

Abstract
DNA damage-binding protein 1 (DDB1) is a large subunit of the heterodimeric DDB complex that recognizes DNA lesions and initiates the nucleotide excision repair process. DDB1 is also a component of the CUL4 E3 ligase complex involved in a broad spectrum of cellular processes by targeted ubiquitination of key regulators. Functions of DDB1 in development have been addressed in several model organisms, however, are not fully understood so far. Here we report an ENU induced mutant ddb1 allele (ddb1m863) identified in zebrafish (Danio rerio), and analyze its effects on development. Zebrafish ddb1 is expressed broadly, both maternally and zygotically, with enhanced expression in proliferation zones. The (ddb1m863 mutant allele affects the splice acceptor site of exon 20, causing a splicing defect that results in truncation of the 1140 amino acid protein after residue 800, lacking part of the β-propeller domain BPC and the C-terminal helical domain CTD. ddb1m863 zygotic mutant embryos have a pleiotropic phenotype, including smaller and abnormally shaped brain, head skeleton, eyes, jaw, and branchial arches, as well as reduced dopaminergic neuron groups. However, early forming tissues develop normally in zygotic ddb1m863 mutant embryos, which may be due to maternal rescue. In ddb1m863 mutant embryos, pcna-expressing proliferating cell populations were reduced, concurrent with increased apoptosis. We also observed a concomitant strong up-regulation of transcripts of the tumor suppressor p53 (tp53) and the cell cycle inhibitor cdkn1a (p21a/bCIP1/WAF1) in proliferating tissues. In addition, transcription of cyclin genes ccna2 and ccnd1 was deregulated in ddb1m863 mutants. Reduction of p53 activity by anti-sense morpholinos alleviated the apoptotic phenotype in ddb1m863 mutants. These results imply that Ddb1 may be involved in maintaining proper cell cycle progression and viability of dividing cells during development through transcriptional mechanisms regulating genes involved in cell cycle control and cell survival.
Introduction
The genetic stability of a cell is constantly challenged by environmental and endogenous factors. 50,000–100,000 different damage events have been estimated to occur each day to the DNA in a single human cell [1]. Within the cell cycle, DNA damage has to be uncovered and repaired before or during genome replication to ensure integrity of the genome. Proper cell cycle progression and DNA repair are meticulously controlled by multiple factors including the DNA damage-binding protein (DDB) complex. One of its large subunits, DDB1, has initially been identified as a critical component of the nucleotide excision repair process (NER) for recognizing and removing DNA lesions induced by various mechanisms including ultraviolet (UV) light, chemical carcinogens, and oxidative stress [2–5]. DDB1 functions in DNA-damage repair via two sub-pathways, global genomic repair (GGR) through a heterodimeric complex of DDB1-DDB2, and transcription-coupled repair (TCR) through the interaction of DDB1 and Cockayne syndrome factor (CSA). The failure of NER may contribute to many diseases, including Down syndrome, Parkinson disease, and Huntington's disease [6]. Other functions of DDB1 beyond its accessory role in DNA repair have been associated with the CUL4 E3 ligase complex.
The CUL4 E3 ligase complex consists of an evolutionarily conserved Cullin4 as a scaffold, at its carboxy-terminus a RING-finger protein (ROC1) to assemble a catalytic core with E2 Ubiquitin-conjugating enzyme, and at its amino-terminus a Cullin-specific adaptor and substrate receptor [7–9]. DDB1, a multi-domain protein with three β-propeller folds (BPA, BPB, and BPC) and a C-terminal helical domain tail [8, 10], is such an adaptor acting in the CUL4 E3 ligase complex. The DDB1 BPB propeller domain binds to the N-terminus of CUL4, as well as to other WD40 proteins containing a DWD box (DDB1-binding and WD40 repeat box) [9, 11, 12]. DDB1 functions in the CUL4 E3 ligase complex to recruit substrate receptors (DCAFs—DDB1 and Cul4-Associated Factors) to CUL4. The resulting (DDB1-DCAF)-CUL4-(ROC1-E2) complexes will ubiquitinate and degrade the targeted protein substrates [13–15]. Substrates identified so far include CDT1 (Dup in Drosophila), p21CIP1/WAF1 (human and C. elegans), polymerase eta (C. elegans), PR-Set7/Set8, E2F1 (Drosophila), spd1 and spd2 (S. pombe), and p27KIP1 (mammalian) [16–23]. These substrates play a role in a wide range of cellular processes. Thus the involvement of DDB1 in CUL4 ligase can regulate, by targeting various substrates, multiple cellular processes such as transcription, cell cycle regulation, proliferation, histone lysine methylation, and the adjustment of cellular levels of the tumor suppressor p53 [24, 25]. Some activities of DDB1 have also been linked to developmental processes. Inactivation of DDB1 in fission yeast results in cell cycle and growth defects, increased rate of spontaneous mutations, and disruption of cell differentiation [14, 26, 27]. In mice, deficiency of DDB1 leads to apoptosis, aberrant accumulation of cell cycle regulators, and increased genomic instability [25, 28]. Similar defects are also observed in human U2OS cells, chicken DT40 B cells, in Drosophila, and Arabidopsis thaliana [29–35]. However, the embryonic lethality caused by complete DDB1 deficiency in model organism such as mice has limited research into potential functions during development. In contrast, due to maternal rescue, deficiency of zygotically expressed Ddb1 in zebrafish (Danio rerio) is only lethal at larval stages, thus facilitating studies of DDB1 function in zebrafish development (this paper).
Here, we report the analysis of Ddb1 functions during the zebrafish development. We isolated in an ENU mutagenesis screen the mutant ddb1 m863allele and characterized its effects on development. Disruption of ddb1 in zebrafish resulted in a pleiotropic phenotype, including reduced size of organs that grow and differentiate during early larval stages, including brain and pharyngeal skeleton. Ddb1 deficiency appears to cause p53-dependent apoptosis of pcna-positive proliferating cells, and deregulation of cell cycle regulators including cyclins and CIP/KIP family members. These results suggest that impairment of Ddb1 contributes to the phenotypic defects of ddb1 m863 mutants by deregulation of programmed cell death and cell cycle.
Results
Morphological and neuronal phenotype of m863 mutant embryos
The m863 allele was isolated during a mutagenesis screen aimed at identifying factors involved in dopaminergic (DA) neuron differentiation in zebrafish embryos and early larvae, using tyrosine hydroxylase (th) expression as a marker. m863 mutant embryos at 3 days post fertilization (dpf) had fewer DA neurons in the pretectum and retina, while early differentiating DA neurons of the posterior tuberculum appeared largely normal (Fig 1A–1F). Furthermore, m863 mutant embryos from 3 dpf on displayed morphological defects, including reduced size of the brain and eyes (Fig 1G–1J). The cartilaginous head skeleton with the jaw and other branchial arch derivatives were smaller and did not develop properly in m863 mutants (Fig 1L, 1M, 1O and 1P). In contrast, early forming tissues, including somites and notochord, developed largely normal in mutant embryos, and the body length of mutant and wild-type larvae was similar (Fig 1G–1J). Cell death, as judged from granular and turbid appearance of tissue, was already apparent in the dorsal midbrain and eyes of life m863 mutants at 36 hpf (unpublished data). At 48 hpf, cell death appeared to be further increased in the retina and fore- and midbrain of m863 mutant embryos, when compared to their wild-type siblings (Fig 1K and 1N). m863 mutant larvae did not grow much beyond the phenotype observed at 4 dpf (Fig 1G–1J) and die between 5 and 10 dpf (unpublished data). The pleiotropic m863 mutant phenotype with smaller size of tissues forming from rapidly proliferating cells during early larval development and enhanced cell death suggest that the affected gene may be involved in control of cell survival and proliferation.
Fig 1. Phenotype of m863 mutant embryos.

(A-F) Reduction of th-expressing dopaminergic neurons in the pretectum and retina of m863 mutants at 72hpf. (A-C) Lateral (A) and dorsal (B-C) views of th expression pattern in wild type siblings. (D-F) Lateral (D) and dorsal (E-F) views of th expression pattern in homozygous mutant m863 larvae. Arrows indicate affected DA groups in the pretectum (D, F) and retina (E'). (G-J) Morphological phenotype of live m863 mutants at 4 dpf. Dorsal (G) and lateral (H) views of wild type larvae. Dorsal (I) and lateral (J) views of homozygous m863 mutants. Compared to wild type siblings, m863 mutants displayed smaller eyes (blue arrow), flattened and smaller head, edema (red triangle), and defective swim bladder (red arrow), but normal body length. (K, N) Lateral views of live wild type (K) and homozygous m863 mutants (N) at 48 hpf. Granular tissue appearance in the midbrain region and retina of mutants indicated elevated cell death. (L,M,O,P) Ventral views of Alcian blue staining of head cartilage in wild type (L, M) and homozygous m863 mutants (O, P) at 4 dpf. The cartilaginous head skeleton of mutants was smaller and underdeveloped compared to wild type siblings. Abbreviations used: Catecholaminergic groups: AAN, arch-associated neurons (noradrenergic); H, hypothalamus; LC, locus coeruleus; MO, medulla oblongata (noradrenergic); OB, olfactory bulb; PO, preoptic region; Pr, pretectum; PT, posterior tuberculum; SP, subpallium; sym, sympathetic neurons (catecholaminergic); VT, ventral thalamus. Cartilage structures: bb, basibranchial; bh, basihyal; cb, ceratobranchials; ch, ceratohyal; ep, ethmoid plate; m, Meckel’s cartilage; pch, parachordal; pq, palatoquadrate; tr, trabecula. Anterior towards the left. Scale bar: 100 μm
Identification of m863 as mutant allele of the ddb1 gene
The mutant allele m863 was genetically mapped to zebrafish linkage group 18 (LG18) between the SSLP markers z13220 (72.4 cM on MGH panel) and z59637 (74.0 cM). This interval was further narrowed down proximally by generating the polymorphic microsatellite marker 18s56, for which no recombinants were detected in a cross representing 2728 meioses (Fig 2A), revealing that the gene affected by m863 must be tightly linked to this marker. Based on the Ensembl Zv8 assembly, in the critical interval zgc:63840 was identified as a candidate gene for the m863 mutant allele (Fig 2A). Sequence analysis by BLAST (Ensembl, Sanger Center) revealed zgc:63840 as homolog of the DDB1 encoding gene of vertebrates, including mouse, human, rat, bovine, and chicken (S1 Fig), the invertebrate Drosophila, and the plant Arabidopsis thaliana. Hence, in the following, zgc:63840 will be named ddb1 in agreement with the current Zv9 genome annotation (www.zfin.org). The ddb1 cDNA was cloned from in vitro reverse transcribed mRNA of individual 2-day-old-embryos using primer pairs (see S1 Table) designed based on the reference sequence (zgc: 63840, NM_200626.1). The resulting zebrafish ddb1 cDNA (submitted to GenBank: JQ692623.1) contained an open reading frame of 3420 bp encoding a polypeptide of 1140 amino acids, which is around 250 amino acid residues longer than the previously predicted protein product NP_956920.1 (S2 Table). The Ddb1 protein deduced from our cloned zebrafish ddb1 (submitted to GenBank: AFI92852.1) is of the same length as previously documented DDB1 proteins from other vertebrate species, including human, mouse, rat, bovine, and chimpanzee (S1 Fig and S2 Table).
Fig 2. Genetic mapping and identification of the m863 gene locus.

(A) Schematic diagram of zebrafish linkage group (LG) 18 and the m863 genomic region (Ensembl Zv8). Numbers in brackets represent recombination event per number of meiosis analyzed. The m863 mutation was mapped to the genomic interval defined by the microsatellite markers z13220 and z59637. The SSLP marker18s56 generated in this study showed one recombinant, while no recombinants were recovered for the SSLP markers 18s18a and z1417. The gene zgc:63840 mapped to the critical interval of the m863 mutation. (B) Chromatogram of partial cDNA sequences from wild type and homozygous mutant m863 embryos. The insertion of 4 bp occurred in m863 embryos at position 2402–2405 (exon 19) of the ddb1 ORF. (C) Chromatogram of genomic sequences of 3'-end of exon 19 and 5'-splice site from wild type, heterozygous embryos, and homozygous m863 mutants. The mononucleotide substitution (T→C) in homozygous m863 mutants affected the second core base of the splice donor consensus sequence, which is highly conserved in eukaryotic splice donor sites. (D) The frame shift by partial intron insertion introduced a stop codon after a stretch of novel 55 variant amino acids in the C-terminal part of the mutant protein. (E) Schematic diagram of the domain structure of Ddb1 protein based on human DDB1 structure prediction [10]. The partial BPC propeller is truncated and C-terminal helical domain absent in the truncated m863 Ddb1 protein. Arrows represent the point mutation (C), the last wild type amino acid (D), and premature stop (E) in mutant m863.
A multi-sequence alignment of vertebrate DDB1 proteins revealed that DDB1 is highly conserved at the amino acid level. Zebrafish Ddb1 has high identity (about 79% identity at DNA and 90% identity at protein level) to mammals, including human, rat, chimpanzee, bovine, and mouse DDB1. A similar percentage of DDB1 identity was also detected for the non-mammalian vertebrates chicken (81% and 91.1% at DNA and protein level, respectively). The domain analysis of the zebrafish Ddb1 confirmed the known conserved features of DDB1 protein: three β-propeller domains (BPA, BPB and BPC) and a C-terminal helical domain (CTD) (S1 Fig) [5, 8, 10, 36].
To determine whether the m863 mutation indeed affects ddb1, cDNA was prepared from individual wild type and m863 -/- 2 dpf old embryos and sequenced. In m863 -/- mutant embryos, an insertion of 4 bp at the end of exon 19 (2402-2405nt of ddb1 ORF) was detected in the ddb1 transcript (Fig 2B). When genomic ddb1 DNA was sequenced, a mononucleotide substitution (T→C) in the conserved GT splice donor site of ddb1 intron 19 was detected (Fig 2C). This point mutation inactivated the correct splice donor site and caused an adjacent cryptic splice site to become active, leading to partial intron insertion into the transcript. As a result, in the ddb1 ORF after amino acid residue 800 (Glu or E) a frameshift occurred, with a stop codon after a stretch of 55 additional non-Ddb1 amino acids (Fig 2D). The truncated m863 Ddb1 protein lacks the C-terminal part of the wild-type sequence, corresponding to the CTD and most of the BPC propeller domain of Ddb1 protein (Fig 2E), based on comparison to human DDB1 domain structure [5, 36]. These domains have been shown to be involved in the formation and function of the BPA-BPC double-propeller fold, which is critical for DDB1-CUL4 mediated specific protein-protein interactions [30, 37, 38].
To confirm that the phenotype caused by the m863 mutation is in the ddb1 gene, ddb1 morpholinos (MO) were used to phenocopy the m863 mutant phenotype. A splice-site (ddb1_MO1) and a translational start-site targeted morpholino (ddb1_MO2) were used, and their knockdown efficiencies tested (S2 Fig). The splice site MO ddb1_MO1 would only affect zygotic expression of ddb1, while the ATG-morpholino ddb1_MO2 would potentially affect translation of both maternal and zygotic transcripts. To test the efficiency of the ddb1_MO2, it was co-injected with a plasmid expressing the ddb1 5' UTR and ATG with morpholino target site linked to the EGFP ORF, using the CMV promoter. ddb1_MO2 mediated elimination of plasmid derived EGFP in 72 hpf larvae (S2D Fig), revealing that the morpholino effectively blocked Ddb1 translation even on the third day of development. Both ddb1_MO1 and ddb1_MO2 mediated knockdown of Ddb1 effectively induced a phenotype with reduced number of th-expressing DA neurons in the pretectum and retina that was similar to the m863 mutant phenotype (Fig 1A–1F; S3A–S3F Fig; Table 1, and unpublished data). To evaluate whether ddb1_MOs may cause p53-dependent off-target effects [39], we co-injected p53_MO into one-cell stage embryos and analyzed th expression at 3 dpf. ddb1_MO and p53_MO coinjected embryos reduced th signal in pretectum and retina similar to ddb1 morphants (S3D–S3F and S3G–S3I Fig). Thus co-injection of p53_MO did not attenuate the DA neuron phenotype of the morphants (S3D–S3I Fig and Table 1), suggesting that the morphant phenotype was not caused by non-specific morpholino induced p53 dependent cell death [39]. The fact that the ddb1_MO2, while potentially eliminating translation from maternal and zygotic messages, did not induce a phenotype significantly stronger than the zygotic mutant or ddb1_MO1 morphant phenotypes may argue that maternal rescue may not solely derive on maternally deposited ddb1 mRNA (S4 Fig and text below), but also on maternally derived Ddb1 protein deposited in the oocyte. We attempted to generate maternal and zygotic mutant embryos by plasmid transgene rescue of ddb1, but did not succeed (unpublished data—we think that a specific amount of Ddb1 may be needed to sustain proper development). Therefore, we cannot evaluate a potential complete loss of Ddb1 activity, both maternal and zygotic, in zebrafish.
Table 1. Dopaminergic phenotype in ddb1_MO and p53_MO knockdown larvae.
| Morpholinos | Embryos | Phenotypic class | ||||||
|---|---|---|---|---|---|---|---|---|
| ddb1 MOs | p53_MO | Total No. | I | II | IIIa | IIIb | IV | |
| ddb1_MO1 | 4ng | 0ng | 39 | 8 | 7 | 10 | 14 | 0 |
| 4ng | 4ng | 62 | 7 | 13 | 17 | 25 | 0 | |
| 6ng | 0ng | 16 | 1 | 0 | 3 | 12 | 0 | |
| 6ng | 6ng | 51 | 1 | 5 | 25 | 17 | 3 | |
| 8ng | 0ng | 39 | 0 | 1 | 0 | 38 | 0 | |
| 8ng | 7ng | 28 | 0 | 0 | 10 | 18 | 0 | |
| ddb1_MO2 | 6ng | 0ng | 29 | 3 | 12 | 14 | 0 | 0 |
| 6ng | 6ng | 31 | 2 | 14 | 15 | 0 | 0 | |
| 8ng | 0ng | 25 | 0 | 0 | 4 | 19 | 2 | |
| 8ng | 7ng | 38 | 3 | 0 | 11 | 21 | 3 | |
Summary of results from ddb1 morpholino knockdown experiments. Amounts of morpholino injected per embryo are given in ng. Injected embryos were allowed to develop until 72 hpf, fixed, and stained by WISH for th expression. Zebrafish larvae from each knockdown experiment were classified into different phenotypic classes according to the effect of the knockdown on the th expressing pattern and on morphology. I, th expression pattern was similar to wild type siblings; II, th expressing cells were slightly reduced in number; IIIa, th expressing cell number was reduced in the pretectum and retina; IIIb, th-expressing cell number clearly reduced plus decreased size of brain and/or retina; IV, dying and decaying embryos. Based on the changes in th-expression pattern, the class IIIa/b phenotype was similar to that of m863 mutants.
ddb1 is expressed broadly in early development and enhanced in proliferation zones
Expression analysis by whole mount in situ hybridization (WISH) revealed that ddb1 mRNA was maternally deposited in the oocyte and detectable from the one cell stage on (S4A–S4F and S4S Fig). It continued to be broadly expressed zygotically in the embryo, but appeared at elevated levels in proliferation regions in larvae at 72 hpf, including the ciliary marginal zone of the retina and the ventricular surfaces of the brain (Fig 3E–3G and S4J–S4R Fig). We performed Vibratome sections from WISH embryos marked by riboprobes for either ddb1 or proliferating cell nuclear antigen (pcna), which encodes a non-histone nuclear protein frequently used as a cell proliferation marker [40]. The results revealed that ddb1 transcripts were detected in both proliferating and non-proliferating tissues in wild type larvae at 72 hpf (Fig 3A and 3E). Although ubiquitously expressed in the brain, ddb1 transcripts were detected at elevated levels in proliferative regions where pcna was also expressed. Regions with enhanced ddb1 WISH signal in the lateral and basal proliferation zone of the optic tectum (Fig 3A–3C and 3E–3G), hypothalamus (Fig 3A–3C and 3E–3G), retina ciliary marginal zone (CMZ) (Fig 3B, 3C, 3F and 3G) were also marked by the expression of pcna. In non-neural tissues such as the pharyngeal endoderm, jaw, and branchial arches, ddb1 expression was also enhanced in regions of pcna expression (Fig 3A, 3C, 3D and 3G–3H).
Fig 3. Expression of ddb1 in proliferation regions in wild type larvae at 72 hpf.

(A-H) Characterization of pcna (A-D) and ddb1 (E-H) expression in wild type larvae at 72 hpf. Lateral views (A, E) and sections (20 μm; B-D, F-H). (A, E) ddb1 was broadly expressed in the brain while pcna was restricted to proliferation zones. (B-D, F-H) In the medial (m), lateral (l) and basal (b) proliferation zones of the tectum opticum (TeO) and the hypothalamic proliferation zone (H), elevated expression of ddb1 was detected compared to other areas of the brain. ddb1 was also highly expressed in the ciliary marginal zone (CMZ) and in proliferation regions of the pharyngeal endoderm (p) (black arrow head), jaw (j) and branchial arches (ba). The expression of ddb1 was also found in pcna-negative regions such as the ganglion cell layer (GCL) and inner nuclear layer (INL) of the retina. Transcripts of ddb1 were detected throughout the medullar oblongata (MO) where pcna was only expressed in the dorsal part, the rhombic lip (rl). Anterior towards the left. Stars mark the otic vesicle. Scale bar: 100 μm.
We also determined ddb1 expression in ddb1 m863 mutant embryos by WISH and semi-quantitative RT-PCR (S5 Fig). The ddb1 WISH signal was strongly reduced in 24 hpf ddb1 m863 mutant embryos, and essentially absent from 72 hpf mutant larvae (S5A–S5I Fig). As the WISH probe efficiency should not be affected by the mutation only at the intron 19 splice donor site, this finding indicates that the aberrantly spliced ddb1 m863 mutant mRNA may be highly instable. We confirmed this finding by ddb1 RT-PCR on wild type, heterozygous, and homozygous mutant m863 siblings and quantification of the PCR bands (S5J and S5K Fig). If the loss of signal is caused by instability of the mutant transcript, the ddb1 residual signal at 24 hpf may represent the maternal transcript. However, we can also not exclude that Ddb1 activity may be required to maintain ddb1 expression.
Reduction of proliferating cells and enhanced apoptosis in ddb1 m863 mutants
ddb1 m863 zygotic mutant embryos showed an increase of cell death from the second day of development onwards and developed smaller brain and eyes at 72 hpf when compared to wild-type siblings. Given that ddb1 transcription is elevated in brain proliferation zones, we wanted to know whether the reduced size of brain and eyes may be due to activation of programmed cell death throughout the brain, or selectively in the proliferation regions. Therefore, we performed pcna WISH and TUNEL assays in ddb1 m863 mutants at different developmental stages.
The WISH assay revealed that pcna expression was differentially affected in homozygous ddb1 m863 mutants during development. At 30 hpf, no obvious differences of pcna expression were detected between homozygous ddb1 m863 mutants and their wild type siblings (Fig 4A and 4B). At 72 hpf, however, the expression pattern of pcna was selectively affected in various brain proliferation zones of ddb1 m863 mutant embryos when compared to wild type siblings. pcna expression was slightly diminished and appeared in a more diffuse way in the retinal CMZ (Fig 4C and 4D), whereas it was severely reduced in the tectal proliferation zones (tpz) and cerebellum of homozygous ddb1 m863 mutants in comparison to wild type siblings (Fig 4E and 4F). No obvious alterations of pcna expression were found in other proliferation regions of ddb1 m863 mutant brain, including the pallial proliferation region, ventricular zone and hypothalamus (Fig 4C–4F). One possible explanation for the differential effect on neural structures may be that persistent maternally derived functional Ddb1 protein may rescues the zygotic ddb1 mutant phenotype of early proliferating brain regions, while late proliferating regions depend on zygotically expressed ddb1.
Fig 4. Enhanced apoptosis in proliferation regions of the ddb1 m863 mutant CNS.
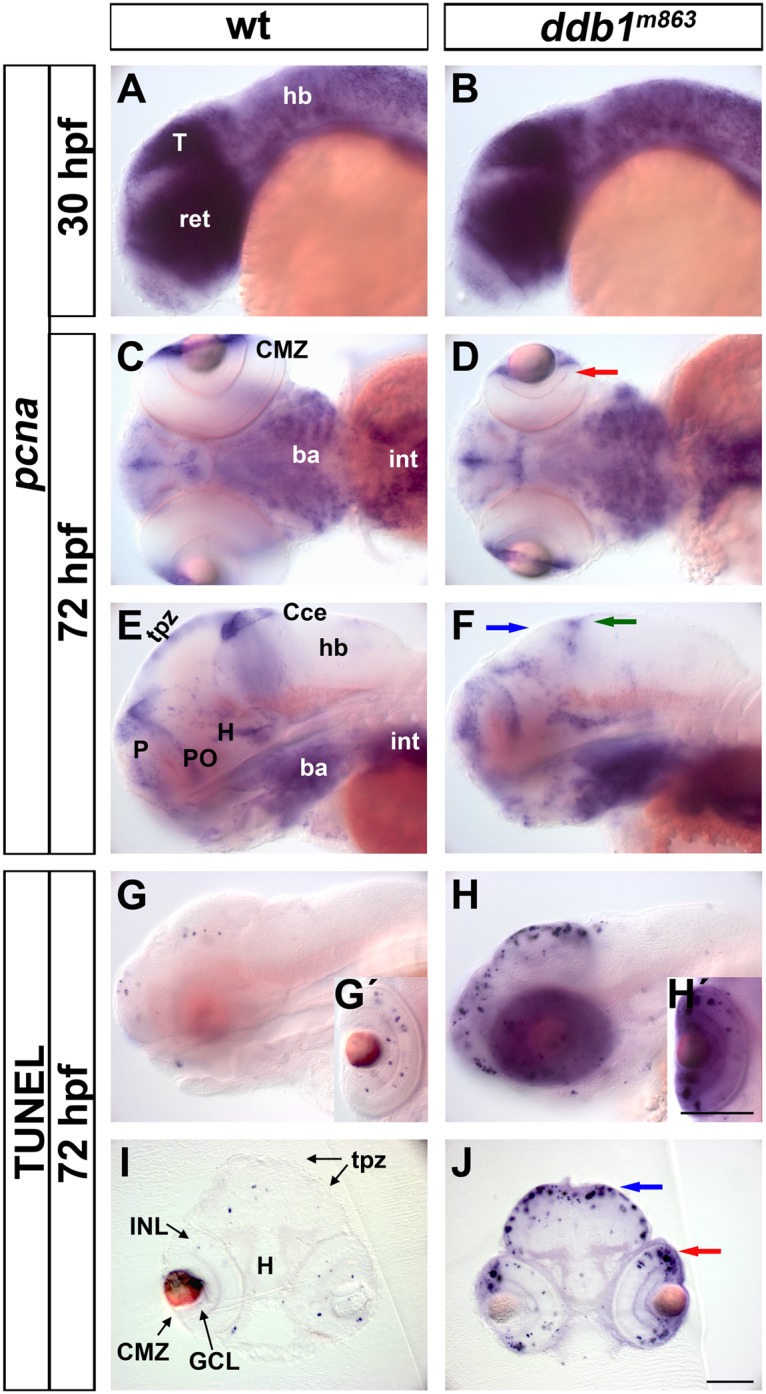
(A-F) The pcna expression pattern in ddb1 m863 mutants and wild type siblings at 30 hpf (A-B) and 72 hpf (C-F). (G-J) TUNEL assay for apoptosis in ddb1 m863 mutants (H-H', J) and wild type siblings (G-G', I) at 72 hpf. Embryos or larvae in lateral (A-B, E-H) and ventral views (C-D). Cross sections (20 μm; I-J) from larvae by TUNEL assay. Arrows represent affected pcna-expressing cells or enhanced apoptotic cells in the retina (D, J; red), pretectal region (F, J; blue), and cerebellum (F; green). Abbreviations used: ba, branchial arches; CMZ, ciliary marginal zone; Cce, cerebellum; GCL, ganglion cell layer; hb, hindbrain; H, hypothalamus; INL, inner nuclear layer; int, intestine; P, pallial proliferation zone; PO, preoptic area; tpz, tectal proliferation zones; Anterior towards the left. Scale bars in H' for G' and H', in J for all of others: 100 μm.
The reduction of pcna expression in the proliferation regions of the tectum, cerebellum, and retina was reminiscent of the apoptotic pattern of ddb1 m863 mutants. TUNEL assay showed that at 72 hpf there were more apoptotic cells in the retinal CMZ, tpz, and cerebellum than in other regions of the brain in homozygous ddb1 m863 mutants compared to wild type siblings (Fig 4G and 4H'). Sections of these TUNEL assay embryos confirmed that the programmed cell death was located prominently in the pcna-expressing tpz and retinal CMZ in ddb1 m863 mutants (Fig 4I and 4J).
Transcriptional activation of p53 in ddb1 m863 mutants
To investigate whether proliferating cells in ddb1 m863 mutant embryos are selectively eliminated by apoptosis, we analyzed the expression of p53 (tp53) [41] in ddb1 m863 mutants during development by WISH. p53 encodes the tumor suppressor protein p53, a transcription factor involved in control of various cellular programs, including cell cycle arrest, apoptosis, DNA repair, and cellular senescence [42].
In 36 hpf wild type embryos, p53 was expressed in the telencephalon, retina, midbrain, cerebellum, hindbrain, otic capsule, branchial arches, pectoral fin bud, and endoderm (Fig 5A and 5B, top). At 48 hpf, the expression level of p53 has declined and was restricted spatially to the proliferation regions, including optic tectum, retina, ventricle zone, branchial arches and other non-brain tissues (Fig 5C and 5D, top). In homozygous ddb1 m863 mutants at 36 hpf, p53 expression levels were strongly increased in the tpz, MHB, cerebellum, and retina, moderately in the ventricular zone, and branchial arches, and slightly in the hindbrain, pectoral fin bud, and endoderm (Fig 5A and 5B, bottom). p53 transcripts were even more severely enhanced in ddb1 m863 mutant embryos at 48 hpf. The increase of p53 transcription in m863 mutants was most prominent throughout the retina, in the dorsal diencephalon, tpz, MHB, cerebellum, pectoral fin bud, and branchial arches, and was moderate in the hindbrain and endoderm when compared to wild type siblings (Fig 5C and 5D). These data suggest that transcription of the tumor suppressor gene p53 was enhanced in ddb1 m863 mutant embryos, with proliferation regions most severely affected.
Fig 5. Increase of p53 transcript levels in ddb1 m863 mutants.

(A-D) Lateral (A-D) and dorsal views (inserts in A, C) of p53 expression pattern in embryos at 36 hpf (wt, n = 30; mut, n = 8) and 48 hpf (wt, n = 25; mut, n = 11). The transcription of p53 was prominently enhanced in ddb1 m863 mutant embryos. Anterior towards the left. Abbreviations used: ba, branchial arches; Cce, cerebellum; end, endoderm, H, hypothalamus, hb, hindbrain; j, jaw; pfb, pectoral fin bud; tel, telencephalon; TeO, tectum opticum. Scale bar: 100 μm
Inactivation of p53 leads to rescue of the ddb1m863 mutant apoptosis phenotype
To determine whether the accumulation of p53 was responsible for the enhanced apoptosis in ddb1 m863 mutants, we inactivated p53 expression by morpholino knockdown in ddb1 m863 mutants. p53 morpholino was microinjected at several concentrations into one cell stage embryos from intercrosses of heterozygous ddb1 m863 parents: group A (non-injected), group B (6 ng p53 MO per embryo) and group C (12 ng p53 MO per embryo). The injected embryos were fixed at 72 hpf, analyzed by TUNEL assay, and genotyped by PCR.
Based on the TUNEL assay results, the phenotypes of the PCR genotyped ddb1 m863 mutant embryos were classified into four classes according to the number of apoptotic cells observed. Based on PCR genotypes, genetically mutant embryos were 20.7% for group A (n = 29), 26.3% for group B (n = 38) and 21.7% for group C (n = 60) (Table 2). At 72 hpf, apoptosis was prominently elevated in the tpz, retinal CMZ and cerebellum in non-injected homozygous ddb1 m863 mutants (class I phenotype) when compared to wild type siblings (group A) (Fig 6A–6F). When p53_MO was injected at an amount of 6 ng per embryo (group B), 30% (n = 10) of mutant embryos showed less apoptotic cells (class II), whereas 70% (n = 10) of them were similar to the mutant phenotype in group A (non-injected) (Fig 6S and Table 2). When 12 ng p53_MO per embryo was injected (group C), the apoptosis in mutant embryos was greatly alleviated compared to group A and group B (Fig 6A–6S and Table 2). In group C, 23.08% (n = 13) of mutant larvae showed reduced numbers of apoptotic cells (class II) (Fig 6J–6L and 6S, Table 2), and 46.15% (n = 13) of the mutants showed strongly reduced numbers of TUNEL stained cells (class III) (Fig 6M–6O and 6S; Table 2). In addition, 30.77% of mutant larvae (class IV, n = 13) in group C showed only very few apoptotic cells and were indistinguishable from wild type siblings (Fig 6G–6I and 6P–6S; Table 2). These data demonstrated that inactivation of p53 rescued apoptosis in ddb1 m863 mutants in a dosage dependent manner, revealing that the apoptotic phenotype of ddb1 m863 mutants depends on p53 activity.
Table 2. Knockdown of p53 by anti-sense morpholino compensates the apoptotic phenotype of ddb1 m863 mutants.
| Morpholino | Embryos Total | Embryos wildtype | No. of embryos per class | |||||
|---|---|---|---|---|---|---|---|---|
| Group | p53_MO | No. | No. | total | class I | class II | class III | class IV |
| A | 0ng | 29 | 23 | 6 | 6 | 0 | 0 | 0 |
| B | 6ng | 38 | 28 | 10 | 7 | 3 | 0 | 0 |
| C | 12ng | 60 | 47 | 13 | 0 | 3 | 6 | 4 |
The ddb1 m863 mutants at one-cell stage were injected with p53 morpholino as indicated (amount of morpholino per embryo provided in ng), and analyzed by TUNEL staining at 72 hpf. Based on the TUNEL staining, genotyped mutant larvae were categorized in the following classes: Class I—severe increase in apoptosis; Class IV weak phenotype with only few apoptotic cells similar to wild type siblings. The apoptosis levels in class II and III were between class I and IV (see Fig 6).
Fig 6. Knock down of p53 rescued the apoptosis phenotype of ddb1 m863 mutants.
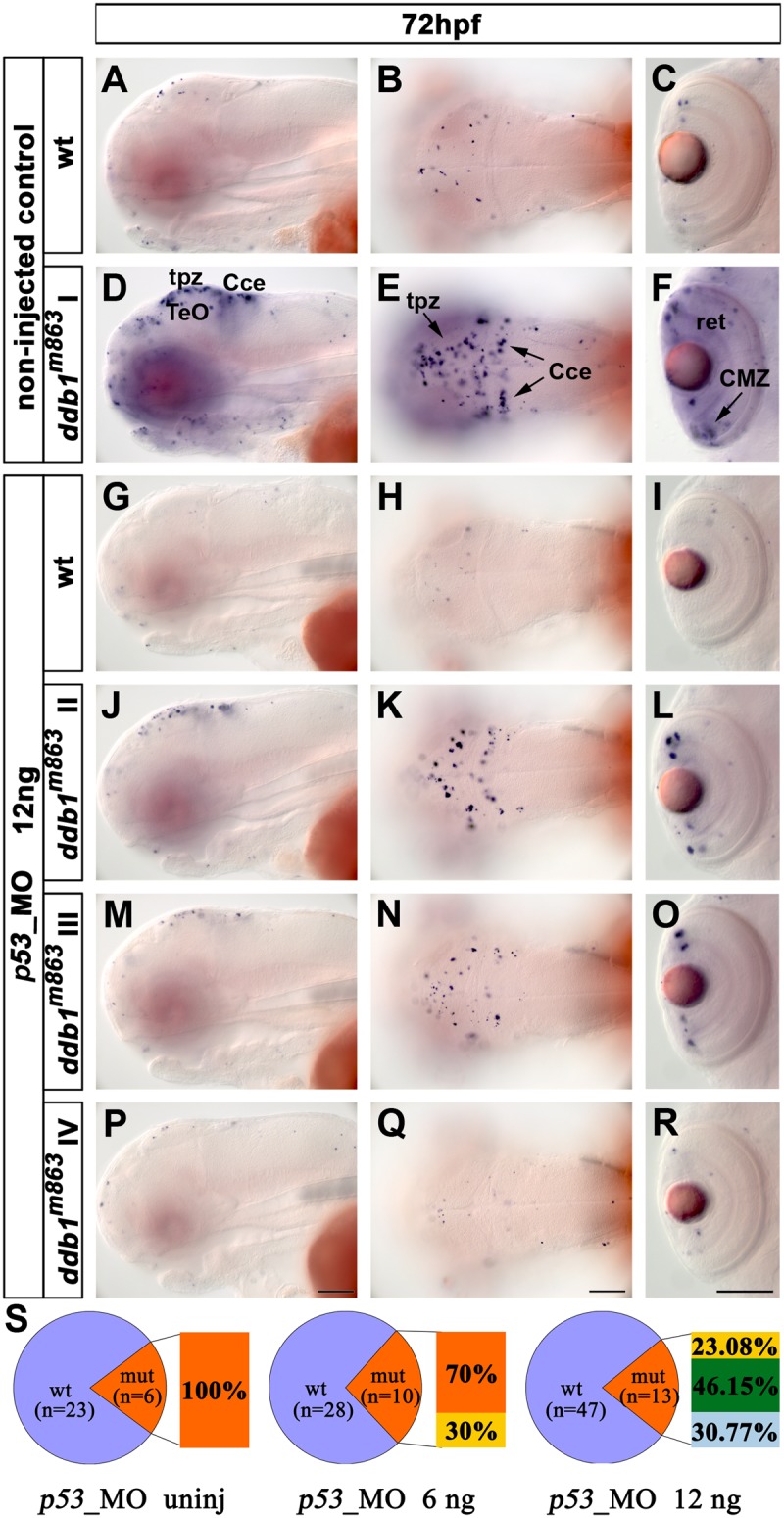
(A-F) Enhanced apoptosis in ddb1 m863 (class I phenotype; D-F) compared to wild type siblings (A-C) without p53_MO injection. (G-R) Partial (class II and class III; J-O) and complete (type IV; P-R) rescue of ddb1 m863 apoptosis phenotype compared to wild siblings (G-I) after injection of 12 ng p53_MO per embryo. (S) Percentage distribution of mutant and rescued mutant phenotypes. Genotypes were determined by PCR. Class I (orange), mutants showed the severest apoptosis phenotype; class IV (light blue) mutants have the weakest phenotype with only a few of apoptotic cells, similar to their wild type siblings; class II (yellow) and III (green) are intermediate phenotypes. Abbreviations used: tpz, tectal proliferation zone; Cce, cerebellum; ret, retina; Anterior towards the left. Scale bar: 100 μm.
Transcriptional deregulation of cell cycle genes in ddb1 m863 mutant embryos
Several cell cycle regulators, including the cyclin-dependent kinase inhibitors p21CIP1/WAF1 (Cdkn1a) and p27KIP1 (Cdkn1b), and the cyclin CcnE, are known to be targeted for degradation by the DDB1-CUL4A ubiquitin ligase [7]. In mice with conditional DDB1 knockout in the CNS, the protein levels of p21CIP1/WAF and p27KIP1 are increased while cyclin E and A are unaltered [25]. However, whether the depletion of ddb1 function may alter the transcription of cell cycle genes is unclear. Therefore, we compared expression of the two paralogues of p21 CIP1/WAF1 (p21 CIP1/WAF1 a and b) and p27 KIP1 (p27 KIP1 a and b), as well as of p57 KIP2 (cdkn1c), ccna2, ccnd1, and ccne2 in ddb1 m863 mutants and wild type siblings by WISH. Mutant and wildtype siblings were stained in one batch for each probe and documented photographically. The genotype of each embryo was determined by PCR using genomic DNA extracted from the fixed and stained material.
The results showed that there were no obvious alterations in WISH signal intensities of p27 KIP1, p57 KIP2 and ccne2 in ddb1 m863 mutants (unpublished data), while ccna2 and ccnd1 WISH signal was significantly reduced (Fig 7). For ccna2, reduced signals were mainly observed in the tectal proliferation zone and CMZ in homozygous ddb1 m863 mutants, whereas WISH signal intensity in hindbrain proliferation zone and outside the brain appeared to be similar to wild type siblings (Fig 7A–7H). Similarly, a reduction in ccnd1 WISH signal was observed in ddb1 m863 mutant embryos in the CMZ and the tectal, cerebellar and ventricular proliferation zones (Fig 7I–7P).
Fig 7. Reduced ccna2 and ccnd1 expression in ddb1 m863 mutant embryos.

(A-D) Expression pattern of ccna2 in wild type siblings. (E-H) Slight reduction of ccna2 expression was detected in the tpz (arrow in E-F) and CMZ (arrow in G) in ddb1 m863 mutants. (I-P) The expression pattern of ccnd1 in wild type and homozygous ddb1 m863 embryos. The transcripts of ccnd1 were slightly decreased in homozygous mutants (M-P). Arrows represent the altered expression of ccnd1 in the tectal proliferation region (M, N), cerebellum (M), and retina (O, P). Abbreviations used: ba, branchial arches; Cce, cerebellum; j, jaw; hb, hindbrain; tel, telencephalon; ret, retina; tpz, tectal proliferation region; VZ, ventricular zone. Anterior towards the left. Scale bars: 100 μm.
In contrast, p21 CIP1/WAF1 WISH signal intensity strongly increased in ddb1 m863 mutants (Fig 8). When analyzing the expression of the two zebrafish paralogues of p21 CIP1/WAF1, we found that the expression pattern of p21a CIP1/WAF1 was broader than that of p21b CIP1/WAF1, which appeared in a more spatial-restricted pattern (Fig 8 and unpublished data). To determine potential effects of Ddb1 on p21 CIP1/WAF1 spatial expression, we focused our analysis on expression of p21b CIP1/WAF1. In 36-hpf wild type embryos, p21b CIP1/WAF1 was highly expressed in several ventral cell groups in the hindbrain and ventral midbrain and in the lens, while weak expression was observed in other brain regions and pectoral fin buds (Fig 8A and 8B). In 36 hpf homozygous ddb1 m863 mutants, however, the expression of p21b CIP1/WAF1 was increased strongly in the dorsal midbrain and slightly in pectoral fin buds, while its WISH signal intensities in other regions were similar to wild type siblings (Fig 8A and 8B). In 48-hpf wild type embryos, p21b CIP1/WAF1 expression in most regions was weaker compared to 36 hpf, and hardly detectable in the forebrain and pectoral fin bud, whereas weak expression persisted in the eyes, ventral midbrain, mid-hindbrain boundary (MHB), hindbrain nuclei, and developing branchial arches (Fig 8C and 8D). In contrast, in 48 hpf ddb1 m863 mutants p21b CIP1/WAF1 expression were strongly increased in the ventricular zone, the tpz, MHB, cerebellum, and retina, as well as elevated moderately in the pectoral fin bud and branchial arches (Fig 8C and 8D).
Fig 8. Increased p21b CIP1/WAF1 expression in homozygous ddb1 m863 mutants.

(A-B) Enhanced expression of p21b CIP1/WAF1 (cdkn1a) in ddb1 m863 homozygous embryos in the proliferation regions of the optic tectum, compared to wild type siblings at 36 hpf. (C-D) At 48 hpf, p21b CIP1/WAF1 expression was strongly elevated in proliferation regions of the optic tectum, cerebellum, telencephalon, retina, branchial arches and pectoral fin bud in ddb1 m863 mutants compared to wild type siblings. In contrast, expression of p21b CIP1/WAF1 in ventral hindbrain nuclei (arrow heads in B, D) was unaltered in ddb1 m863 mutants. All views are lateral except for inserts (dorsal views). Abbreviations used: Cce, cerebellum; H, hypothalamus; L, lens; pfb, pectal fin bud; ret, retina; tel, telencephalon; TeO, tectum opticum. Anterior towards the left. Scale bar: 100 μm.
Discussion
We analyzed the zebrafish homologue of the human DDB1 gene for its activities during development. DDB1 has been demonstrated to contribute to several essential cellular mechanisms, including DNA-damage repair, cell proliferation, survival, and genomic stability [2–5]. DDB1 is also involved in formation of CUL4-RING E3 ligase complexes, which are associated with targeting and proteolysis of specific substrates protein using the ubiquitin-dependent pathway [7–9]. DDB1 is a large multidomain CUL4-adaptor protein containing three β-propellers (BPA, BPB, and BPC) and a CTD domain [36]. The BPB domain mediates predominantly the formation of the DDB1-CUL4 complex, whereas a large clam-shaped double β-propeller pocket BPA-BPC is associated with substrate protein recruitment [8, 10, 25]. Zebrafish Ddb1 shares high sequence identity with its orthologs in mammalian and non-mammalian vertebrates (about 90% protein sequence identity to human, rat, chimpanzee, bovine, mouse, chicken, and Xenopus DDB1), invertebrates (Drosophila 61%) and plant species (Arabidopsis 54%). The high sequence conservation also suggests a strong functional conservation.
Ddb1 in zebrafish development
We identified a new ddb1 m863 mutant allele from a chemical ENU mutagenesis screen, and analyzed its functions in zebrafish early development. The mutation in the ddb1 m863 allele affects proper splicing of exon 19 to exon 20, causing Ddb1 protein to be truncated after amino acid 800, lacking half of the BPC and the whole CTD conserved domains. ddb1 m863 zygotic mutant embryos and early larvae were morphologically similar to wildtype siblings on the first and second day. On the third day, however, when organogenesis and growth from local proliferation zones prevail, a prominent pleiotropic phenotype with reduced size and abnormal structure of the eyes, brain, and head skeleton can be observed. To understand the function of ddb1 in zebrafish, we analyzed its expression pattern throughout embryonic and early larval stages. Both RT-PCR and whole mount in situ hybridization revealed that ddb1 was maternally expressed and mRNA deposited in oocytes. Thus, maternally derived Ddb1 protein may compensate Ddb1 loss in ddb1 m863 mutants during early developmental stages and rescue potential developmental defects through cleavage, gastrulation and somitogenesis stages.
Similar to the expression profiles in mouse [25], ddb1 mRNA was ubiquitously detected in early zebrafish embryos. In embryos at post-somitogenesis stages during the third day of development, however, ddb1 expression became more restricted to the CNS, head skeleton, pharyngeal region, and endoderm. We found that areas with elevated ddb1 transcription levels were correlated with pcna expression and therefore with regions of cell proliferation. When analyzing the larval brain at 3 dpf, this correlation was most pronounced in the proliferation zones in the tectum, the cerebellum, the subventricular zone, and the CMZ of the retina. The zebrafish CNS and retina grow over its lifetime, and new cells are constantly added from proliferation zones [43, 44]. The non-proliferation zone areas of the brain and retina consist largely of differentiated cells, which have lost their proliferating capacity. Hence, we presume that cells with a continued potential to proliferate may rely on high levels of ddb1 expression.
In other animal model systems, including Drosophila melanogaster [32, 33, 45], and mouse [25, 46], deficiency of DDB1 leads to early embryonic lethality, while tissue specific knockouts affect proliferation and development in the studied tissues. In zebrafish, recent studies revealed that the malformation of pectoral fins and otic vesicle after treatment with the drug thalidomide are caused by its binding to Crbn, a substrate receptor of DDB1-CUL4 E3 complexes, and inhibition of CUL4 E3 ubiquitin ligase activity [47]. The resulting aberrant accumulation of DDB1-CUL4 ligase substrates and abnormal proliferation and differentiation by deregulated FGF8, at least partially, have been implied to be responsible for the thalidomide developmental defects. The importance of CUL4 E3 system in fin development is further supported by data from zebrafish cul4a morphants [48]. In contrast to these publications, our data did not reveal morphologic fin defects in ddb1 m863 mutants. However, our data showed that the brain and eyes are affected in ddb1 m863 mutants, which have been reported to develop normally in zcrbn and cul4a morphants [47]. Interestingly, recent data indicate increased apoptosis and decreased PCNA-positive proliferating cells in cul4a morphants [48], which is similar to our observations of ddb1 m863 mutants, albeit studies focused on different tissues. It appears that the DDB1-CUL4 E3 complex has important functions in cellular apoptosis and proliferation. Given the reported differences in PCNA expression in different tissues, additional tissue-specific factors are likely involved. This is in line with DDB1-CUL4 E3 complexes containing basic DDB1-CUL4-ring plus various substrate receptors, and affecting diverse downstream targets. Such tissue selectivity has been reported with fgf8 as target of CUL4-CRBN in fins [47], and tbx5 as target of Cul4a in cardiovascular development [48]. Thus, the discrepancy between phenotypes reported could be caused by combination of DDB1-CUL4-Ring with tissue specific substrate receptors and downstream targets [47]. Given that the ddb1 m863 mutant phenotype is again different from CRBN or CUL4, we speculate that distinct pathways with distinct substrate receptors and targets may be associated with DDB1 function in brain development.
Ddb1 and apoptosis
We observed enhanced apoptosis in zebrafish ddb1 m863 mutants, most pronounced in active proliferation zones within the brain. We also observed a dramatic upregulation of p53 transcript levels in ddb1 m863 mutant zebrafish. p53 encodes the tumor suppressor protein p53 which is activated by myriad stressors such as DNA damage and oxidative stress [49, 50] and its activation can lead to apoptosis [51]. Our data revealed that enhanced expression of p53 predominantly occurred in the proliferation zones of ddb1 m863 mutant embryos. Morpholino-mediated knockdown of p53 in zebrafish ddb1 m863 mutants rescued the apoptosis phenotype, implying that p53 mediates apoptosis in Ddb1-deficient proliferating cells. Our findings that ddb1 m863 mutant larvae displayed an increase in p53-mediated apoptosis in conjunction with the specific reduction of pcna expression in proliferation zones prompt the notion that proliferating cells require ddb1 function. This is in line with the data from conditional deletion of DDB1 in the CNS and retina of mice [25], demonstrating that loss of DDB1 in the mouse brain causes p53-dependent elimination of proliferating cells. PCNA may not only act as a marker for this process, but may be directly involved, as DDB1-CUL4A has been shown to target destruction of Cdt1, a DNA replication licensing factor, only when bound to PCNA [52]. DDB1-CUL4A mediated degradation and cycling of Cdt1 has also been shown to be conserved in zebrafish [53].
The zebrafish ddb1 m863 mutant was initially identified by its phenotype of loss of specific dopaminergic neural groups in early larval zebrafish. Previous studies have shown that oxidative DNA damage accumulation is induced in mutant mice lacking DDB1 function in the brain and retina [25]. Similar mechanisms have been previously associated with neurodegenerative disease including Alzheimer's disease (AD) [54], PD and Amyotrophic Lateral Sclerosis (ALS) [55]. However, other types of DNA damage may occur in our ddb1 mutants. In addition, an impairment of the ubiquitin-proteasome system (UPS), which leads to accumulation and aggregation of abnormal proteins may be a common mechanism in neurodegenerative disorders [56, 57]. In ddb1 m863 mutant embryos, however, we could not detect dopaminergic neurons that would have initially developed and were then lost by apoptosis. Thus, the most likely explanation for the loss of specific late differentiating dopaminergic groups in ddb1 m863 mutant embryos may be that programmed cell death of proliferating cells may have eliminated the neural precursor pools that give rise to these dopaminergic groups.
Zebrafish Ddb1 and cell cycle regulators
The proper progression through each phase of the cell cycle is monitored by specific checkpoints [58, 59]. A large regulatory network controlling synthesis, phosphorylation status, and stability of checkpoint regulators guarantees proper cell cycle progression (Fig 9), and responds to incomplete cell cycle events, damaged DNA, or unfavorable extracellular environment [60]. The DDB1-CUL4 E3 ligase participates in cell cycle control by destruction of members of the cyclin dependent kinase inhibitor (CKI) CIP/KIP family, including p21CIP1/WAF1 and p27KIP1 [14, 16, 19, 61, 62], the licensing factors Cdt1 [63–65], and Set8 [22, 66–68]. Here, we observed that expression of p21a/b CIP1/ WAF1 was dramatically increased in zebrafish ddb1 m863 mutants, whereas p27a/b kIP1 and p57 kIP2 expression was not altered. These results suggest that loss of Ddb1 may also lead to enhanced p21b CIP1/WAF1 transcript levels. Previous studies have already shown that p21CIP1/WAF1 is dramatically accumulated in the epidermis of DDB1-deficient mice [28]. Since DDB1 can transcriptionally regulate UV-induced genes [69, 70] and may function as a transcriptional partner of E2F1 [71], it could be speculated that Ddb1 is not only involved in the post-translational regulation of p21 CIP1/ WAF1 family members, but also in their transcriptional regulation as observed in our zebrafish ddb1 m863 mutants. However, since p21 CIP1/WAF1 is a well-established direct transcriptional target of p53 [51, 72], and p53 transcription was upregulated in ddb1 m863 mutants, it seems more likely that the transcriptional activation of p21a/b CIP1/ WAF1 may be mediated through elevated p53 levels in ddb1 m863 mutants. In addition, the p53 repressed genes CCNA2 [73] and CCND1 [74] were also downregulated in ddb1 m863 mutants. Further, the DDB1-CUL4A complex together with PCNA interact physically with p53 and the inactivation of this complex leads to p53 stabilization and cell cycle arrest in vitro [75]. This supports the idea that the altered transcriptional regulation of cell cycle genes in ddb1 m863 mutant embryos may be mediated by the upregulation of p53.
Fig 9. Model for DDB1 effects on regulators of cell cycle, cell survival and proliferation.

Ddb1, as a component of the CUL4-DDB1 ubiquitin ligase complex, may control, among other targets and potentially in an indirect way, the expression and activity of p53 (tp53). Loss of DDB1 results in enhanced p53 activity. This in turn may cause upregulation of p21 CIP1/WAF1 (cdkn1a), which again contributes to downregulation of ccnd and ccna in proliferating cells. As a result, the cell cycle is deregulated and the equilibrium shifted from proliferation to apoptosis. Whether DDB1 deficiency contributes to genome instability has not been analyzed in our study.
In summary, our studies revealed the requirement of Ddb1 for proper cell cycle progression and survival of proliferating cells in our zebrafish in vivo model. Previous studies showed that DDB1 guards the genomic stability in the cell division cycle by its DNA repair function, and coordinates cell cycle progression through the DDB1-CUL4A complex [76, 77], while loss of DDB1 leads to apoptosis and loss of genome integrity by regulating the activity of downstream targets in mice [25, 28]. Thus our data, together with the findings in mice, corroborate the concept of a ubiquitous requirement for DDB1 to maintain the correct cell division cycle and survival of proliferating cells. In this process, DDB1 may directly and indirectly regulate the proper activity of targets, at both post-transcriptional and transcriptional levels. Our zebrafish data also strengthen the notion of an involvement of Ddb1 in the transcriptional regulation of p53 (Fig 9). Loss of DDB1 in other systems has been shown to lead to p53 protein stabilization [28]. Transcriptional upregulation of p53 may be caused either by inactivation of the DDB1-CUL4A complex or by a stress signal from accumulated DNA damage (genomic instability). DNA damage can activate checkpoint kinases including Ataxia-Telangiectasia mutated (ATM) and Ataxia-Telangi-ectasia (ATR) [78–80], which have highly conserved and interconnected functions [81–83], and may affect p53 [42], we cannot excluded that upregulation of p53 in ddb1 m863 mutants might be caused by interference with these signaling networks. In turn, the p53 activation/upregulation may lead to cell cycle arrest by upregulating CKIs and down regulating cyclins. Furthermore, the accumulation of p53 initiates the apoptosis pathway (Fig 9). The zebrafish ddb1 m863 mutant offers a new in vivo model to study the function of DDB1 and could contribute to unravel mechanisms by which DDB1 malfunction may contribute to neurodegeneration, aging, or carcinogenesis.
Materials and Methods
Fish strains
The zebrafish strains used in this project were the wild type (WT) strains AB, EK, WIK, and IND for mutagenesis and genetic mapping, ABTL and AB for expression analysis and knockdown experiments, and the mutant line carrying the m863 allele. Embryos were obtained by natural spawning and raised at 28.5°C with a 14-hour light/10-hour dark cycle. Developmental stages were determined both by developmental time (hours/days post fertilization, hpf/dpf) and morphological characterization [84]. Melanin pigment formation was inhibited by incubating embryos in egg-water containing 0.2 mM 1-phenyl-2-thiourea (PTU, Sigma) [85]. The use of zebrafish in this research was approved by Regierungspräsidium Freiburg AZ 35–9185.81/G-12/49.
Genomic DNA preparation from zebrafish
Genomic DNA was prepared from individual whole zebrafish embryos or from fin-clips. Briefly, zebrafish embryos or fins biopsies were transferred into 96-well PCR plate containing lysis buffer (10 mM Tris-Cl, pH = 8.0; 50 mM KCl; 0.3% Tween 20; 0.3% NP40; 1 mM EDTA) plus proteinase K (3.4 mg/ml). The plate was sealed with film (Microseal “A” film, Bio-Rad, USA) and samples were digested at 50°C overnight in a humid chamber. Then, Proteinase K was inactivated by heating samples for 10 min at 95°C. The resulting genomic DNA was stored at -80°C and diluted in ddH2O or 0.1xTE at proper ratio for PCR reaction.
Mapping, cloning and genotyping of ddb1 m863
A genetic map cross was carried out between AB/EK strain carriers of the m863 allele with WIK or IND wild type lines (G0) as previous described [86]. Initial mapping was performed using the bulked segregant analysis [87]. Potentially linked markers were confirmed by PCR on single map-cross embryos, and significance of a potential linkage was assumed values of logarithm of odds (LOD) scores higher than 3. High resolution mapping was performed by testing additional SSLP markers to define the critical region. Additional new polymorphic markers within the critical region were generated using the zebrafish SSR online tools (http://danio.mgh.harvard.edu/markers/ssr.html). The candidate gene zgc: 63840 (ddb1) was cloned from in vitro transcribed cDNA of individual two-day-old zebrafish by PCR (primer pairs see S1 Table). The ddb1 m863 embryos were genotyped by genomic PCR using a common reverse primer and mismatch forward primers that hybridize preferentially with mutant or wild type sequences (wild type forward primer, 5'-TGGTGGACCAGCACACGTTTGAGGG-3'; mutant forward primer, 5'-TGGTGGACCAGCACACGTTTGAGCC-3'; reverse primer, 5'-CACCGTCAGTATAGTGGAAGACGATGATGC-3').
In situ hybridization and apoptosis assay
Whole-mount in situ hybridization (WISH) was performed as described [88]. The gene specific probe fragments, including tumor protein p53 (NM_131327.1), cyclin-dependent kinase inhibitor 1A (p21a/b CIP1/WAF1, OTTDART00000030315), cyclin A2 (ccna2, NM_152949.1), cyclin D1 (ccnd1, NM_131025.3), and ddb1 (zgc: 63840), were obtained by PCR (primer pairs see S1 Table) from cDNA of 2 dpf zebrafish wild type embryos, cloned into pCRII-TOPO plasmid, and verified by sequencing. The resulting plasmids pCRII-p21a/b, pCRII-p53, pCRII-ccna2, pCRII-ccnd1, and pCRII-ddb1, as well as pBS-II-ccne2, pBS-II-p57 KIP2 (Zebrafish International Resource Center (ZIRC), Oregon), and plasmids containing pcna [40] and p27a/b KIP1 [89] were used for synthesis of Digoxiginin-labelled riboprobes (DIG RNA-labelling reagents, Roche Biochemicals). Apoptosis of zebrafish embryos was detected by 3´-end labeling of DNA fragments in situ using the ApopTag In Situ Apoptosis Detection Kit (S7100, Chemicon) as previously described [90] Whole mount stained embryos were sectioned using a vibratome as described [90].
Microinjection
To generate synthetic capped Tol2 transposase mRNA, we used the pCS2FA-transposase plasmid [91] as template (mMessage mMachine kit, Ambion). To make ddb1_egfp fusion construct, a ddb1 fragment of 220bp 5´ UTR nucleotides (primer see S1 Table) containing the ddb1_MO2 ATG Morpholino targeting sequence was cloned in frame upstream of the EGFP ORF. The construct was confirmed by sequencing
The Morpholinos ddb1_MO1 (5´---CCACCCTAAAGTGTGCTCACCTGGA---3), ddb1_MO2 (5---CGGTCACCACGTAGTTGTAGGACAT---3), and p53_MO (`)[92] were obtained from GeneTools (Philomath, USA). The mRNAs and DNA injection solutions were prepared in H2O containing 0.05% phenol red. The injection solutions were loaded into glass capillaries using Microloader Pipettes (Eppendorf) just prior to the injection. The injection volume was controlled by measuring the diameter of one drop of injection solution in halocarbon oil (Halocarbon Products Corporation, Series 27) on a micrometer slide. Embryos at one-cell stage were used for injection.
Supporting Information
The DDB1 protein sequences used for alignment are from human (NP_001914.3), Chimpanzee (XP_508472.2), Bovine (NP_001073731.1), mouse (NP_056550.1), rat (NP_741992.1), chicken (NP_989547.1), and zebrafish (AFI92852.1, this work). Three β-propeller domains of BPA, BPB and BPC and a C-terminal domain CTD were marked according to the crystal structure of human DDB1 protein [10]. Homo, Homo sapiens; Pan, Pan troglodytes; Bos, Bos taurus; Mus, Mus musculus; Rattus, Rattus norvegicus; Gallus, Gallus gallus; Danio, Danio rerio.
(TIF)
(A-B) RT-PCR to test the efficiency of ddb1 splice site targeted morpholino (ddb1_MO1). (A) Schematic diagram of ddb1_MO1 targeting the 5th exon-intron junction. (B) Efficiency assay of ddb1_MO1 by RT-PCR using two pairs of ddb1-specific primers. The amount of RNA and cDNA used for RT-PCR was the same for the different samples and for the internal control β-actin. Negative control (con) contained no cDNA. (C-D) Evaluation of the knockdown efficiency of ddb1 translation start site morpholino (ddb1_MO2). (C) Schematic representation of ddb1-egfp fusion construct used for ddb1_MO2 efficiency assay. (D) The expression of EGFP in larvae injected with ddb1-egfp fusion construct together with or without ddb1_MO2. Anterior towards the left. Abbreviations used: E4/E5/E6, 4th /5th /6th exon; I4/I5/I6, 4th / 5th / 6th intron; ddb1_LF/R, the forward and reverse primers of longer ddb1-specific fragment; ddb1_SF/R, forward and reverse primers of shorter ddb1-specific fragment; 1 and 2, different samples; uninj, uninjected embryos.
(TIF)
(A-I) The th expression pattern in wild type control larvae (A-C), in larvae injected with ddb1_MO1 (4 ng/embryo) (D-F), and larvae injected with ddb1_MO1 (4 ng/embryo) and the same amount of p53_MO (G-I) was analyzed by WISH. (A, D, G) lateral views and (B-C, E-F, H-I) dorsal views. Red arrows point at affected th-expressing neurons in the pretectum (D-E, G-H) and retina (F, I). Abbreviations used: AAN, arch-associated neurons; H, hypothalamus; LC, locus coeruleus; MO, medulla oblongata; OB, olfactory bulb; Pr, pretectum; PT, posterior tuberculum; SP, subpallium; sym, sympathetic neurons; VT, ventral thalamus. Anterior towards the left. Scale bar: 100 μm.
(TIF)
(A-I') The WISH signal of ddb1 antisense probe (A-I) and its sense control (A'-I') in embryos before 24 hpf. ddb1 transcript was ubiquitously detected in all blastomeres (A-F') before mid-blastula transition (MBT) when zygotic transcription starts, revealing that ddb1 was expressed maternally. The ubiquitous expression was continued in subsequent stages including sphere, shield and 80% epiboly (G-I, G'-I'). (J-R) The expression of ddb1 at 24 hpf (J-L), 48 hpf (M-O), and 72 hpf (P-R). From 24 hpf onwards, ddb1 mRNA was observed to be expressed broadly and at high levels in the brain and somites (J-L), then spatially restricted to the brain, retina (high in the CMZ but moderate in the GCL and INL, M), the branchial arches, and endoderm at 48 hpf (N-O), followed at 72 hpf by downregulation and more distinct spatial expression pattern (P-R). ddb1 was detected at moderate levels in the telencephalic proliferation region, tectal proliferation region, cerebellum, CMZ, and branchial arches, whereas in other regions the signal was weak (P-Q). (S) ddb1 was maternally expressed as revealed by RT-PCR from two separate cDNA templates prepared from one cell stage zygotes (lanes 1 and 2; lane 3—control with water only as template) using a ddb1 specific primer pair.
(TIF)
(A-I) WISH analysis of ddb1 transcripts revealed reduced expression in heterozygous (B, E, H) and near-absent expression in homozygous ddb1 m863 mutants (C, F, I) compared to wild type siblings (A, D, G) at 24 hpf (A-C) and 72 hpf (D-I). Planes focus on the retina of heterozygous embryos (B') and homozygous mutants (C'). Lateral views (A-C; G-I) and dorsal views (D-F). Abbreviations used: Cce, cerebellum; dc, diencephalon; hb, hindbrain; j, jaw; I, lens; li, liver; mb, midbrain; ret, retina; sc, spinal cord; T, tectum; tc, telencephalon. Anterior is towards the left. Scale bars in C' for B'-C', in I for others: 100 μm. (J) RT-PCR analysis of ddb1 transcript levels during development stages from 24 hpf to 96 hpf in wild type, heterozygous and homozygous m863 mutants. β-actin was used as an internal control, and the negative control was template-free. (K) The RT-PCR gel was quantified densitometrically and traces of ddb1 PCR signal and of internal β-actin control were shown together with the wild type, heterozygous and homozygous m863 mutant samples adjacent to each other within each frame showing one developmental stage. Transcription of ddb1 was progressively downregulated in wild type embryos along the development. Compared to wild type siblings, the transcription level of ddb1 was slightly decreased in heterozygous embryos/larvae, but strongly reduced in homozygous ddb1 m863 mutants.
(TIF)
(PDF)
(PDF)
Acknowledgments
We are grateful to H. Löhr for advice on mapping, to A. Filippi and D. Onichtchouk for advice and technical support, to S. Götter for expert zebrafish care.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by a fellowship of the LGFG (State of Baden-Württemberg; www.mwk.baden-wuerttemberg.de) (ZH), by the EC programs EU-FP6 IP ZF-MODELS and EU-FP7 IP ZF-HEALTH (www.http://ec.europa.eu/research/), and the Excellence Initiative of the German Federal and State Governments (DFG BIOSS - EXC 294; www.dfg.de). The article processing charge was funded by the German Research Foundation (DFG) and the Albert Ludwigs University Freiburg in the funding programme Open Access Publishing. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, et al. (2006) DNA repair: from molecular mechanism to human disease. DNA Repair (Amst) 5: 986–96. . [DOI] [PubMed] [Google Scholar]
- 2. Cleaver JE. (2005) Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 5: 564–73. 10.1038/nrc1652 . [DOI] [PubMed] [Google Scholar]
- 3. Chu G, Chang E. (1988) Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242: 564–7. . [DOI] [PubMed] [Google Scholar]
- 4. Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S. (1995) Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics 29: 62–9. 10.1006/geno.1995.1215 . [DOI] [PubMed] [Google Scholar]
- 5. Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, et al. (2006) DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res 66: 8590–7. 10.1158/0008-5472.CAN-06-1115 . [DOI] [PubMed] [Google Scholar]
- 6. Subba Rao K. (2007) Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol 3: 162–72. 10.1038/ncpneuro0448 . [DOI] [PubMed] [Google Scholar]
- 7. Jackson S, Xiong Y. (2009) CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends in biochemical sciences 34: 562–70. 10.1016/j.tibs.2009.07.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443: 590–3. 10.1038/nature05175 . [DOI] [PubMed] [Google Scholar]
- 9. He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. (2006) DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev 20: 2949–54. 10.1101/gad.1483206 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, et al. (2008) Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 135: 1213–23. 10.1016/j.cell.2008.10.045 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bondar T, Ponomarev A, Raychaudhuri P. (2004) Ddb1 is required for the proteolysis of the Schizosaccharomyces pombe replication inhibitor Spd1 during S phase and after DNA damage. J Biol Chem 279: 9937–43. 10.1074/jbc.M312570200 . [DOI] [PubMed] [Google Scholar]
- 12. Fukumoto Y, Dohmae N, Hanaoka F. (2008) Schizosaccharomyces pombe Ddb1 recruits substrate-specific adaptor proteins through a novel protein motif, the DDB-box. Mol Cell Biol 28: 6746–56. 10.1128/MCB.00757-08 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. (2009) Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol Cell 36: 642–53. 10.1016/j.molcel.2009.09.035 . [DOI] [PubMed] [Google Scholar]
- 14. Bondar T, Kalinina A, Khair L, Kopanja D, Nag A, Bagchi S, et al. (2006) Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol Cell Biol 26: 2531–9. 10.1128/MCB.26.7.2531-2539.2006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hotton SK, Callis J. (2008) Regulation of cullin RING ligases. Annu Rev Plant Biol 59: 467–89. 10.1146/annurev.arplant.58.032806.104011 . [DOI] [PubMed] [Google Scholar]
- 16. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 22: 2496–506. 10.1101/gad.1676108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim Y, Starostina NG, Kipreos ET. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev 22: 2507–19. 10.1101/gad.1703708 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soria G, Gottifredi V. (2010) PCNA-coupled p21 degradation after DNA damage: The exception that confirms the rule? DNA Repair (Amst) 9: 358–64. 10.1016/j.dnarep.2009.12.003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. (2008) CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem 283: 29045–52. 10.1074/jbc.M806045200 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim SH, Michael WM. (2008) Regulated proteolysis of DNA polymerase eta during the DNA-damage response in C. elegans. Mol Cell 32: 757–66. 10.1016/j.molcel.2008.11.016 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shibutani ST, de la Cruz AF, Tran V, Turbyfill WJ 3rd, Reis T, Edgar BA, et al. (2008) Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev Cell 15: 890–900. 10.1016/j.devcel.2008.10.003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. (2010) CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 40: 9–21. 10.1016/j.molcel.2010.09.014 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vejrup-Hansen R, Fleck O, Landvad K, Fahnoe U, Broendum SS, Schreurs AS, et al. (2014) Spd2 assists Spd1 in the modulation of ribonucleotide reductase architecture but does not regulate deoxynucleotide pools. Journal of cell science 127: 2460–70. 10.1242/jcs.139816 . [DOI] [PubMed] [Google Scholar]
- 24. Lee J, Zhou P. (2007) DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell 26: 775–80. 10.1016/j.molcel.2007.06.001 . [DOI] [PubMed] [Google Scholar]
- 25. Cang Y, Zhang J, Nicholas SA, Bastien J, Li B, Zhou P, et al. (2006) Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 127: 929–40. 10.1016/j.cell.2006.09.045 . [DOI] [PubMed] [Google Scholar]
- 26. Holmberg C, Fleck O, Hansen HA, Liu C, Slaaby R, Carr AM, et al. (2005) Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev 19: 853–62. 10.1101/gad.329905 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zolezzi F, Fuss J, Uzawa S, Linn S. (2002) Characterization of a Schizosaccharomyces pombe strain deleted for a sequence homologue of the human damaged DNA binding 1 (DDB1) gene. J Biol Chem 277: 41183–91. 10.1074/jbc.M207890200 . [DOI] [PubMed] [Google Scholar]
- 28. Cang Y, Zhang J, Nicholas SA, Kim AL, Zhou P, Goff SP. (2007) DDB1 is essential for genomic stability in developing epidermis. Proc Natl Acad Sci U S A 104: 2733–7. 10.1073/pnas.0611311104 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernhardt A, Lechner E, Hano P, Schade V, Dieterle M, Anders M, et al. (2006) CUL4 associates with DDB1 and DET1 and its downregulation affects diverse aspects of development in Arabidopsis thaliana. Plant J 47: 591–603. 10.1111/j.1365-313X.2006.02810.x . [DOI] [PubMed] [Google Scholar]
- 30. Bernhardt A, Mooney S, Hellmann H. (2010) Arabidopsis DDB1a and DDB1b are critical for embryo development. Planta 232: 555–66. 10.1007/s00425-010-1195-9 . [DOI] [PubMed] [Google Scholar]
- 31. Shimanouchi K, Takata K, Yamaguchi M, Murakami S, Ishikawa G, Takeuchi R, et al. (2006) Drosophila damaged DNA binding protein 1 contributes to genome stability in somatic cells. J Biochem 139: 51–8. 10.1093/jb/mvj006 . [DOI] [PubMed] [Google Scholar]
- 32. Takata K, Shimanouchi K, Yamaguchi M, Murakami S, Ishikawa G, Takeuchi R, et al. (2004) Damaged DNA binding protein 1 in Drosophila defense reactions. Biochem Biophys Res Commun 323: 1024–31. 10.1016/j.bbrc.2004.08.182 . [DOI] [PubMed] [Google Scholar]
- 33. Takata K, Yoshida H, Yamaguchi M, Sakaguchi K. (2004) Drosophila damaged DNA-binding protein 1 is an essential factor for development. Genetics 168: 855–65. 10.1534/genetics.103.025965 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wakasugi M, Matsuura K, Nagasawa A, Fu D, Shimizu H, Yamamoto K, et al. (2007) DDB1 gene disruption causes a severe growth defect and apoptosis in chicken DT40 cells. Biochem Biophys Res Commun 364: 771–7. 10.1016/j.bbrc.2007.10.063 . [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y, Feng S, Chen F, Chen H, Wang J, McCall C, et al. (2008) Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell 20: 1437–55. 10.1105/tpc.108.058891 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li T, Chen X, Garbutt KC, Zhou P, Zheng N. (2006) Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell 124: 105–17. 10.1016/j.cell.2005.10.033 . [DOI] [PubMed] [Google Scholar]
- 37. Zimmerman ES, Schulman BA, Zheng N. (2010) Structural assembly of cullin-RING ubiquitin ligase complexes. Current opinion in structural biology 20: 714–21. 10.1016/j.sbi.2010.08.010 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lv XB, Xie F, Hu K, Wu Y, Cao LL, Han X, et al. (2010) Damaged DNA-binding protein 1 (DDB1) interacts with Cdh1 and modulates the function of APC/CCdh1. The Journal of biological chemistry 285: 18234–40. 10.1074/jbc.M109.094144 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, et al. (2007) p53 activation by knockdown technologies. PLoS genetics 3: e78 10.1371/journal.pgen.0030078 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee KS, Yang WI. (1992) Comparison of brain tumor growth kinetics by proliferating cell nuclear antigen (PCNA) and bromodeoxyuridine (BrdU) labeling. Yonsei Med J 33: 265–71. . [DOI] [PubMed] [Google Scholar]
- 41. Cheng R, Ford BL, O'Neal PE, Mathews CZ, Bradford CS, Thongtan T, et al. (1997) Zebrafish (Danio rerio) p53 tumor suppressor gene: cDNA sequence and expression during embryogenesis. Mol Mar Biol Biotechnol 6: 88–97. . [PubMed] [Google Scholar]
- 42. Harris SL, Levine AJ. (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24: 2899–908. 10.1038/sj.onc.1208615 . [DOI] [PubMed] [Google Scholar]
- 43. Cid E, Velasco A, Ciudad J, Orfao A, Aijon J, Lara JM. (2002) Quantitative evaluation of the distribution of proliferating cells in the adult retina in three cyprinid species. Cell and tissue research 308: 47–59. 10.1007/s00441-002-0529-8 . [DOI] [PubMed] [Google Scholar]
- 44. Grandel H, Kaslin J, Ganz J, Wenzel I, Brand M. (2006) Neural stem cells and neurogenesis in the adult zebrafish brain: origin, proliferation dynamics, migration and cell fate. Dev Biol 295: 263–77. 10.1016/j.ydbio.2006.03.040 . [DOI] [PubMed] [Google Scholar]
- 45. Takata K, Ishikawa G, Hirose F, Sakaguchi K. (2002) Drosophila damage-specific DNA-binding protein 1 (D-DDB1) is controlled by the DRE/DREF system. Nucleic Acids Res 30: 3795–808. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamaji S, Zhang M, Zhang J, Endo Y, Bibikova E, Goff SP, et al. (2010) Hepatocyte-specific deletion of DDB1 induces liver regeneration and tumorigenesis. Proc Natl Acad Sci U S A 107: 22237–42. 10.1073/pnas.1015793108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. (2010) Identification of a primary target of thalidomide teratogenicity. Science 327: 1345–50. 10.1126/science.1177319 . [DOI] [PubMed] [Google Scholar]
- 48. Zhao X, Jiang B, Hu H, Mao F, Mi J, Li Z, et al. (2015) Zebrafish cul4a, but not cul4b, modulates cardiac and forelimb development by upregulating tbx5a expression. Human molecular genetics 24: 853–64. 10.1093/hmg/ddu503 . [DOI] [PubMed] [Google Scholar]
- 49. Reinhardt HC, Schumacher B. (2012) The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends in genetics: TIG 28: 128–36. 10.1016/j.tig.2011.12.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vousden KH, Prives C. (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137: 413–31. 10.1016/j.cell.2009.04.037 . [DOI] [PubMed] [Google Scholar]
- 51. Vogelstein B, Lane D, Levine AJ. (2000) Surfing the p53 network. Nature 408: 307–10. 10.1038/35042675 . [DOI] [PubMed] [Google Scholar]
- 52. Arias EE, Walter JC. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nature cell biology 8: 84–90. 10.1038/ncb1346 . [DOI] [PubMed] [Google Scholar]
- 53. Sugiyama M, Sakaue-Sawano A, Iimura T, Fukami K, Kitaguchi T, Kawakami K, et al. (2009) Illuminating cell-cycle progression in the developing zebrafish embryo. P Natl Acad Sci USA 106: 20812–7. 10.1073/pnas.0906464106 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Butterfield DA, Drake J, Pocernich C, Castegna A. (2001) Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med 7: 548–54. S1471-4914(01)02173-6 [pii]. . [DOI] [PubMed] [Google Scholar]
- 55. Simonian NA, Coyle JT. (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36: 83–106. 10.1146/annurev.pa.36.040196.000503 . [DOI] [PubMed] [Google Scholar]
- 56. Hyun DH. (2010) Effect of the overexpression of mutant ubiquitin (K48R) on the cellular response induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation. Neurosci Lett 477: 115–20. 10.1016/j.neulet.2010.04.045 . [DOI] [PubMed] [Google Scholar]
- 57. Rogers N, Paine S, Bedford L, Layfield R. (2010) Review: the ubiquitin-proteasome system: contributions to cell death or survival in neurodegeneration. Neuropathol Appl Neurobiol 36: 113–24. 10.1111/j.1365-2990.2010.01063.x . [DOI] [PubMed] [Google Scholar]
- 58. Harashima H, Dissmeyer N, Schnittger A. (2013) Cell cycle control across the eukaryotic kingdom. Trends in cell biology 23: 345–56. 10.1016/j.tcb.2013.03.002 . [DOI] [PubMed] [Google Scholar]
- 59. Bertoli C, Skotheim JM, de BRA. (2013) Control of cell cycle transcription during G1 and S phases. Nature reviews Molecular cell biology 14: 518–28. 10.1038/nrm3629 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Elledge SJ. (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274: 1664–72. . [DOI] [PubMed] [Google Scholar]
- 61. Iovine B, Iannella ML, Bevilacqua MA. (2011) Damage-specific DNA binding protein 1 (DDB1) is involved in ubiquitin-mediated proteolysis of p27Kip1 in response to UV irradiation. Biochimie 93: 867–75. 10.1016/j.biochi.2010.12.017 . [DOI] [PubMed] [Google Scholar]
- 62. Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, et al. (2003) Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A 100: 10231–6. 10.1073/pnas.1831009100 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. (2003) Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol 5: 1008–15. 10.1038/ncb1061 . [DOI] [PubMed] [Google Scholar]
- 64. Hu J, McCall CM, Ohta T, Xiong Y. (2004) Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 6: 1003–9. 10.1038/ncb1172 . [DOI] [PubMed] [Google Scholar]
- 65. Zhong W, Feng H, Santiago FE, Kipreos ET. (2003) CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 423: 885–9. 10.1038/nature01747 . [DOI] [PubMed] [Google Scholar]
- 66. Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, et al. (2010) CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell 40: 22–33. 10.1016/j.molcel.2010.09.015 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, et al. (2010) Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 40: 364–76. 10.1016/j.molcel.2010.10.011 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, et al. (2010) The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12: 1086–93. 10.1038/ncb2113 . [DOI] [PubMed] [Google Scholar]
- 69. Bevilacqua MA, Iovine B, Zambrano N, D'Ambrosio C, Scaloni A, Russo T, et al. (2005) Fibromodulin gene transcription is induced by ultraviolet irradiation, and its regulation is impaired in senescent human fibroblasts. J Biol Chem 280: 31809–17. 10.1074/jbc.M414677200 . [DOI] [PubMed] [Google Scholar]
- 70. Iovine B, Nino M, Irace C, Bevilacqua MA, Monfrecola G. (2009) Ultraviolet B and A irradiation induces fibromodulin expression in human fibroblasts in vitro. Biochimie 91: 364–72. 10.1016/j.biochi.2008.10.017 . [DOI] [PubMed] [Google Scholar]
- 71. Nichols AF, Itoh T, Zolezzi F, Hutsell S, Linn S. (2003) Basal transcriptional regulation of human damage-specific DNA-binding protein genes DDB1 and DDB2 by Sp1, E2F, N-myc and NF1 elements. Nucleic Acids Res 31: 562–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jin S, Levine AJ. (2001) The p53 functional circuit. Journal of cell science 114: 4139–40. . [DOI] [PubMed] [Google Scholar]
- 73. Desdouets C, Ory C, Matesic G, Soussi T, Brechot C, Sobczak-Thepot J. (1996) ATF/CREB site mediated transcriptional activation and p53 dependent repression of the cyclin A promoter. FEBS Lett 385: 34–8. 0014-5793(96)00330-4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 74. Guo Z, Wang J, Yang J, Wu NH, Zhang Y, Shen YF. (2009) An inhibitory role of p53 via NF-kappaB element on the cyclin D1 gene under heat shock. Biochim Biophys Acta 1789: 758–62. 10.1016/j.bbagrm.2009.09.011 . [DOI] [PubMed] [Google Scholar]
- 75. Banks D, Wu M, Higa LA, Gavrilova N, Quan J, Ye T, et al. (2006) L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle 5: 1719–29. . [DOI] [PubMed] [Google Scholar]
- 76. Abbas T, Dutta A. (2011) CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 10: 241–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Iovine B, Iannella ML, Bevilacqua MA. (2011) Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions. Int J Biochem Cell Biol 43: 1664–7. 10.1016/j.biocel.2011.09.001 . [DOI] [PubMed] [Google Scholar]
- 78. Shiloh Y. (2003) ATM and related protein kinases: safeguarding genome integrity. Nature reviews Cancer 3: 155–68. 10.1038/nrc1011 . [DOI] [PubMed] [Google Scholar]
- 79. Smith J, Tho LM, Xu N, Gillespie DA. (2010) The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in cancer research 108: 73–112. 10.1016/B978-0-12-380888-2.00003-0 . [DOI] [PubMed] [Google Scholar]
- 80. Zhou BB, Elledge SJ. (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–9. 10.1038/35044005 . [DOI] [PubMed] [Google Scholar]
- 81. Bartek J, Bartkova J, Lukas J. (2007) DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 26: 7773–9. 10.1038/sj.onc.1210881 . [DOI] [PubMed] [Google Scholar]
- 82. Bartek J, Lukas J. (2007) DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 19: 238–45. 10.1016/j.ceb.2007.02.009 . [DOI] [PubMed] [Google Scholar]
- 83. Hurley PJ, Bunz F. (2007) ATM and ATR: components of an integrated circuit. Cell cycle 6: 414–7. . [DOI] [PubMed] [Google Scholar]
- 84. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. 10.1002/aja.1002030302 . [DOI] [PubMed] [Google Scholar]
- 85. Westerfield M. THE ZEBRAFISH BOOK. Eugene, USA: University of Oregon Press; 1995. [Google Scholar]
- 86. Knapik EW, Goodman A, Ekker M, Chevrette M, Delgado J, Neuhauss S, et al. (1998) A microsatellite genetic linkage map for zebrafish (Danio rerio). Nature genetics 18: 338–43. 10.1038/ng0498-338 . [DOI] [PubMed] [Google Scholar]
- 87. Michelmore RW, Paran I, Kesseli RV. (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. P Natl Acad Sci USA 88: 9828–32. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hauptmann G, Gerster T. (2000) Multicolor whole-mount in situ hybridization. Methods in molecular biology 137: 139–48. 10.1385/1-59259-066-7:139 . [DOI] [PubMed] [Google Scholar]
- 89. Masai I, Yamaguchi M, Tonou-Fujimori N, Komori A, Okamoto H. (2005) The hedgehog-PKA pathway regulates two distinct steps of the differentiation of retinal ganglion cells: the cell-cycle exit of retinoblasts and their neuronal maturation. Development 132: 1539–53. 10.1242/dev.01714 . [DOI] [PubMed] [Google Scholar]
- 90. Ryu S, Holzschuh J, Erhardt S, Ettl AK, Driever W. (2005) Depletion of minichromosome maintenance protein 5 in the zebrafish retina causes cell-cycle defect and apoptosis. P Natl Acad Sci USA 102: 18467–72. 10.1073/pnas.0506187102 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, et al. (2007) The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236: 3088–99. 10.1002/dvdy.21343 . [DOI] [PubMed] [Google Scholar]
- 92. Langheinrich U, Hennen E, Stott G, Vacun G. (2002) Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Current biology: CB 12: 2023–8. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The DDB1 protein sequences used for alignment are from human (NP_001914.3), Chimpanzee (XP_508472.2), Bovine (NP_001073731.1), mouse (NP_056550.1), rat (NP_741992.1), chicken (NP_989547.1), and zebrafish (AFI92852.1, this work). Three β-propeller domains of BPA, BPB and BPC and a C-terminal domain CTD were marked according to the crystal structure of human DDB1 protein [10]. Homo, Homo sapiens; Pan, Pan troglodytes; Bos, Bos taurus; Mus, Mus musculus; Rattus, Rattus norvegicus; Gallus, Gallus gallus; Danio, Danio rerio.
(TIF)
(A-B) RT-PCR to test the efficiency of ddb1 splice site targeted morpholino (ddb1_MO1). (A) Schematic diagram of ddb1_MO1 targeting the 5th exon-intron junction. (B) Efficiency assay of ddb1_MO1 by RT-PCR using two pairs of ddb1-specific primers. The amount of RNA and cDNA used for RT-PCR was the same for the different samples and for the internal control β-actin. Negative control (con) contained no cDNA. (C-D) Evaluation of the knockdown efficiency of ddb1 translation start site morpholino (ddb1_MO2). (C) Schematic representation of ddb1-egfp fusion construct used for ddb1_MO2 efficiency assay. (D) The expression of EGFP in larvae injected with ddb1-egfp fusion construct together with or without ddb1_MO2. Anterior towards the left. Abbreviations used: E4/E5/E6, 4th /5th /6th exon; I4/I5/I6, 4th / 5th / 6th intron; ddb1_LF/R, the forward and reverse primers of longer ddb1-specific fragment; ddb1_SF/R, forward and reverse primers of shorter ddb1-specific fragment; 1 and 2, different samples; uninj, uninjected embryos.
(TIF)
(A-I) The th expression pattern in wild type control larvae (A-C), in larvae injected with ddb1_MO1 (4 ng/embryo) (D-F), and larvae injected with ddb1_MO1 (4 ng/embryo) and the same amount of p53_MO (G-I) was analyzed by WISH. (A, D, G) lateral views and (B-C, E-F, H-I) dorsal views. Red arrows point at affected th-expressing neurons in the pretectum (D-E, G-H) and retina (F, I). Abbreviations used: AAN, arch-associated neurons; H, hypothalamus; LC, locus coeruleus; MO, medulla oblongata; OB, olfactory bulb; Pr, pretectum; PT, posterior tuberculum; SP, subpallium; sym, sympathetic neurons; VT, ventral thalamus. Anterior towards the left. Scale bar: 100 μm.
(TIF)
(A-I') The WISH signal of ddb1 antisense probe (A-I) and its sense control (A'-I') in embryos before 24 hpf. ddb1 transcript was ubiquitously detected in all blastomeres (A-F') before mid-blastula transition (MBT) when zygotic transcription starts, revealing that ddb1 was expressed maternally. The ubiquitous expression was continued in subsequent stages including sphere, shield and 80% epiboly (G-I, G'-I'). (J-R) The expression of ddb1 at 24 hpf (J-L), 48 hpf (M-O), and 72 hpf (P-R). From 24 hpf onwards, ddb1 mRNA was observed to be expressed broadly and at high levels in the brain and somites (J-L), then spatially restricted to the brain, retina (high in the CMZ but moderate in the GCL and INL, M), the branchial arches, and endoderm at 48 hpf (N-O), followed at 72 hpf by downregulation and more distinct spatial expression pattern (P-R). ddb1 was detected at moderate levels in the telencephalic proliferation region, tectal proliferation region, cerebellum, CMZ, and branchial arches, whereas in other regions the signal was weak (P-Q). (S) ddb1 was maternally expressed as revealed by RT-PCR from two separate cDNA templates prepared from one cell stage zygotes (lanes 1 and 2; lane 3—control with water only as template) using a ddb1 specific primer pair.
(TIF)
(A-I) WISH analysis of ddb1 transcripts revealed reduced expression in heterozygous (B, E, H) and near-absent expression in homozygous ddb1 m863 mutants (C, F, I) compared to wild type siblings (A, D, G) at 24 hpf (A-C) and 72 hpf (D-I). Planes focus on the retina of heterozygous embryos (B') and homozygous mutants (C'). Lateral views (A-C; G-I) and dorsal views (D-F). Abbreviations used: Cce, cerebellum; dc, diencephalon; hb, hindbrain; j, jaw; I, lens; li, liver; mb, midbrain; ret, retina; sc, spinal cord; T, tectum; tc, telencephalon. Anterior is towards the left. Scale bars in C' for B'-C', in I for others: 100 μm. (J) RT-PCR analysis of ddb1 transcript levels during development stages from 24 hpf to 96 hpf in wild type, heterozygous and homozygous m863 mutants. β-actin was used as an internal control, and the negative control was template-free. (K) The RT-PCR gel was quantified densitometrically and traces of ddb1 PCR signal and of internal β-actin control were shown together with the wild type, heterozygous and homozygous m863 mutant samples adjacent to each other within each frame showing one developmental stage. Transcription of ddb1 was progressively downregulated in wild type embryos along the development. Compared to wild type siblings, the transcription level of ddb1 was slightly decreased in heterozygous embryos/larvae, but strongly reduced in homozygous ddb1 m863 mutants.
(TIF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.