z. ardabiliae. For the orthologous transcripts Zp80707 in Z. pseudotritici and Za80707 in Z. ardabiliae, the transcription start sites are 69 and 75 nucleotides downstream of the start site in Z. tritici, respectively. The differing transcripts lengths between Zymoseptoria species were confirmed using RT-PCR analysis (S4 Fig). Interestingly, based on our RACE-PCR results, we found that the N-terminal end of the gene Zt80707 encodes a predicted signal peptide transcribed only in Z. tritici. We computationally predicted the signal peptide with low probability scores using SignalP v3 [14]. Only the C score (the raw cleavage site score recognizing signal peptide cleavage sites) supported the presence of a signal peptide at the N-terminal end of the gene Zt80707 (maximal value of C score = 0.581). This weakly predicted signal peptide corresponds exactly to the upstream sequence that is not transcribed in Z. pseudotritici or Z. ardabiliae. To experimentally confirm that this signal peptide is transcribed in Zt80707 and targets the translated protein for secretion, we designed an in vitro secretion assay. We used quantitative PCR to measure the expression of Zt80707 and its orthologs in Z. pseudotritici and Z. ardabiliae in vitro, and found that the gene is only weakly expressed during axenic growth. Therefore, the constitutive glyceraldehyde-3-phosphate dehydrogenase (gpdA) promoter from Aspergillus nidulans [15] was used to express Zt80707 and Zp80707 in vitro in Z. tritici cells. Furthermore, we fused both genes with a C-terminal green fluorescent protein (GFP) tag. As a positive control for protein secretion, we used the well-characterized Lysine motif (LysM) effector protein Zt111221 [16,17] and as a negative control, we used the non-secreted protein Zt77228 (a predicted member of the intramitochondrial-sorting protein family). The two genes encoding Zt111221 and Zt77228 were expressed as Zt80707 and Zp80707 with a C-terminal GFP tag under the control of the gpdA promoter. Western blot analyses confirmed that Zt80707 is present in both the pellet and supernatant fraction of axenically grown Z. tritici cells, as also shown for the secreted LysM-positive control (Fig 2). On the other hand, the orthologous protein from Z. pseudotritici and the non-secreted negative control Zt77228 were only detectable in the pellet fraction, revealing that the Z. pseudotritici ortholog is not secreted. Together, these results supported an extracellular function of the protein uniquely in Z. tritici.
Fig 2. Zt80707 encodes a signal peptide targeting the protein for cell secretion in Z. tritici.
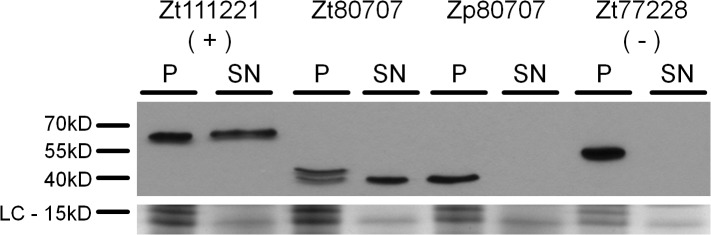
Western blot analyses of the proteins Zt111221-GFP (secreted LysM effector, positive control (+)), Zt80707-GFP (Z. tritici), Zp80707-GFP (Z. pseudotritici) and Zt77228-GFP (non-secreted protein, lysis control (-)) detected in the pellet fraction (P) and the cell culture supernatant (SN) with an anti-GFP antibody. The presence of the GFP tagged gene products in the supernatant demonstrates the secretion of the Zt80707 protein and the functioning of the predicted signal peptide of Zt80707. As Loading Control (LC) a Coomassie stained SDS gel is included to show comparable amounts of protein loaded in the lanes of the different samples.
Reconstruction of protein structure
To link signatures of positive selection to protein structure and function, we applied a computational method for protein structure prediction; I-TASSER (Iterative Threading ASSEmbly Refinement) [18]. Due to the absence of any homologous entries for Zt80707, Zt103264 and Zt110804 in the relevant databases it was not possible to predict the structures of the proteins encoded by these three genes. However, the structural conservation of Zt89160 allowed us to predict the protein structure (S5 Fig). Homologs of Zt89160 can be found in other Dothideomycete fungi, however a pathogenicity-related function has never been shown. The predicted structure of Zt89160 resembles a ring-like RCC1 β-propeller structure containing multiple lateral loops [10]. We related the predicted structure of Zt89160 to a well characterized RCC1 protein of Drosophila melanogaster known to interact with nucleosomes (S5 Fig) [10]. The conserved structure of the predicted RCC1 protein in Z. tritici supports a similar DNA or protein-binding function as described for other RCC1 proteins. Based on the structure of the protein we could assign each site to either central or surface regions. In general we find an excess of substitutions on the surface of the Zt89160 protein; 15.6% in surface regions vs. 4.8% in central regions (Fisher’s exact test, p-value = 0.00422). This effect is essentially due to an excess of non-synonymous substitutions; 61.1% of all non-synonymous substitutions are in surface regions vs. 38.9% in central regions (Fisher’s exact test, p-value = 0.000405), compared to 20% of all synonymous substitutions in surface regions vs. 80% in central regions (Fishers’ exact test, p-value = 1) (S5 Fig). The fact that majorities of amino acid differences between Zymoseptoria species locate on the surface of the protein suggests that positive selection was driven by divergent DNA sequences or proteins interacting with the different RCC orthologs in Z. tritici, Z. pseudotritici and Z. ardabiliae.
Zt80707, Zt89160, Zt103264 and Zt110804 are differentially expressed during hemibiotrophic growth
To assess the expression of Zt80707, Zt89160, Zt103264 and Zt110804 in planta, we performed a quantitative RT-PCR (qRT-PCR) experiment. RNA was extracted from Z. tritici from axenic cultures and from infected wheat leaves at 4, 7, 14 and 28 days post infection (dpi). These time points correspond to initial infection (4 dpi), biotrophic growth (7 dpi), a metabolic switch from biotrophic to necrotrophic growth (14 dpi) and necrotrophic growth (28 dpi). In general, all four genes were upregulated in wheat seedlings during the entire infection cycle, indicating that their function relates to in-planta growth (Fig 3). However, the four genes exhibited different expression patterns during biotrophic and necrotrophic growth and may be involved in distinct processes during development of infectious hyphae in planta. Zt80707 is upregulated during all phases of infection except for the initial infection stage at 4 dpi. The three genes Zt89160, Zt103264 and Zt110804 show the highest expression during biotrophic growth.
Fig 3. The four positively selected candidate genes are expressed during host infection.

In planta expression data for the four positively selected candidate genes Zt80707, Zt89160, Zt103264 and Zt110804 of Z. tritici, relative to expression during axenic growth. Values are normalized to the expression of the gene encoding GAPDH, a constitutively expressed housekeeping control. Error bars indicate the standard error of the mean (SEM) of three independent biological replicates per sample (** p<0.01; p-values were calculated using the Mann-Whitney U-test).
Deletion of candidate genes
To investigate the functional role of Zt80707, Zt89160, Zt103264 and Zt110804, we generated independent deletion mutants in the Z. tritici isolate IPO323 using an Agrobacterium tumefaciens mediated transformation (ATMT) approach [16]. We verified the correct integration of hygromycin deletion constructs by homologous recombination with Southern blot analyses (S6 Fig).
To determine any putative non-pathogenicity-related functional role we first conducted an in vitro phenotypic characterization of the deletion mutants. For each gene, four independent deletion strains were used in an in vitro stress assay using NaCl (1.5 M), H2O2 (2 mM), Congored (500 μg/ml), Calcofluor (200 μg/ml) and 28°C temperature stress. We found no difference in the sensitivity to osmotic, oxidative and cell wall stresses between the wild-type and the four deletion mutants, further supporting an in-planta related role of the genes (S7 Fig). Next, we performed plant experiments to determine the pathogenicity of the Z. tritici deletion mutants on the susceptible wheat variety Obelisk (Wiersum Plantbreeding, Winschoten, Netherlands). Disease development was evaluated 28 dpi by assessing the percentage of the leaf area covered with asexual fruiting bodies (pycnidia) of the Z. tritici strains (Figs 4 and S8). The formation of pycnidia was significantly reduced on leaves infected with the IPO323ΔZt80707 and IPO323ΔZt103264 mutants. Interestingly, the IPO323ΔZt89160 mutant caused a significantly higher amount of pycnidia, consistent with a hypervirulent phenotype. Pycnidia formation was however not affected in the IPO323ΔZt110804 mutant. To verify that the observed phenotypic differences were solely caused by the deletion of our candidate genes, we reintroduced the Zt80707, Zt89160 and Zt103264 open reading frames (ORFs) into the deletion strains at the endogenous locus of each gene. We confirmed that the inserted wild-type genes were all expressed as wild-type during early host infection using qRT-PCR (S9 Fig). Plant infections of these complementation strains showed that wild-type virulence could be restored for all three genes by re-insertion of the respective wild-type genes at their native locus and confirm a virulence-related role of Zt80707, Zt89160 and Zt103264 in Z. tritici (Fig 4).
Fig 4. In planta phenotypic assay demonstrates impact of gene deletion in Z. tritici.

Mutant phenotypes of Z. tritici are characterized by measures of leaf area covered with pycnidia on the susceptible wheat variety Obelisk. To compare quantitative differences in pycnidia levels at 28 dpi, six categories ranging from 0 (without any visible symptoms), 1 (1–20%), 2 (21–40%), 3 (41–60%), 4 (61–80%) and 5 (81–100%) are used. Significant deviations in pycnidia levels between the wild-type strain and mutants are observed for the IPO323ΔZt80707, IPO323ΔZt89160 and IPO323ΔZt103264 mutants (** p<0.01), but not the IPO323ΔZt110804 mutant. Wild-type pycnidia levels for IPO323ΔZt80707, IPO323ΔZt89160 and IPO323ΔZt103264 are restored by insertion of the wild-type gene at the respective native loci in the mutants. P-values were calculated using the Mann-Whitney-U-Test.
Replacement of Z. tritici genes with the orthologous genes of Z. pseudotritici and Z. ardabiliae
We hypothesize that positively selected amino acid changes in Z. tritici have played a role during speciation and adaptation to the wheat host. To test the importance of species-specific substitutions in the three candidate genes Zt80707, Zt89160 and Zt103264, we replaced them with the respective orthologous genes from Z. pseudotritici and Z. ardabiliae. We confirmed that the inserted genes, still under the control of the native Z. tritici promoters, were expressed as in wild-type using qRT-PCR (S9 Fig).
For Zt80707, we replaced the gene in Z. tritici with either the full-length ortholog from Z. pseudotritici (Zp13) Zp80707 or the full-length ortholog from Z. ardabiliae (Za17). The 3’ end of Za80707 in Z. ardabiliae is 21 aa shorter than the orthologs of Z. tritici and Z. pseudotritici (Fig 1). The replacement with both the Z. ardabiliae and Z. pseudotritici orthologs in Z. tritici thereby also allowed us to assess the importance of the different transcription stop sites for virulence of Z. tritici on wheat. Furthermore, we generated a fusion construct with the Zt80707 signal peptide (until the cleavage site at aa position 21) and the ORF of the Zp13 ortholog (S10 Fig). Our aim was to assess whether the Z. pseudotritici ortholog could complement the Z. tritici gene if secreted as the native Z. tritici protein. Replacing the Z. tritici genes with orthologs from Z. pseudotritici or Z. ardabiliae showed that wild-type virulence could not be restored in any of the replacement strains (Fig 5). However, with the fusion construct of the Zt80707 signal peptide and the Zp13 ORF, it was possible to partially restore wild-type virulence levels, since the resulting amount of pycnidia produced by the mutant ΔZt80707::sp+Zp80707 was higher than pycnidia produced by the deletion strain IPO323ΔZt80707. This result suggests that the protein encoded by Zt80707 plays an essential role in the extracellular space of Z. tritici and that the amino acid substitutions in Zp80707, acquired since the divergence of Z. tritici and Z. pseudotritici, to some extent allow the protein to fulfill the same function as Zt80707.
Fig 5. In planta phenotypic assay demonstrates impact of gene replacement in Z. tritici.

To assess the functional relevance of non-synonymous changes in Zt80707, Zt89160 and Zt103264, the orthologous genes from Z. pseudotritici and Z. ardabiliae were inserted at the respective native loci in the deletion mutants IPO323ΔZt80707, IPO323ΔZt89160 and IPO323ΔZt103264. For the gene Zt80707, the Zp80707 ortholog was inserted with (IPO323ΔZt80707::sp+Zp80707) and without (IPO323ΔZt80707::Zp80707) the signal peptide of Zt80707. Quantitative differences in pycnidia levels were measured according to six categories: 0 (without any visible symptoms), 1 (1–20%), 2 (21–40%), 3 (41–60%), 4 (61–80%) and 5 (81–100%). Significant differences in pycnidia levels (** p<0.01) were documented using a Mann-Whitney-U-Test for the Zt80707 and Zt89160 mutants. Only the replacement strain IPO323ΔZt103264::Za103264 could restore the wild-type phenotype of the Z. tritici isolate IPO323.
Replacement of Zt89160 with the orthologous gene from Z. pseudotritici (isolate Zp13) could not restore wild-type virulence in Z. tritici (Fig 5). The replacement strain showed the same hyper-virulent phenotype as the mutant, suggesting that adaptive substitutions in Zt89160 indeed have been important for the divergent specialization of Z. tritici. The deletion of Zt103264 was replaced with the ortholog of Z. ardabiliae (Za17). The introduction of this ortholog could, in contrast to the orthologs of Zt80707 and Zt89160, restore virulence levels of the wild-type Z. tritici isolate, suggesting that adaptive substitutions in this gene do not directly relate to host specialization in Z. tritici.
IPO323ΔZt80707 and IPO323ΔZt103264 mutants are impaired in pycnidia maturation
As shown in Fig 4, the IPO323ΔZt80707 and IPO323ΔZt103264 deletion mutants caused reduced amounts of pycnidia on wheat leaves 28 dpi. To further understand the impact on pycnidia production of gene products of these two genes and Zt89160, we conducted a more detailed comparison of pycnidia development and size. We first of all observed a delayed development of pycnidia and a delayed release of pycnidia spores in the two deletion mutants IPO323ΔZt80707 and IPO323ΔZt103264 compared to the wild-type strain. Twenty-eight dpi pycnidiospores were exuded from pycnidia on wild-type-infected leaves, but not from mutant-infected leaves (Fig 6A–6C). To evaluate the viability of these two mutants pycnidiospores, we conducted a qualitative assay comparing the pycnidia from leaf samples infected with the wild-type, the IPO323ΔZt89160 mutant and the two deletion mutants impaired in pycnidia production. We harvested infected leaves and induced oozing (release of spores) from pycnidia under high-humidity conditions (S11A Fig). Pycnidiospores were released from pycnidia after seven days. To assess and compare the viability of spores of wild-type and mutant pycnidia, we isolated spores from each leaf sample and prepared a dilution series to determine the proportions of germinating spores (S11B Fig). We observed no significant difference in the viability of spores from wild-type and the three deletion mutants, suggesting that there is no qualitative effect of the gene deletion of Zt80707 or Zt103264 on pycnidiospores.
Fig 6. Mutant IPO323ΔZt80707 and IPO323ΔZt103264 produce fewer pycnidiospores per pycnidium.

Macroscopic images of infected wheat leaves 28 dpi with wild-type IPO323 (A) and the two mutants IPO323ΔZt80707 (B) and IPO323ΔZt103264 (C). Pycnidiospores are oozing from the pycnidia of IPO323 (A, white arrow), but not from the pycnidia of the two mutants. D) A quantitative comparison of pycnidiospores produced by the wild-type and the three mutant strains IPO323ΔZt80707, IPO323ΔZt89160 and IPO323ΔZt103264 measured as counts of spores isolated from a known number of pycnidia. ** p<0.01; p-values were calculated using a Mann-Whitney U test. Whiskers indicate standard deviations.
To further investigate the quantitative differences in pycnidiospore production between deletion mutants of Zt80707, Zt89160 and Zt103264 and wild-type that we observe macroscopically, we assessed the amount of pycnidiospores per pycnidium. To do so, we also induced oozing from pycnidia on harvested leaves. We first counted the number of pycnidia per leaf, and then used a “Neubauer-improved” counting chamber to count the amount of pycnidiospores isolated from the oozing pycnidia. The number of pycnidiospores in the spore suspensions was divided by the number of pycnidia on each leaf from which the spores were isolated. This process enabled us to calculate the number of pycnidiospores per pycnidium. Our findings showed that the pycnidia of the two deletion strains IPO323ΔZt80707 and IPO323ΔZt103264 contained significantly fewer pycnidiospores than the wild-type pycnidia (Fig 6D). However, for the hypervirulent mutant IPO323ΔZt89160 we observed no quantitative difference of pycnidiospores compared to wild-type. The effect on pycnidiospore production could be restored for both IPO323ΔZt80707 and IPO323ΔZt103264 mutants by reintroducing the respective gene into the deletion strain (Fig 6D).
We next confirmed the developmental defect of the pycnidia formation in IPO323ΔZt80707 and IPO323ΔZt103264 in comparison to wild-type and the hypervirulent mutant IPO323ΔZt89160 using confocal microscopy. We stained infected leaf samples at 14 and 28 dpi using a wheat germ agglutinin—fluorescein isothiocyanate / propidium iodide double staining to visualize both the fungus and plant cells. We measured the width of the pycnidia (n = 50 for each strain and time point) on wheat leaves infected by the wild-type strain and mutants. We find that the size of the pycnidia generated by wild-type Z. tritici and the mutant IPO323ΔZt89160 is almost unchanged from 14 dpi to 28 dpi at 50–60 μm (Fig 7). Furthermore, we found that the mean pycnidia size of IPO323ΔZt80707 and IPO323ΔZt103264 is significantly smaller than the wild-type pycnidia at both time points of infection, 14 and 28 dpi (Fig 7). Pycnidia produced by the hypervirulent mutant IPO323ΔZt89160 on the other hand did not deviate from wild-type pycnidia. We confirmed that the effect found in the IPO323ΔZt80707 and IPO323ΔZt103264 mutants is due to the deletion of the two genes, since reintroduction of the respective genes could restore wild-type pycnidia development.
Fig 7. Confocal laser scanning microscopy image analyses of pycnidia of wild and mutant Z. tritici.

WGA-FITC / Propidium iodide double-stained pycnidia on infected wheat leaves 28 dpi of wild-type (A), mutant Z. tritici isolates; IPO323ΔZt80707 (B) and IPO323ΔZt103264 (C). Fungal cell walls (green) and fungal and plant nuclei (red) are visible in the infected plant tissue (scale bar: 50 μm). D) Average size of pycnidia measured 14 and 28 dpi. For each strain and time point, 50 pycnidia were measured. ** p<0.01; p-values were calculated using a Mann-Whitney U test. Whiskers indicate standard deviations.
Discussion
Recent speciation of the wheat pathogen Z. tritici entailed adaptation to a new host and involved a strong effect of natural selection during divergence from a common ancestor of Z. tritici, Z. pseudotritici and Z. ardabiliae [8]. We hypothesize that signatures of positive selection in the genomes of these pathogens reflect those traits that have been important for divergent host specialization. The functional analyses of four positively selected genes in Z. tritici allowed us to identify three genes with significant impact on disease development in wheat. For two of these three genes, Zt80707 and Zt103264, we could furthermore show that Z. tritici-specific amino acid changes are crucial for virulence in wheat. Rapid evolution and positive selection of pathogenicity related genes have been documented in other filamentous plant pathogens [5,19,20]. Aguileta and colleagues used genome data and EST libraries from different species of Botrytis and Sclerotinia to search for genes with signatures of positive selection in a dataset of 642 orthologs [21]. Of 21 positively selected genes, four genes were further tested for virulence related functions, but proved non-essential for disease development of Botrytis at least as tested under laboratory conditions. The four genes studied by Aguileta and colleagues and the gene Zt110804 included in our functional analyses demonstrate that the relevance of positively selected amino acid changes cannot always be detected with standard experimental assays. Other functional assays could include a variety of host genotypes or different environmental conditions to determine eventual fitness effects in mutant strains.
For two of the genes studied here, Zt80707 and Zt10324, we not only find variation at the level of nucleotide substitutions, but also at the level of reading frame structure. To our knowledge this is the first report of large variation of reading frame structure variation in virulence related genes of a fungal plant pathogen. A dramatic consequence of the different transcript start of Zt80707 in Z. tritici is a signal peptide that is only transcribed and translated in Z. tritici. Thus, in Z. tritici, the protein encoded by Zt80707 acts in the extracellular space and may interact with host-produced proteins while orthologous proteins in Z. pseudotritici and Z. ardabiliae act intracellularly. In Z. ardabiliae, but not in Z. pseudotritici, there is a methionine at the corresponding site of the transcription start in Z. tritici. However, our 5’ RACE-PCR experiment clearly shows that the transcription start of Za80707 is initiated 25 amino acids downstream (S4 Fig). Either, transcription of the signal peptide in Z. pseudotritici and Z. ardabiliae was lost after divergence of the Z. tritici lineage, or, more plausible, the earlier transcription start originated more recently in the Z. tritici lineage. We speculate that non-synonymous substitutions in the Z. tritici gene relate to a novel function of the protein in the extra-cellular space. Indeed the function of Zt80707 could not be fully restored by integration of the orthologs from Z. pseudotritici and Z. ardabiliae with or without the secretion signal (Fig 5). Further characterization of protein function in Z. pseudotritici, Z. ardabiliae and the more distantly related sister species Z. passerinii, will be necessary to clarify the ancestral structure and function of the protein.
The deletion mutants of both Zt80707 and Zt103264 are still able to infect and reproduce asexually in wheat, however the mutants are significantly impaired in the development of pycnidia. We measured this as a reduced number of pycnidia and as significantly smaller pycnidia in the mutant-infected plants. Zt103264 is upregulated during early host colonization and may therefore play a role in the early establishment of biotrophic growth and defeat of host defenses. The observed effect on pycnidia production would thereby be a secondary effect following lower biomass and impaired pathogen development. Zt80707 is expressed during necrotrophic growth and pycnidia formation. In other ascomycete fungi, asexual spore formation has been linked to primary and secondary metabolite production [22, 23]. Consistent with an extracellular function of Zt80707 in Z. tritici, we did not detect any difference in pigment production during an in vitro cell wall and temperature stress assay. Nor did we observe differences in the growth morphology of yeast-like cells or hyphae. We speculate that Zt80707 instead plays a role in host-fungus signaling and interaction and indirectly affects the development of pycnidia in Z. tritici.
The quantitative impact on disease development by the two mutants IPO323ΔZt80707 and IPO323ΔZt103264 suggest that multiple gene products contribute to virulence in Z. tritici. A single determining virulence factor has also been described in Z. tritici [16]. Deletion of the Mg3LysM effector causes full virulence defect in Z. tritici. LysM effectors are known to interfere with chitin-triggered immunity in plants and have been described in a number of fungal plant pathogens [17]. It is likely that Z. tritici encodes an arsenal of effector proteins during early biotrophic colonization of wheat. These genes may also evolve by positive selection driven by an antagonistic arms-race evolution between effectors and their target proteins. However, the genes picked up by our comparative genome analyses and further investigated here reflect signatures of past positive selection related to speciation and divergent host specialization. Sequencing of more Z. tritici genomes will allow inference of ongoing positive selection in the genome of the wheat pathogen.
Our third candidate, Zt89160, exhibited a hypervirulent phenotype. Mutant hypervirulence has only been described in a few examples of fungal plant pathogens [24–27]. Zt89160 contains two RCC1-like domains. RCC homologues have been characterized as nuclear proteins in many eukaryotes, including Saccharomyces cerevisiae and Schizosaccharomyces pombe [28–31]. Disruption of the gene in different organisms affects RNA processing and transport, mating, initiation of mitosis and chromatin condensation. So far RCC1 proteins have never been studied in a fungal plant pathogen or connected to fungal pathogenicity. We speculate that Zt89160 could play a central role in the regulation of virulence-related genes in Z. tritici consistent with the increased pycnidia formation in the deletion mutant. The hemibiotrophic nature of Z. tritici requires a fine-tuned regulation of transcription during host infection and the switch from biotrophic feeding to necrotrophic feeding, and this regulation may be affected in the IPO323ΔZt89160 mutant. Positive selection acting on the gene could reflect changes in the binding sites of Zt89160 relative to the orthologs in Z. pseudotritici and Z. ardabiliae. This hypothesis is supported by the fact that most amino acid substitutions locate on the outer surface of the protein (S5 Fig).
In summary, we here show a strong correlation between evolutionary predictions and virulence function in a plant-pathogenic fungus. Previous studies of prokaryote and eukaryote pathogenic species have likewise demonstrated accelerated evolution of virulence-related genes [3,32]. The focus of these studies has been the evolution of effector-encoding genes, typically small, secreted proteins. Our selection of candidate genes has however been determined only on the basis of evolutionary predictions and no a priori information about gene function or structure. We demonstrate that adaptive evolution during host specialization also strongly affects non-secreted proteins without a putative effector function (Zt89160, Zt103264 and Zt110804). Even so, several of these genes may play a central role in virulence through the regulation of other genes or impact on in planta development of hyphae and spore production. The findings presented here suggest that the integration of evolutionary predictions and functional analyses provide a strong framework for the identification of new pathogenicity-related traits and host species determinants of pathogens.
Materials and Methods
Positively selected candidate genes
The four genes, Zt80707, Zt89160, Zt103264 and Zt110804, were selected according to their elevated rates of non-synonymous substitutions in Zymoseptoria tritici. Signatures of strong positive selection in these four genes were previously demonstrated using the approach of Nei and Gojobori [9] in a comparative genome study, including full genome sequences of Z. tritici and two closely related species, Z. pseudotritici and Z. ardabiliae [8]. The genomic coordinates of the candidate genes in the Z. tritici reference genome [33] are provided in Table 1.
Validation of gene structures by RACE-PCR
To validate gene structure, including start and stop codons and intron-exon boundaries, we conducted 5'-RACE-PCR in Z. tritici, Z. pseudotritici and Z. ardabiliae for the genes Zt80707 and Zt103264 and their orthologs in Z. pseudotritici and Z. ardabiliae. RNA extraction was conducted as described below from axenic cultures grown in YMS medium from the Z. tritici isolate IPO323, the Z. pseudotritici isolate STIR04_2.2.1 (Zp13) and the Z. ardabiliae isolate STIR04_1.1.1 (Za17) [8]. We used the 5’ RACE System for Rapid Amplification of cDNA Ends Kit (Invitrogen, Karlsruhe, Germany) and designed three gene-specific primers (GSPs) for each gene for the first strand cDNA synthesis, for a first PCR GSP2 and a nested GSP3 for a nested PCR (S1 Table). The resulting PCR products were cloned into the TOPO (Invitrogen, Karlsruhe, Germany) backbone and subsequently sequenced.
Analyses of positive selection in candidate genes
Gene sequences were aligned in Seaview [34] using the program muscle [35]. We re-analyzed gene alignments of Zt80707, Zt89160, Zt103264 and Zt110804 to assess d N/d S ratios (the non-synonymous substitutions rate divided by the synonymous substitutions rate) using the codeml program from the Phylogenetic Analysis by Maximum Likelihood (PAML) package [36]. The approach used by Nei and Gojobori allowed us to identify an excess of non-synonymous mutations; however, it did not provide information about the non-homogenous rates of evolution among the three Zymoseptoria species. With the new gene alignments, we asked whether any of the four genes in particular had experienced accelerated evolution in the wheat pathogen Z. tritici. Using the program PhyML [37], we generated a phylogenetic tree for each gene alignment, including sequence data from the more distantly related species Z. passerinii. We calculated d N/d S ratios for the individual branches of the trees [36]. Branch-specific d N/d S ratios above 1 are indicative of positive selection.
Prediction of the protein structures
Signal P v3 was used for prediction of secretion signals [14]. For the prediction of the protein structure we used the I-TASSER program [18]. The program is based on a composite approach of many threading (fold recognition) programs to structure alignments. The quality of the predicted protein structure is evaluated using the C-score (-0.4) and the TM-score (0.66) which both indicate an evaluation of the structure prediction. For the three protein products of Zt80707, Zt103264 and Zt110804 we were not able to obtain significant structure predictions. We could however obtain a reliable prediction for the protein structure of Zt89160. For this protein, we determined the surface accessible amino acids using the SwissPDB-Viewer [38]. The protein structure was visualized using the PyMOL Molecular Graphics System [39].
Fungal and bacterial strains
For all in planta and transformation experiments, we used the reference isolate of Z. tritici IPO323 [40]. The genome sequence of IPO323 is available from http://genome.jgi-psf.org/Mycgr3/Mycgr3.home.html [33]. The Z. tritici gene IDs that we used here correspond to the Mgr gene IDs used in the JGI genome database. Furthermore, we used the Z. pseudotritici isolate ST04IR_2.2.1 (Zp13) and the Z. ardabiliae isolate ST04IR_1.1.1 (Za17), for which genome sequences are available from the National Center for Biotechnology Information (NCBI) database (taxonomy ID: 985140 and 985147, respectively) [8]. The isolates were inoculated from glycerol stocks onto solid yeast-malt-sucrose agar plates at 18°C. Yeast-like cells grown on these plates were used as inoculum for all experiments. All plasmids used in our study were maintained in E. coli Top10 cells (Invitrogen, Karlsruhe, Germany). The Agrobacterium tumefaciens strain AGL1 was used for Agrobacterium tumefaciens mediated transformation (ATMT) of the fungal cells. Both bacterial strains were grown on double yeast tryptone (dYT) medium. For the maintenance of the plasmids already present in the AGL1 strain, 50 μg/ml Rifampicin (Sigma, Taufkirchen, Germany) and 100 μg/ml Carbenicillin (Sigma, Taufkirchen, Germany) were added to the dYT medium.
ATMT of fungal cells
To assess the functional role of the four candidate genes, Zt80707, Zt89160, Zt103264 and Zt110804, we generated four deletion mutants in Z. tritici. For targeted gene deletion, we amplified a DNA fragment for each gene, including the ORF and 1 kb upstream and downstream sequence by PCR. Each amplification product was fused with a hygromycin resistance cassette [41] using an overlap PCR approach [42]. Deletion constructs were ligated into the pES22 plasmid via two restriction sites added by outer primers (S6 Fig and S1 Table). The plasmid pES22 is a derivate of the binary vector pNOV-ABCD previously developed for targeted gene deletion in Z. tritici [41]. We furthermore generated constructs for complementation tests of Zt80707, Zt89160 and Zt103264. For each construct, a DNA fragment, including the coding sequence of the candidate gene and 1 kb of upstream and downstream sequence, was fused together with a Geneticin (G418) resistance, cloned into the pES22 plasmid cassette using Gibson assembly [43] and introduced into the respective deletion strain. The same approach was conducted to replace the Z. tritici genes with orthologs from Z. pseudotritici or Z. ardabiliae. As for the complementation strains, orthologous genes were fused in frame to the respective Z. tritici promoter and introduced in the deletion mutants using ATMT. In total we generated five replacement strains including IPO323ΔZt80707::sp+Zp80707 IPO323ΔZt80707::Zp80707 IPO323ΔZt80707::Za80707, IPO323ΔZt89160::Zp89160, IPO323ΔZt103264::Za103264. Electro-competent cells of the A. tumefaciens strain AGL1 were transformed with the final plasmids (S2 Table) using standard procedures. For ATMT of Z. tritici, we used the protocol described by Zwiers and De Waard (2001) [44]. Transformed fungal colonies were visible on hygromycin- or Geneticin-containing plates two weeks after transformation. Colonies obtained from single cells were propagated in YMS medium for further DNA extraction, PCR and Southern blot analysis.
Isolation of fungal genomic DNA and Southern blot analysis
Fungal DNA was extracted using a standard phenol-chloroform extraction protocol [45]. We first screened transformed strains using a PCR-based approach, amplifying the hygromycin resistance cassette and the endogenous locus using the outer primers of the deletion constructs (S1 Table and S6 Fig). To confirm homologous recombination and correct transformation, we performed a Southern blot analysis using standard procedures [46]. Probes were generated using the PCR Digoxigenin (DIG) Labeling Mix (Roche, Mannheim, Germany) according to the manufacturer’s instructions.
Plant infections
For plant infections, we used 15-day-old wheat (Triticum aestivum) seedlings of the cultivar Obelisk (Wiersum Plantbreeding). A spore solution of 1 × 107 cells/ml containing 0.1% Tween 20 (Roth, Karlsruhe, Germany) was brushed onto an 8–10 cm marked area of the second leaf of each plant. After an initial 48-h incubation period at 100% humidity, the infected plants were incubated at 22°C with a 16-h light period at 75% humidity for another 26 days. For all experiments two independent strains (biological replicates) were used.
Phenotypic assays
We performed an in vitro assay of wild-type and deletion mutants to test whether any observed phenotype relates to the host-pathogen interaction or to basic growth performance of the mutants. First, we investigated single cells of the wild-type and the deletion mutants microscopically using a light microscope (Leica DM750, Wetzlar, Germany). Next, we conducted a stress assay to investigate whether deletion mutants were affected in their response to cell stress reagents. Four μl of a spore suspension containing 1 × 107 spores/ml and 6 1:10 dilutions of a dilution series were pipetted on stress plates and incubated for six days at 18°C. We grew fungal cells on plates containing NaCl (1.5 M), H2O2 (2 mM), Congored (500 μg/ml) and Calcofluor (200 μg/ml) to compare the sensitivity of strains to osmotic and oxidative cell wall stresses. We also incubated fungal colonies at 28°C to assess temperature sensitivity. Wild-type, mutant and complementation strains were all assayed for their in vitro phenotypes. To investigate the effect of gene deletion in planta, we compared disease development of wild-type and mutant-infected plant leaves 28 dpi. Symptoms recognized as pycnidia were evaluated. To quantify disease levels, we used a scoring scheme of six categories (0%, 1–20%, 21–40%, 41–60%, 61–80% and 81–100%) representing the percentage of the leaf area covered with pycnidia. 48 wheat leaves infected with wild-type were compared to 59–96 leaves infected with deletion mutants, complementation or replacement strains (Figs 4 and 5). Disease scoring was done by eye always by the same person. A Mann-Whitney-U-test was applied to test the statistical significance of the observed differences between wild-type all and mutant strains.
We evaluated and compared pycnidia spore viability from wild-type and the Zt80707- and Zt103264-deletion mutant-infected leaves. The infected leaves were harvested four weeks after infection and surface sterilized using 5% sodium hypochlorite and 70% ethanol. The infected leaves were incubated under high-humidity conditions for seven days on a metal grid in a sealed Petri dish in the phytochamber with the same light settings as the infection experiment (see above). The high-humidity conditions in the Petri dish induce the oozing of pycnidia and the release of spores. The whole-leaf samples were vortexed gently in 500 μl sterile H2O and 1:10 dilution series were made with three steps. Three μl of every dilution was pipetted on YMS and YMS-hygromycin plates. The proportions of germinating spores were compared between the wild-type and the deletion strains (S11 Fig).
To investigate the quantitative difference between deletion mutants of Zt80707, Zt89160 and Zt103264 and the wild-type strain, we estimated the number of pycnidiospores per pycnidium. Therefore, we also induced oozing from pycnidia on harvested leaves and then used a “Neubauer-improved” counting chamber to count the amount of pycnidiospores isolated from the oozing pycnidia. The number of pycnidiospores in the spore suspensions was divided by the number of pycnidia on each leaf from which the spores were isolated to estimate the number of pycnidiospores per pycnidium.
WGA-FITC / Propidium iodide staining
Harvested leaf samples from two independent plant experiments were de-stained overnight (or longer) in 2 ml Eppendorf (Hamburg, Germany) tubes in 100% ethanol. Ethanol was exchanged if necessary. Next, the leaves were incubated in 10% KOH at 85°C for 5 min. The samples were then washed 3–4 times with PBS (pH 7.4) and the staining solution was added. The samples were vacuum infiltrated at 100 mbar using a cvc3000 vacuum controller (Vacuubrand, Wertheim, Germany). The staining solution was collected for reuse and the samples were de-stained in PBS and stored in the dark at 4°C. The staining solution was prepared using 20 μg/ml Propidium iodide, 10 μg/ml WGA-FITC, and 0.02% Tween 20 in 1× PBS (pH 7.4). Microscopy was conducted using a Leica SP5 confocal microscope. The filter wavelengths used for the detection were 488 nm (for FITC) and 561 nm (for Propidium iodide). The fluorophors were excited by an argon and a diode-pumped solid-state (DPSS) laser.
RNA isolation and quantitative RT-PCR
We analyzed gene expression patterns of Zt80707, Zt89160, Zt103264 and Zt110804 in Z. tritici using a qRT-PCR experiment. Total RNA was extracted from fungal axenic cultures (grown for 72 h in YMS medium at 18°C and 140 rpm) and from freeze-dried leaf tissue infected with Z. tritici (4, 7, 14 and 28 dpi) using the TRIZOL reagent (Invitrogen), following the manufacturer’s instructions. Three biological replicates were sampled from axenically grown cultures and from each time point of infection. The samples were crushed in liquid nitrogen and 100 mg was used for RNA extraction and cDNA synthesis. The cDNA samples were used in a qRT-PCR experiment employing the iQ SYBR Green Supermix Kit (Bio-Rad, Munich, Germany), GSPs (S1 Table) and an annealing temperature of 59°C. PCR was conducted in a CFX96 RT-PCR Detection System (Bio-Rad) with the constitutively expressed control gene Zt99044, a Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [33]. A Mann-Whitney U test was applied to test the significance of different gene expression levels.
Secretion assay for Zt80707
To investigate the presence of a putative signal peptide at the 5’ end of the gene Zt80707, we analyzed the transcribed sequence using SignalP 3.0 [14]. SignalP only returned weak evidence for a secretion signal and we therefore aimed to experimentally verify the putative signal peptide. To do this, we designed a construct for stable expression of Zt80707 in a non-coding region of chromosome 1 (Chr1: 464436–466636) in Z. tritici. Zt80707 is only weakly expressed in axenic culture and we therefore expressed it under the control of the constitutively induced gpdA promoter from Aspergillus nidulans [15]. To verify secretion in a Western blot-based experiment, we also fused a 3’ GFP tag to the Zt80707 sequence. In addition to Zt80707, we also generated a construct with the ortholog Zp80707 of Z. pseudotritici. The ORFs were cloned into the plasmid pES150 by Gibson assembly [43]. The positive and negative controls, Zt111221 and Zt77228, respectively, were also expressed under control of the gpdA promoter and tagged with a 3’ GFP. The constructs furthermore included a Geneticin resistance cassette and 1 kb flanking region of the non-coding locus at chromosome 1 in order to allow for correct integration of the constructs at this site via homologous recombination. After transformation of the four constructs in IPO323, positive transformants were grown in 50 ml YMS medium at 200 rpm and 18°C for 72 h until an OD600 of 1. For protein extraction, the cultures were centrifuged at 10000× g for 15 min. Protein extraction from the cells was conducted using a peqGOLD TriFast kit (Peqlab, Erlangen, Germany), according to the manufacturer’s instructions. For precipitation of the proteins in the supernatant, 40 ml of each culture was lyophilized and dissolved again in 1 ml H20. This same methodology was used for a TCA precipitation [45] and the resulting protein pellet was dissolved in 50 μl 1% sodium dodecyl sulfate (SDS). The protein concentrations were estimated using the Bradford reagent in order to load similar amounts on a 15% SDS gel. This was confirmed by a Coomassie staining of the SDS gels to show similar protein amounts for the pellet and supernatant lanes. Hereafter, a Western blot analysis was performed using electrophoretic transfer. Finally, the proteins of interest were detected using an a-GFP primary antibody (Roche), together with a horseradish peroxidase (HRP) linked secondary antibody (New England Biolabs, Frankfurt, Germany) and the Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Freiburg, Germany).
Supporting Information
(DOCX)
(DOCX)
Protein alignment of Zt89160, Zt103264 and Zt110804 of different isolates of Z. tritici (Zt), Z. pseudotritici (Zp) and Z. ardabiliae (Za).
(TIF)
Schematic illustration of conserved gene order in genomic regions encoding Zt80707, Zt89160, Zt103264 and Zt110804 in Z. tritici (Zt09), Z. pseudotritici (Zp13) and Z. ardabiliae (Za17). The four candidate genes are shown in green, neighboring genes are shown in dark grey, inverted genes are shown in light grey and transposable elements are shown in red.
(TIF)
Phylogenetic trees were made for Z. tritici genes Zt80707 (A), Zt89160 (B), Zt103264 (C) and Zt110804 (D) and the respective orthologs of Z. pseudotritici, Z. ardabiliae and Z. passerinii. A branch model was used to estimate the ω (d N/d S ratio) for each branch. Branch-specific ω values above 1 are indicative of positive selection.
(TIF)
Rapid Amplification of cDNA Ends (RACE-PCR) of Zt80707 and Zt103264 revealed the ORFs (black arrows) schematically illustrated, including 5’UTR (white rectangles) and start codon positions. The lengths of the ORFs were confirmed using the primer combinations “A” and “C” and “B” and “C” with cDNA of Zt80707 and D-E and D-F of Zt103264. As template for the PCRs genomic DNA (g) of the three strains, Zt09, Zp14 and Za17 were used, as well as cDNA (c) obtained from infected wheat leaves.
(TIF)
A) Top view, side view and bottom view of the Zt89160 protein structure predicted with I-TASSER. Beta sheets are depicted in yellow and the accessible amino acids on the surface of the protein are shown in red. B) Top view, side view and bottom view of the structure predicted by I-TASSER for the Drosophila melanogaster protein CG6678, a protein homologous to Zt89160. Beta sheets are depicted in yellow and alpha helices are shown in red. The table shows that the significant majority of positively selected amino acids is located on the surface of the protein Zt89160. ** p<0.01. p-value calculated using a Fishers exact test.
(TIF)
Schematic illustration of the gene deletion approach in Z. tritici (example Zt80707). A) Gene deletion by homologous recombination using Agrobacterium tumefaciens-mediated transformation. B) PCR screening to identify correctly transformed strains using the primer combination oES19 and oES22. C) Correctly transformed strains were finally confirmed via Southern blot analysis using the oES19- and oES22-amplified sequence as a probe for hybridization.
(TIF)
Three independent mutants of each generated deletion and complementation strain for the genes Zt80707, Zt89160 and Zt103264 were tested under multiple abiotic stress conditions. The deletion of Zt110804 was not complemented. The following conditions were used: heat stress (28°C), NaCl (1.5 M), Congored (500 μg/ml), H2O2 (2 mM) and Calcofluor (200 μg/ml).
(TIF)
Representative images for the six categories that were used to score disease levels at 28 dpi in plant assays. The number of pycnidia per leaf (ppl) is shown for every leaf. Areas encircled by red lines illustrate the proportion of necrotic areas with pycnidia.
(TIF)
Expression levels of the three genes in the respective complementation- and replacement strains at 4 dpi have been compared to wild-type Z. tritici infected leaves (WT). Values are normalized to the expression of the gene encoding GAPDH, a constitutively expressed housekeeping control. Error bars indicate the standard error of the mean (SEM) of three independent biological replicates per sample.
(TIF)
The promoter (light green) and signal peptide (light grey) of Zt80707 from Z. tritici were fused with the ORF of Zp80707 from Z. pseudotritici (beginning of the ORF shown in dark grey) to obtain the secretion of Zp80707 in Z. tritici.
(TIF)
A) Macroscopic picture of oozing pycnidia of an infected wheat leaf (IPO323 wild-type) after surface sterilization and one week of incubation in high humidity. B) Exuded fungal spores were isolated and added in different concentrations to YMS media to test the viability of wild-type and mutant spores.
(TIF)
Acknowledgments
The authors thank Gert Kema for providing the IPO323 strain, M. Csuki, Syngenta for sharing plasmids, Shilpi Chaurasia for advice regarding protein structure prediction, Jonathan Grandaubert for assistance with repeat analyses and M. Boelker, R. Kahmann and J. Dutheil for helpful discussions and comments on a previous version of this manuscript.
Data Availability
Data are available from the Joint Genome Institute (http://genome.jgi-psf.org/Mycgr3/Mycgr3.home.html) and from the NCBI database (http://www.ncbi.nlm.nih.gov/genome/browse/).
Funding Statement
This study was funded by intramural funding to EHS by the Max Planck Society (http://www.mpg.de/en). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Giraud T, Gladieux P, Gavrilets S (2010) Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol Evol 25: 387–395. 10.1016/j.tree.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stukenbrock EH (2013) Evolution, selection and isolation: a genomic view of speciation in fungal plant pathogens. New Phytol. [DOI] [PubMed] [Google Scholar]
- 3. Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, et al. (2010) Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330: 1540–1543. 10.1126/science.1193070 [DOI] [PubMed] [Google Scholar]
- 4. Schardl CL, Young CA, Hesse U, Amyotte SG, Andreeva K, et al. (2013) Plant-Symbiotic Fungi as Chemical Engineers: Multi-Genome Analysis of the Clavicipitaceae Reveals Dynamics of Alkaloid Loci. PLoS Genet 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stukenbrock EH, McDonald BA (2007) Geographical variation and positive diversifying selection in the host-specific toxin SnToxA. Mol Plant Pathol 8: 321–332. 10.1111/j.1364-3703.2007.00396.x [DOI] [PubMed] [Google Scholar]
- 6. Brunner PC, Torriani SFF, Croll D, Stukenbrock EH, McDonald BA (2013) Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol Biol Evol 30: 1337–1347. 10.1093/molbev/mst041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponomarenko A, Goodwin SB, Kema GHJ (2011) Septoria tritici blotch (STB) of wheat Septoria tritici blotch (STB) of wheat. Plant Heal Instr: 1–7. [Google Scholar]
- 8. Stukenbrock EH, Bataillon T, Dutheil JY, Hansen TT, Li R, et al. (2011) The making of a new pathogen: insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res 21: 2157–2166. 10.1101/gr.118851.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426. [DOI] [PubMed] [Google Scholar]
- 10. Makde RD, England JR, Yennawar HP, Tan S (2010) Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467: 562–566. 10.1038/nature09321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williamson MP (1994) The structure and function of proline-rich regions in proteins. Biochem J 297: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stukenbrock EH, McDonald BA (2008) The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol 46: 75–100. 10.1146/annurev.phyto.010708.154114 [DOI] [PubMed] [Google Scholar]
- 13. Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- 14. Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795. [DOI] [PubMed] [Google Scholar]
- 15. Mikkelsen L, Sarrocco S, Lübeck M, Jensen DF (2003) Expression of the red fluorescent protein DsRed-Express in filamentous ascomycete fungi. FEMS Microbiol Lett 223: 135–139. [DOI] [PubMed] [Google Scholar]
- 16. Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, et al. (2011) Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol 156: 756–769. 10.1104/pp.111.176347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, et al. (2010) Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329: 953–955. 10.1126/science.1190859 [DOI] [PubMed] [Google Scholar]
- 18. Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stergiopoulos I, De Kock MJD, Lindhout P, De Wit PJGM (2007) Allelic variation in the effector genes of the tomato pathogen Cladosporium fulvum reveals different modes of adaptive evolution. Mol Plant Microbe Interact 20: 1271–1283. [DOI] [PubMed] [Google Scholar]
- 20. Dong S, Stam R, Cano LM, Song J, Sklenar J, et al. (2014) Effector specialization in a lineage of the Irish potato famine pathogen. Science 343: 552–555. 10.1126/science.1246300 [DOI] [PubMed] [Google Scholar]
- 21. Aguileta G, Lengelle J, Chiapello H, Giraud T, Viaud M, et al. (2012) Genes under positive selection in a model plant pathogenic fungus, Botrytis. Infect Genet Evol 12: 987–996. 10.1016/j.meegid.2012.02.012 [DOI] [PubMed] [Google Scholar]
- 22. Gummer JPA, Trengove RD, Oliver RP, Solomon PS (2013) Dissecting the role of G-protein signalling in primary metabolism in the wheat pathogen Stagonospora nodorum. Microbiology 159: 1972–1985. 10.1099/mic.0.067009-0 [DOI] [PubMed] [Google Scholar]
- 23. Calvo AM, Wilson RA, Bok JW, Keller NP (2002) Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66: 447–459, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baldwin TK, Winnenburg R, Urban M, Rawlings C, Koehler J, et al. (2006) The pathogen-host interactions database (PHI-base) provides insights into generic and novel themes of pathogenicity. Mol Plant Microbe Interact 19: 1451–1462. [DOI] [PubMed] [Google Scholar]
- 25. Degrassi G, Devescovi G, Bigirimana J, Venturi V (2010) Xanthomonas oryzae pv. oryzae XKK.12 contains an AroQgamma chorismate mutase that is involved in rice virulence. Phytopathology 100: 262–270. 10.1094/PHYTO-100-3-0262 [DOI] [PubMed] [Google Scholar]
- 26. Kamper J, Kahmann R, Bolker M, Ma L- JJ, Brefort T, et al. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444: 97–101. [DOI] [PubMed] [Google Scholar]
- 27. Meir S, Amsellem Z, Al-Ahmad H, Safran E, Gressel J (2009) Transforming a NEP1 toxin gene into two Fusarium spp. to enhance mycoherbicide activity on Orobanche—failure and success. Pest Manag Sci 65: 588–595. 10.1002/ps.1736 [DOI] [PubMed] [Google Scholar]
- 28. Aebi M, Clark MW, Vijayraghavan U, Abelson J (1990) A yeast mutant, PRP20, altered in mRNA metabolism and maintenance of the nuclear structure, is defective in a gene homologous to the human gene RCC1 which is involved in the control of chromosome condensation. Mol Gen Genet 224: 72–80. http://www.ncbi.nlm.nih.gov/pubmed/2277633. Accessed 13 November 2014. [DOI] [PubMed] [Google Scholar]
- 29. Kadowaki T, Goldfarb D, Spitz LM, Tartakoff AM, Ohno M (1993) Regulation of RNA processing and transport by a nuclear guanine nucleotide release protein and members of the Ras superfamily. EMBO J 12: 2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clark KL, Sprague GF (1989) Yeast pheromone response pathway: characterization of a suppressor that restores mating to receptorless mutants. Mol Cell Biol 9: 2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sazer S, Nurse P (1994) A fission yeast RCC1-related protein is required for the mitosis to interphase transition. EMBO J 13: 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, et al. (2011) Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog 7: e1002132 10.1371/journal.ppat.1002132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goodwin SB, Ben M’barek S, Dhillon B, Wittenberg AH, Crane CF, et al. (2011) Finished Genome of the Fungal Wheat Pathogen Mycosphaerella graminicola Reveals Dispensome Structure, Chromosome Plasticity, and Stealth Pathogenesis. PLoS Genet 7: e1002070 10.1371/journal.pgen.1002070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27: 221–224. 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- 35. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang Z, Nielsen R (1998) Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol 46: 409–418. [DOI] [PubMed] [Google Scholar]
- 37. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 38. Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30 Suppl 1: S162–S173. 10.1002/elps.200900140 [DOI] [PubMed] [Google Scholar]
- 39.Schrödinger, LLC (2010) The {PyMOL} Molecular Graphics System, Version~1.3r1.
- 40. Kema GH, van Silfhout CH (1997) Genetic Variation for Virulence and Resistance in the Wheat-Mycosphaerella graminicola Pathosystem III. Comparative Seedling and Adult Plant Experiments. Phytopathology 87: 266–272. [DOI] [PubMed] [Google Scholar]
- 41. Bowler J, Scott E, Tailor R, Scalliet G, Ray J, et al. (2010) Technical advance New capabilities for Mycosphaerella graminicola research. Mol Plant Pathol 11: 691–704. 10.1111/j.1364-3703.2010.00629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shuldiner AR, Tanner K, Scott LA, Moore CA, Roth J (1991) Ligase-free subcloning: a versatile method to subclone polymerase chain reaction (PCR) products in a single day. Anal Biochem 194: 9–15. [DOI] [PubMed] [Google Scholar]
- 43. Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, et al. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 44. Zwiers LH, De Waard MA (2001) Efficient Agrobacterium tumefaciens-mediated gene disruption in the phytopathogen Mycosphaerella graminicola. Curr Genet 39: 388–393. [DOI] [PubMed] [Google Scholar]
- 45. Sambrook J, Russell DW, Laboratory CSH (2001) Molecular cloning: a laboratory manual 3rd. ed. Cold Spring Harbor, N. Y: Cold Spring Harbor Laboratory. [Google Scholar]
- 46. Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 503–517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
Protein alignment of Zt89160, Zt103264 and Zt110804 of different isolates of Z. tritici (Zt), Z. pseudotritici (Zp) and Z. ardabiliae (Za).
(TIF)
Schematic illustration of conserved gene order in genomic regions encoding Zt80707, Zt89160, Zt103264 and Zt110804 in Z. tritici (Zt09), Z. pseudotritici (Zp13) and Z. ardabiliae (Za17). The four candidate genes are shown in green, neighboring genes are shown in dark grey, inverted genes are shown in light grey and transposable elements are shown in red.
(TIF)
Phylogenetic trees were made for Z. tritici genes Zt80707 (A), Zt89160 (B), Zt103264 (C) and Zt110804 (D) and the respective orthologs of Z. pseudotritici, Z. ardabiliae and Z. passerinii. A branch model was used to estimate the ω (d N/d S ratio) for each branch. Branch-specific ω values above 1 are indicative of positive selection.
(TIF)
Rapid Amplification of cDNA Ends (RACE-PCR) of Zt80707 and Zt103264 revealed the ORFs (black arrows) schematically illustrated, including 5’UTR (white rectangles) and start codon positions. The lengths of the ORFs were confirmed using the primer combinations “A” and “C” and “B” and “C” with cDNA of Zt80707 and D-E and D-F of Zt103264. As template for the PCRs genomic DNA (g) of the three strains, Zt09, Zp14 and Za17 were used, as well as cDNA (c) obtained from infected wheat leaves.
(TIF)
A) Top view, side view and bottom view of the Zt89160 protein structure predicted with I-TASSER. Beta sheets are depicted in yellow and the accessible amino acids on the surface of the protein are shown in red. B) Top view, side view and bottom view of the structure predicted by I-TASSER for the Drosophila melanogaster protein CG6678, a protein homologous to Zt89160. Beta sheets are depicted in yellow and alpha helices are shown in red. The table shows that the significant majority of positively selected amino acids is located on the surface of the protein Zt89160. ** p<0.01. p-value calculated using a Fishers exact test.
(TIF)
Schematic illustration of the gene deletion approach in Z. tritici (example Zt80707). A) Gene deletion by homologous recombination using Agrobacterium tumefaciens-mediated transformation. B) PCR screening to identify correctly transformed strains using the primer combination oES19 and oES22. C) Correctly transformed strains were finally confirmed via Southern blot analysis using the oES19- and oES22-amplified sequence as a probe for hybridization.
(TIF)
Three independent mutants of each generated deletion and complementation strain for the genes Zt80707, Zt89160 and Zt103264 were tested under multiple abiotic stress conditions. The deletion of Zt110804 was not complemented. The following conditions were used: heat stress (28°C), NaCl (1.5 M), Congored (500 μg/ml), H2O2 (2 mM) and Calcofluor (200 μg/ml).
(TIF)
Representative images for the six categories that were used to score disease levels at 28 dpi in plant assays. The number of pycnidia per leaf (ppl) is shown for every leaf. Areas encircled by red lines illustrate the proportion of necrotic areas with pycnidia.
(TIF)
Expression levels of the three genes in the respective complementation- and replacement strains at 4 dpi have been compared to wild-type Z. tritici infected leaves (WT). Values are normalized to the expression of the gene encoding GAPDH, a constitutively expressed housekeeping control. Error bars indicate the standard error of the mean (SEM) of three independent biological replicates per sample.
(TIF)
The promoter (light green) and signal peptide (light grey) of Zt80707 from Z. tritici were fused with the ORF of Zp80707 from Z. pseudotritici (beginning of the ORF shown in dark grey) to obtain the secretion of Zp80707 in Z. tritici.
(TIF)
A) Macroscopic picture of oozing pycnidia of an infected wheat leaf (IPO323 wild-type) after surface sterilization and one week of incubation in high humidity. B) Exuded fungal spores were isolated and added in different concentrations to YMS media to test the viability of wild-type and mutant spores.
(TIF)
Data Availability Statement
Data are available from the Joint Genome Institute (http://genome.jgi-psf.org/Mycgr3/Mycgr3.home.html) and from the NCBI database (http://www.ncbi.nlm.nih.gov/genome/browse/).