

Abstract
It has been widely accepted that tumor cells and normal stromal cells in the host environment coordinately modulate tumor progression. Mitogen-activated protein kinase pathways are the representative stress-responsive cascades that exert proper cellular responses to divergent environmental stimuli. Genetically engineered mouse models and chemically induced tumorigenesis models have revealed that components of the MAPK pathway not only regulate the behavior of tumor cells themselves but also that of surrounding normal stromal cells in the host environment during cancer pathogenesis. The individual functions of MAPK pathway components in tumor initiation and progression vary depending on the stimuli and the stromal cell types involved in tumor progression, in addition to the molecular isoforms of the components and the origins of the tumor. Recent studies have indicated that MAPK pathway components synergize with environmental factors (e.g. tobacco smoke and diet) to affect tumor initiation and progression. Moreover, some components play distinct roles in the course of tumor progression, such as before and after the establishment of tumors. Hence, a comprehensive understanding of the multifaceted functions of MAPK pathway components in tumor initiation and progression is essential for the improvement of cancer therapy. In this review, we focus on the reports that utilized knockout, conditional knockout, and transgenic mice of MAPK pathway components to investigate the effects of MAPK pathway components on tumor initiation and progression in the host environment.
Keywords: Carcinogenesis, genetic engineering, metastasis, proliferation/differentiation, signal transduction
Eukaryotic cells sense a wide range of biological and physicochemical stressors from both external and internal environments. Stress-responsive signaling cascades convert these stressors to appropriate physiological cellular responses (e.g. apoptosis and proliferation). Dysfunction in stress-responsive signaling cascades may often result in the acquired hallmarks of cancer, which are necessary for tumor progression;1 thus, the components of stress-responsive signaling cascades could be very promising drug targets for cancer therapy.
Mitogen-activated protein kinase pathways are one of these stress-responsive cascades. In response to various stressors, upstream MAP3K phosphorylates and activates MAP2K, and MAP2K subsequently activates MAPK. Activated MAPK regulates the dynamics of intranuclear transcription factors that induce physiological responses (Fig.1).2,3 In mammals, MKK4 (MAP2K4) and MKK7 (MAP2K7) phosphorylate and activate JNK, whereas MKK3 (MAP2K3), MKK4, and MKK6 (MAP2K6) activate p38. Both JNK and p38 regulate the expression of numerous genes related to cell survival (e.g. Bcl-2 and BAX), cell cycle (e.g. p53 and cyclin D1), and cell proliferation (e.g. JUN and MYC).4
Fig 1.
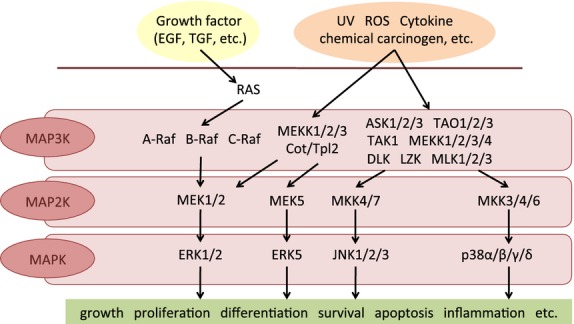
In mammalian MAPK pathways, upstream MAPK kinase kinase (MAP3K) phosphorylates and activates MAPK kinase (MAP2K) and MAP2K sequentially activates MAPK. Activated MAPK regulates the dynamics of intranuclear transcription factors that induce physiological responses. EGF, epidermal growth factor; ROS, reactive oxygen species; TGF, transforming growth factor.
Stress-responsive signaling cascades have been shown to regulate features of tumor cells themselves, such as hyperproliferation, replicative immortality, and resistance to cell death.1 Meanwhile, it has been recently revealed that tumors are more than homogenous masses of proliferating malignant cells. Rather, they consist of heterogeneous cell types, including recruited monocytes, lymphocytes, and fibroblasts, interacting with one another in the host environment.1,5 Moreover, stress-responsive signaling cascades also play pivotal roles in these heterotypic interactions, and a plethora of evidence has been accumulated that suggests the involvement of MAPK pathways in these interactions.
This review outlines the reports that establish the impact of MAPK pathways on tumor progression, focusing not only on malignant cells but also on the host environment, including stromal cells. We summarize the findings obtained by using KO, cKO, and Tg mice of MAPK pathway components.
MAPK pathways and tumorigenesis
A wide variety of GEMMs and chemically induced tumorigenesis models have been developed because they are useful to assess the functions of specific proteins in tumorigenesis. In this section, we will classify the results from current studies of the roles of MAPK pathway components in tumorigenesis based on the tissues and the organs of origin.
Skin tumorigenesis
Various chemically induced tumorigenesis models as well as GEMMs have been established for the analysis of skin tumorigenesis. The most widely used model is the two-stage skin tumorigenesis model. In this chemically induced tumorigenesis model, murine dorsal skin is treated once with DMBA to induce DNA damage in keratinocytes (initiation), followed by repeated applications of TPA, which enhances the inflammatory responses (promotion).
Several MAPK pathway components have been reported to regulate skin tumorigenesis in this model. Cot/Tpl2 (Map3k8) KO mice developed a higher number of tumors than WT mice through the induction of keratinocyte hyperproliferation triggered by elevated expression of inflammatory cytokines.6 In contrast, Erk1 (Mapk3) KO mice,7 p38δ (Mapk13) KO mice,8 and tamoxifen-inducible, keratinocyte-restricted Mkk4 cKO mice9 showed resistance to skin tumorigenesis in this model due to defects in keratinocyte proliferation.
Here, we introduce one of the MAPK pathway components: the ASK family. The ASK family is a member of the MAP3Ks in the JNK and p38 MAPK pathways and is activated in response to various stressors, such as cytokines and oxidative stress.10,11 The mammalian ASK family is composed of three isoforms, ASK1 (MAP3K5), ASK2 (MAP3K6), and ASK3 (MAP3K15), which have high homology, especially in the serine/threonine kinase domain.12 We have previously investigated the roles of ASK1 and ASK2 in skin tumorigenesis with the DMBA/TPA model. Ask2 KO mice formed more skin tumors than WT mice, whereas Ask1 KO mice showed comparable number of skin tumors to WT mice. The DMBA-induced apoptosis of epidermal keratinocytes was attenuated both in Ask1 KO and Ask2 KO mice to a similar extent. In contrast, TPA-dependent inflammatory responses, such as epidermal thickening and cytokine production, were impaired in Ask1 KO mice but not in Ask2 KO mice. These results suggest that ASK1 and ASK2 in keratinocytes cooperatively inhibit tumorigenesis by inducing apoptosis triggered by DNA damage, whereas only ASK1 is distinctively pro-tumorigenic by evoking inflammation through the production of pro-inflammatory cytokines (Fig.2).13 Considering the fact that Ask1;Ask2 doubleKO mice showed a similar phenotype to Ask2 KO mice, the antitumorigenic role of ASK1 is thought to compete with its tumorigenic role. This may explain why there was no difference in the extent of skin tumorigenesis between WT and Ask1 KO mice.
Fig 2.
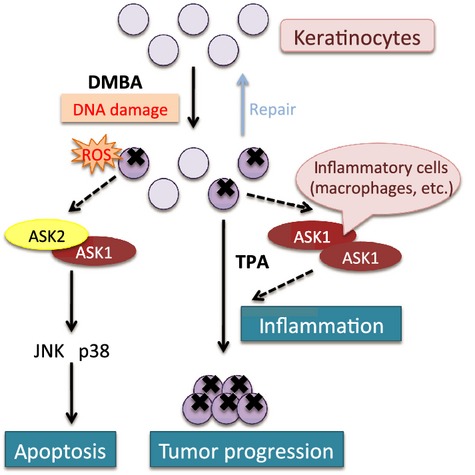
Apoptosis signal-regulating kinase 1 (ASK1) and ASK2 regulate skin tumorigenesis. The ASK2–ASK1 heterocomplex exerts a tumor-suppressive function by inducing apoptosis through activation of JNK and p38 during the initiation stage. In contrast, the ASK1 homocomplex in inflammatory cells evokes pro-inflammatory cytokine production in the promotion stage, accelerating tumorigenesis. DMBA, 7,12-dimethylbenz(a)anthracene; ROS, reactive oxygen species.
In addition to the DMBA/TPA model, models using gene manipulation and the induction of skin tumorigenesis with other chemicals show that several other MAPK pathway components also have pro-tumorigenic or antitumorigenic functions. Keratinocyte-specific Mek1 (Map2k1) cKO mice had fewer and smaller papillomas after DMBA/TPA treatment compared with WT or Mek2 (Map2k2) KO mice.14 Meanwhile, keratinocyte-specific overexpression of a constitutively active Mek1 mutant showed spontaneous skin tumor development out of hyperplasia.15 These reports suggest that MEK1 in the host environment has essential and isoform-specific pro-tumorigenic roles in skin tumorigenesis. Alternatively, Jnk1 (Mapk8) deficiency promoted skin tumorigenesis in the DMBA/TPA model16 as well as the DMBA/UVA exposure model, probably due to reduced apoptosis.17 By contrast, Jnk2 deficiency suppressed skin tumorigenesis in both models, presumably owing to a defect in cell proliferation and tumor vascularization.17,18 Collectively, individual JNK isoforms play unique roles in skin tumorigenesis. Epidermis-specific c-Raf (Raf-1) cKO mice were resistant to the formation of skin tumors in the DMBA/TPA model through reduced proliferation and enhanced apoptosis of keratinocytes.19 DMBA evokes skin tumorigenesis by introducing mutations to the ras gene.20 However, most human squamous skin carcinomas have elevated oncogenic Ras signaling without the presence of activating mutations in ras.21 Thus, Ehrenreiter et al. used another model of skin tumorigenesis to imitate the progressive stages of skin tumorigenesis in humans. When crossed with the mice that express a dominant active form of SOS-F in the epidermis in a 4-OHT-inducible manner, c-Raf cKO showed resistance to skin tumorigenesis by inducing the differentiation of keratinocytes. In addition, keratinocyte-restricted overexpression of a dominant negative form of p38α impaired skin tumorigenesis in a UV-induced skin tumorigenesis model.22,23 Dickinson et al.22 suggested that the attenuation of UV B-induced skin tumorigenesis in these mice was due to a reduction in the chronic hyperproliferation of keratinocytes. Liu et al.23 suggested that solar UV-induced skin tumorigenesis in these mice was suppressed through defects in inflammation.
As described above, different isoforms of MAPK pathway components have the following features during the course of skin tumorigenesis: (i) isoform specificity (i.e. MEK1, but not MEK2 has pro-tumorigenic roles); (ii) bidirectionality (JNK1 functions as a tumor suppressor, whereas JNK2 functions as an oncogene); and (iii) distinctiveness (both ASK1 and ASK2 induce apoptosis, whereas only ASK1 evokes inflammatory responses). In summary, different MAPK pathway components, including different isoforms, play diverse roles in skin tumorigenesis. A previous clinical report suggested that, for example, a combination of trametinib, a MEK inhibitor, and dabrafenib, a Raf inhibitor, has significant clinical benefits for skin cancer patients.24 This is a convincing example showing the effectiveness to target MAPK pathway components for cancer therapy.
Lung tumorigenesis
Oncogenic K-ras mutations have been identified in many lung cancer patients, and lung tumorigenesis GEMMs have been established by expressing these mutants in mice. Various KO or cKO mice of MAPK pathway components were crossed with the mice that have conditional expression of oncogenic K-ras (G12D or G12V, for example) to investigate the involvement of MAPK pathway components in lung tumorigenesis. For example, 4-OHT-inducible KO of p38α in adult mice fostered lung tumorigenesis triggered by K-RasG12V by interrupting maturation and promoting hyperproliferation of lung epithelium.25 By contrast, p38δ deletion attenuated lung tumorigenesis triggered by K-RasG12D through an unknown mechanism,8 illustrating the distinct roles of p38 isoforms in lung tumorigenesis. Bronchial epithelium-specific deletion of Mkk426 and epidermis-specific ablation of Mkk727 were revealed to deteriorate lung tumorigenesis combined with K-RasG12D. Schramek et al. further investigated whether MKK7 affects lung tumorigenesis through its well-known downstream effectors, JNKs. Jnk1+/−; Jnk2−/− mice were also sensitive to K-rasG12D-induced lung tumorigenesis. Although there is still a need for further detailed analysis, it is conceivable that MKK7 modulates lung tumorigenesis through JNK1 and JNK2 and that the MKK7–JNK signaling axis acts as an “anticancer barrier.”
However, Blasco et al.28 showed that the c-Raf–MEK–ERK axis is oncogenic during lung tumorigenesis in the same manner as in skin tumorigenesis. K-rasG12V-induced lung tumorigenesis was mitigated when crossed with c-Rafflox/flox, Mek1flox/flox; Mek2−/−, and Erk1−/−; Erk2 (Mapk1)flox/flox mice that were treated with Cre recombinase. The Cre-mediated recombination was induced by intratracheal infusion of a non-replicative adenovirus that encodes Cre recombinase (Ad-Cre). Intriguingly, Ad-Cre-induced ablation of c-Raf, but not B-Raf, specifically attenuated lung tumorigenesis evoked by K-RasG12D.29 Combined with the fact that the mice with lung-selective overexpression of c-Raf spontaneously developed lung adenomas,30 it is possible that c-Raf may be a suitable therapeutic target for lung cancer.
Mice overexpressing the B-Raf V600E mutant, which has constitutive kinase activity and a transforming ability, were shown to spontaneously develop hyperproliferative lung adenocarcinoma.31,32 The transgene was induced with Ad-Cre31 or with DOX32 in a lung-specific manner. As mentioned above, however, B-Raf deletion did not affect oncogenic K-ras-induced lung tumorigenesis, even though both c-Raf and B-Raf are downstream signaling components of ras. Collectively, ras may dominantly cultivate lung cancer in a c-raf-dependent manner. However, excessive activation of B-Raf caused by gene mutations may also trigger lung tumorigenesis.
Lung tumors can also be induced with chemical carcinogens. When Cot/Tpl2 KO mice were exposed to urethane, a chemical carcinogen, the mice were susceptible to the development of lung tumors exhibiting hyperproliferation, aggressive invasion, and cytologic atypia of lung epithelial cells through a defect in JNK–p53 activation.33 Based on the fact that lung cancers are caused by environmental factors, such as tobacco smoke and asbestos, a more detailed understanding of the function of MAPK pathway components should be explored in chemically induced lung tumorigenesis models.
Mammary tumorigenesis
Manipulation of gene expression, such as overexpression of oncogenes and downregulation of tumor suppressor genes, can drive mammary tumors. Mammary tumorigenesis driven in mice by overexpression of NeuT, an activated form of human epidermal growth factor receptor 2, was facilitated when Mkk7 was ablated in mammary epithelial cells.27 Further analysis suggested that MKK7 maintains p53 protein stability and p53-mediated responses to genotoxic stresses, such as cell senescence. Alternatively, deficiency in either Jnk1 or Jnk2 reduced breast tumor-free survival when crossed with Trp53+/− mice, which exhibit mild mammary tumorigenesis compared with Trp53−/− mice.34 Moreover, the absence of Jnk2 exacerbated mammary tumorigenesis triggered by the PyVmT transgene through the dysregulation of DNA damage responses and a consequent increase in tumor aneuploidy.35 Previous reports proposed that JNK regulates protein phosphorylation and the stability of p53.36 Considering that p53 is one of the genes responsible for mammary tumors and that mutations in p53 are found most frequently in human breast cancer patients,37 we can speculate that the MKK7–JNK axis has critical roles in mammary tumorigenesis through the regulation of p53 dynamics (e.g. protein phosphorylation and stability) and subsequent cellular responses (e.g. cell senescence).
Colon tumorigenesis
Mutations in the Apc gene are observed in almost all colon tumor patients in the relatively early stage of tumorigenesis, and these mutations are known to induce the formation of premalignant adenomatous polyps.38 Thus, GEMMs harboring mutations in the Apc gene are used to explore the mechanism of colon tumorigenesis. Cot/Tpl2 KO mice crossed with Apcmin/+ mice, which express a dysfunctional truncated Apc protein without sufficient tumor suppressive functions, formed more aggressive colon tumors compared with control Apcmin/+ mice. This may be because Apcmin/+; Tpl2−/− mice showed enhanced intestinal inflammation associated with a decreased number of immunosuppressive regulatory T cells.39 Jnk2 deletion in Apc1638+/− mice, which express undetectable amount of Apc protein, promoted colon tumorigenesis through accelerated inflammation and aberrant β-catenin expression. Intriguingly, Jnk2 deletion enhanced the burden of colon tumors only if combined with a “high-risk, Western-style” diet containing high fat, high phosphate, low calcium, and low vitamin D.40 These results imply that JNK2 and dietary factors may cooperatively regulate colon tumorigenesis under certain circumstances.
Chemically induced tumorigenesis models have also been widely used for studying colon tumorigenesis. One typical model of colon tumorigenesis is established by single i.p. injection of AOM and continuous administration of DSS through drinking water. It has been already shown that deletion of p38α in IECs fostered colon tumorigenesis in this model through an unknown mechanism.41 However, using this model, Gupta et al.42 recently showed that p38α has both a tumor-suppressive role in colon tumorigenesis and a tumor-promoting role in tumor progression. Deficiency in p38α in IECs augmented sensitivity to colitis-associated colon cancer due to enhanced infiltration of inflammatory cells and increased apoptosis of IECs followed by compensatory hyperproliferation. Notably, p38α ablation in IEC triggered colon tumorigenesis when the mice received only DSS treatment. These data also indicate that p38α has a tumor-suppressive role in colon tumorigenesis. However, 4-OHT-inducible, IEC-specific cKO of p38α in tumor-established mice alleviated tumor burden, suggesting that p38α also has a supportive effect on tumor cell proliferation. Considering the dual functions of p38α in the course of colon cancer progression, targeting p38α should be examined carefully.4 In addition, other p38 isoforms, p38δ and p38γ (MAPK12), have been revealed to regulate colon tumorigenesis in the same model. It was reported that p38δ−/−, p38γ−/− and p38δ−/−; p38γ−/− mice showed mitigation in colon tumorigenesis.43 Deletion of p38δ and/or p38γ in mice attenuated IEC proliferation and enhanced IEC apoptosis. It also reduced the expression of pro-inflammatory chemokines and cytokines such as monocyte chemotactic and activating factor-1 and TNF-α, resulting in defective recruitment of macrophages and neutrophils. Importantly, p38δ−/−; p38γ−/− mice were more resistant to colon tumorigenesis compared with p38δ−/− or p38γ−/− mice, which is indicative of the synergistic impact of p38δ and p38γ on colon tumorigenesis.
Ask1 KO mice were reported to show susceptibility to AOM/DSS colon tumorigenesis coinciding with severe inflammation.44 In vitro experiments have revealed that macrophages of Ask1 KO mice had impaired cytotoxicity against enteric bacteria and were vulnerable to bacteria-induced apoptosis owing to a defect in p38 activation. These phenomena may be attributed to decreased messenger RNA expression of anti-apoptotic genes, such as cellular inhibitor of apoptosis protein 1 and 2 and serpin B2.
It was also revealed that JNK1 regulates colon tumorigenesis. Overexpression of a constitutively active form of Jnk1 in an IEC-specific manner increased colitis-associated colon cancer susceptibility to AOM/DSS-induced colon tumorigenesis owing to enhanced proliferation of progenitor cells.45 However, there is also a report that Jnk1 ablation can spontaneously induce colon tumorigenesis.46 Although compensation by other JNK isoforms and the cell type-specific roles of JNK1 may account for this discrepancy, further analysis is needed to clarify the divergent functions of JNK1 and p38α in colon tumorigenesis. Compared with other organs, the colon is incessantly exposed to innumerable enteric bacteria, which can trigger divergent inflammatory responses. This peculiarity may explain why MAPK pathway components in the host environment have contrasting functions in colon tumorigenesis.
Liver tumorigenesis
There are many mouse models for liver tumorigenesis that recapitulate human liver cancers. Single i.p. injection of DEN is a common chemically induced model of liver tumorigenesis. Loss of Jnk1 mitigated liver tumorigenesis with a reduction of DEN-induced cell death, compensatory proliferation, and neovascularization.47,48 Moreover, cell type-dependent effects of JNKs in liver tumorigenesis were investigated using Jnk2−/− mice crossed with hepatocyte-specific (Alb−Cre+/−; Jnk1flox/flox), and hepatocyte/non-parenchymal cell-specific (Mx1−Cre+/−; Jnk1flox/flox) Jnk1 cKO mice,49 hereafter referred to as HΔJNK and MxΔJNK mice, respectively. HΔJNK mice developed more tumors, whereas MxΔJNK mice developed fewer tumors compared with control mice, which express only Cre recombinase in the corresponding tissues. In-depth analysis of these opposing phenotypes by Das et al. revealed that the expression and release of inflammatory cytokines, such as IL-6, IL-1α, IL-1β, and TNF-α, were enhanced in HΔJNK mice but were decreased in MxΔJNK mice. Consequently, hepatocyte death and compensatory proliferation were accelerated in HΔJNK mice but attenuated in MxΔJNK mice.
Meanwhile, hepatocyte-specific p38α cKO resulted in elevated HCC with this model due to an increase in reactive oxygen species production, liver damage, and compensatory proliferation.50 Sakurai et al.51 further demonstrated that p38α inhibited liver fibrogenesis and subsequent HCC in another liver tumorigenesis model with thioacetamide added to drinking water. Hui et al.52 also reported the suppressive functions of p38α in liver tumorigenesis with another similar tumorigenesis model; liver-specific p38α-deficient mice were injected with DEN i.p. and subsequently fed with phenobarbital mixed with their diet. Notably, p38α is hypothesized to modulate hepatocyte proliferation by antagonizing the JNK–c-Jun pathway,52 suggesting the crosstalk between MAPK pathway components in liver tumorigenesis.
It has also been proposed that ASK1 regulates liver tumorigenesis; Ask1 KO mice formed more tumors in DEN-induced liver tumorigenesis model.53 ASK1 appeared to have an influence on death receptor-mediated apoptosis through JNK activation and DNA damage responses by p38 activation.
In addition to chemically induced tumorigenesis models, manipulating gene expressions of MAPK pathway components have been shown to induce spontaneous formation of liver tumors. Ablation of Tak1 (Map3k7) in hepatocytes generated HCC through various mechanisms: dysregulated hepatic inflammation and hepatocyte injury that cause liver fibrosis, enhancement of TNF-α-dependent hepatocyte death, and compensatory proliferation.54,55 Alternatively, Tak1 ablation specifically in liver parenchymal cells also led to spontaneous formation of liver tumors with hepatic inflammation, liver fibrosis, cholestasis, and ductopenia.56 Furthermore, TAK1 was shown to regulate hepatocyte apoptosis and necrosis through a nuclear factor-κB-dependent and -independent manner, respectively. It is conceivable that gene manipulation of other MAPK pathway components may affect liver tumorigenesis, and further study is warranted to comprehensively understand the mechanism of how these components contribute to it. Taking p38α and JNK as an example, we have to pay close attention to the interaction between MAPK pathway components and their distinct functions according to cell types.
Gastric tumorigenesis
Similar to lung and liver tumorigenesis, gastric tumorigenesis can be evoked by environmental factors such as Helicobacter pylori and alcohol intake. A typical chemically induced model of gastric tumorigenesis that mimics those environmental factors is to challenge mice with N-methyl-N-nitrosourea in drinking water. Genetic disruption of Jnk1 attenuated the growth of GC, probably owing to decreases in apoptosis and in compensatory cell proliferation in a reactive oxygen species-dependent manner.57
Ask1 KO mice were reported to be resistant to gastric tumorigenesis induced with N-methyl-N-nitrosourea.58 ASK1 was shown to participate in a positive feedback loop with cyclin D1 through JNK activation and to promote the proliferation of gastric cells. It was further shown that K811, an ASK1 inhibitor, prevented the growth of GC, suggesting that ASK1 might be a promising drug target for GC therapy.59 As MAPKs, such as p38, JNK, and ERK, were shown to be activated by H. pylori,60 investigating the effect of MAPK pathway components on gastric tumorigenesis induced by environmental factors would clarify the clinical significance of MAPK pathway components.
Pancreatic tumorigenesis
Human pancreatic cancer has a linear progression associated with accumulated gene mutations. Its premalignant lesion is referred to as PanIN and is classified into multiple stages based on cellular morphology and polarity, micropapillary structure, and chromosomal composition. Some GEMMs spontaneously generate PanIN in the course of developing pancreatic cancers.61 For example, RIP1Tag2 Tg mice express Simian vacuolating virus 40 large T antigen transgene under RIP1 and spontaneously develop pancreatic islet carcinoma. The impact of genetic deletion of MAPK pathway components has been investigated in these models. In RIP1Tag2 Tg mice, pancreatic β cell-specific deletion of B-Raf delayed tumor progression with attenuated cell proliferation and suppressed angiogenesis.62 Pancreas-specific ablation of Mkk4 and Mkk7 drastically facilitated PanIN progression in KrasG12D-triggered pancreatic tumorigenesis.63 The detailed molecular mechanism is yet to be elucidated, but these results were presumably due to additional effects of MKK4 and MKK7 on the activity of JNK. Davies et al. further suggested that MKK4 and MKK7 may promote the transdifferentiation of pancreatic cell types from acinar to ductal cells and that they have synergistic impacts on pancreatic tumorigenesis. As pancreatic tumors are known to have a very poor clinical outcome, it will be important to identify the precise molecular mechanisms involved in pancreatic tumor progression, including the roles of MKK4 and MKK7, to develop effective therapies for pancreatic cancer.
Hematological malignancies and tumorigenesis in other tissues
The MAPK pathway components are known to govern non-epithelial cancers, such as hematological malignancies, as well as epithelial solid tumors. Manipulating the gene expression of specific MAPK pathway components can spontaneously trigger leukemia. Lymphocyte-specific overexpression of a Cot/Tpl2 mutant, which has a truncation in the carboxyl terminus leading to increased catalytic activity and hyperactivation of the MAPK pathway, gave rise to spontaneous lymphoma in mice.64 By contrast, Cot/Tpl2 deficiency resulted in T-cell lymphoma by promoting T cell proliferation when crossed with TCR2C Tg mice, which express the specific T cell receptors to MHC class I.65 This discrepancy is informative because it suggests that appropriate kinase activity of Cot/Tpl2 is vital for inhibition of lymphoma and that either excessive or defective kinase activity results in the formation of lymphoma.
Myeloid lineage-specific deletion of Tak1 resulted in the development of myelomonocytic leukemia with splenomegaly due to a lack of myeloid cell maturation and genomic instability.66 B-Raf is oncogenic also in hematological malignancies, and interferon-inducible overexpression of the B-Raf V600E mutant in somatic tissues led to the spontaneous development of non-lymphocytic leukemia.67
B-Raf is thought to be an oncogene in other epithelial solid tumors,68 including thyroid69 and prostate70 cancers. Overexpression of the B-Raf V600E mutant in thyroid cells evoked poorly differentiated papillary thyroid carcinomas.69 Similarly, DOX-induced overexpression of the B-Raf V600E mutant in prostate basal epithelial cells led to the development of invasive prostate adenocarcinomas with aberrant cell proliferation.70 Jeong et al. also showed that DOX withdrawal eliminated the expression of B-Raf within established adenocarcinomas, but those established tumors did not regress. From these data, Jeong et al. suggested that stimulation of B-Raf is sufficient to initiate prostate adenocarcinoma growth, but unnecessary to maintain these tumors once they were established. As we have previously mentioned, p38α also has distinctive roles during the establishment and the maintenance phases of colon cancer growth.42 These reports suggest that the divergent roles of MAPK pathway components in tumor progression should be analyzed separately prior to and after the establishment of tumors.
Mitogen-activated protein kinase pathways and tumor metastasis
Tumor cells from primary lesions gradually invade into the local surrounding stroma, enter circulation (intravasate) and disseminate, exit circulation (extravasate), and finally adapt to foreign environments of distant sites (colonize).71 This sequential transition is called tumor metastasis. It is of paramount importance from a clinical perspective to elucidate the mechanism of tumor metastasis because over 90% of tumor mortality is attributed to metastatic spread. However, deciphering the complexity of tumor metastasis is a challenging task because this process is very inefficient. Only a tiny fraction of tumor cells can adapt to foreign sites and form metastatic foci. Although there are a few reports that show that MAPK pathway components in the host environment participate in tumor metastasis, we will present the collection of studies on the relationships between MAPK pathway components and tumor metastasis using our unpublished data as well.
Mekk1 (Map3k1) KO mice were examined in a PyVmT transgene-induced mammary tumorigenesis model, which is also known to develop lung metastasis.72 Ablation of Mekk1 did not affect the frequency and growth of primary mammary tumors, but it mitigated tumor cell dissemination and lung metastasis. Melanocyte-specific Tg mice of the B-Raf V600E mutant developed melanoma and tumor metastasis to draining lymph nodes.73 They even developed lung metastases when crossed with Cdkn2a/p16+/− mice. There are apparently few molecules other than B-Raf that trigger both tumorigenesis and tumor metastasis. Hence, the impact of B-Raf on tumor progression may be overwhelming and it is agreeable to target B-Raf for cancer therapy.
The most common models for experimental tumor metastasis are to inject tumor cells into the tail vein or into the heart. Intravenously injected tumor cells mainly metastasize to the lung, whereas intracardiac injection enables tumor cells to spread systemically, including to the bone.74 Tumor cells inoculated into the systemic circulation are transported to metastatic sites such as the lung and the bone, form emboli within the vasculature, and adhere to endothelial cells. Only a small fraction of the surviving tumor cells can extravasate from the vasculature and form micrometastases, which gradually proliferate into macroscopic metastases. The experimental tumor metastasis models can recapitulate the stages from transport of tumor cells to formation of macroscopic metastases.
Jnk1 KO mice were sensitive to lung metastasis of B16F0 melanoma and EL-4 thymoma cell lines injected i.v. because the cytotoxic functions of CD8+ T cells were suppressed.75 In addition, Jnk2 KO mice showed resistance to lung and bone metastasis caused by intracardiac injection of 4T1 mammary tumor cell line.76 Through osteolysis mediated by mature osteoclasts, a variety of growth factors, such as transforming growth factor-β, fibroblast growth factors, and bone morphogenetic proteins, are released and stimulate the proliferation of tumor cells. Nasrazadani and Van Den Berg suggested that Jnk2 ablation might lead to inhibition of osteoclast differentiation and subsequent growth arrest of tumor cells.
Once intravasated, circulating tumor cells face several risks for death, such as the shear stress of the blood or the lymphatic flow and immunosurveillance by antitumor immune cells. Platelets can shield tumor cells from those threats in the case of hematogenous metastasis by forming platelet–tumor cell (that sometimes include leukocytes) aggregates. The formation of these aggregates is mediated by the release of factors (e.g. chemokines and growth factors) and adhesive molecules (e.g. integrins and selectins).77 These aggregates support tumor cell attachment to and interaction with endothelial cells, and aid tumor cells in crossing endothelial walls to extravasate.78
Subcutaneous injection of B16F10 melanoma and Lewis lung carcinoma cell lines into p38α+/− mice did not affect the penetrance or growth of primary tumors compared with WT mice. However, when p38α+/− mice were challenged with i.v. injection of B16F10 and Lewis lung carcinoma, lung metastasis was reduced in p38α+/− mice.79 Matsuo et al.79 claimed that tumor cell-dependent upregulation of P-selectin in platelets and E-selectin in endothelial cells was attenuated in p38α+/− mice, resulting in weaker interactions between platelets, endothelial cells, and tumor cells.
As previously mentioned, collaborative researches, including our own, suggest the involvement of ASK1 and ASK2 in tumorigenesis through various biological mechanisms including inflammation.13,44,53,58,59 Inflammation and tumor metastasis have been revealed to be closely related,80 but whether the ASK family is also involved in tumor metastasis is unknown. We subjected Ask1 KO mice to i.v. injection of 3LL-Luc2 cells, which have constitutive luciferase expression so that lung micrometastases can be detected by measuring the luciferase activity of lung lysates. As a result, Ask1 KO mice had a markedly reduced number of tumor nodules and decreased luciferase activity in lung lysates compared with WT mice (unpublished data, M.K., I.S. and H.I.). These data indicate that Ask1 deficiency attenuates tumor metastasis in the experimental lung metastasis model. Our data also suggest that ASK1 is involved in the relatively early stage of tumor metastasis.
In summary, the reports shown above imply that MAPK pathway components in the host environment could perform multifaceted functions in tumor metastasis, sometimes with little effect on tumor initiation. Thus, it is necessary to dissect the functions of MAPK pathway components in tumor initiation and metastasis separately while keeping the comprehensive system in mind.
Conclusion
Tumor cells and normal stromal cells in the host environment are revealed to communicate each other, and their complex network appears to influence every aspect of tumor progression.5 Although the fully detailed picture of this heterotypic interaction is still enigmatic, ever-progressing technologies such as high-resolution single-cell imaging, high accuracy gene manipulation both in vivo and in vitro (e.g. CRISPR/Cas9, TALEN), and GEMMs with temporally or spatially increased specificity will reveal the comprehensive frameworks of tumor biology.
A myriad of evidence has been accumulated to suggest that MAPK pathway components in tumor cells as well as normal stromal cells in the host environment greatly influence tumor progression. Some MAPK pathway components such as B-Raf and MKK7 seem to have pro-tumorigenic or antitumorigenic functions, respectively (Table1).68 However, many of MAPK pathway components seem to have both pro-tumorigenic and antitumorigenic functions, depending on the context (Table2). In addition, these components have redundant functions and crosstalk with one another. From a research standpoint, we have to keep in mind the importance of investigating the divergent functions of MAPK pathway components in tumorigenesis and tumor progression of various cancers. Specifically, we must take into account the tissue, cell type, and isoform-specificity, as well as the degree of tumor progression (i.e. in tumor initiation or after the establishment of tumors). Although individual MAPK pathway components have been shown to take part in tumor progression through various biological responses, such as apoptosis and inflammation, we should also investigate the combined effects on these responses in vivo. Manipulating multiple MAPK pathway components allows us to examine these combined effects, and several reports have indeed clarified these effects (Table3). This research will be instrumental for understanding the true regulatory networks of MAPK pathway components and to develop more effective, more specific, and less harmful drugs for cancer therapy.
Table 1.
Mitogen-activated protein kinase pathway components reported to be either pro-tumorigenic or antitumorigenic
| Molecule | Gene manipulation of MAPK pathway components | Tumorigenesis model | Tumor type | Phenotype and concomitant phenomena | Role | References |
|---|---|---|---|---|---|---|
| B-Raf | Lung-Tg (V600E, adenovirus with Cre-inducible) | Spontaneous | Lung adenocarcinoma | Enhanced (mechanism unknown) | Pro | 30 |
| Lung-Tg (V600E, DOX-inducible) | Spontaneous | Lung adenocarcinoma | Enhanced (mechanism unknown) | Pro | 31 | |
| Pancreatic β cell-cKO | RIP1Tag2 Tg | Pancreatic adenoma | Enhanced (cell proliferation ↓, angiogenesis ↓) | Pro | 61 | |
| Prostate basal epithelial-Tg (V600E, DOX-inducible) | Spontaneous | Prostate adenocarcinoma | Enhanced (aberrant cell proliferation) | Pro | 69 | |
| Thyroid cell-Tg (V600E) | Spontaneous | Papillary thyroid carcinoma | Enhanced (mechanism unknown) | Pro | 68 | |
| Somatic tissues-Tg (IFN-inducible) | Spontaneous | Nonlymphoid leukemia | Enhanced (mechanism unknown) | Pro | 66 | |
| C-Raf (Raf-1) | Lung-Tg | Spontaneous | Lung adenoma | Enhanced (mechanism unknown) | Pro | 29 |
| cKO (adenovirus with Cre-inducible) | K-rasG12V Tg | NSCLC | Attenuated (mechanism unknown) | Pro | 27 | |
| cKO (adenovirus with Cre-inducible) | K-rasG12D Tg | Lung adenocarcinoma | Attenuated (mechanism unknown) | Pro | 28 | |
| Epidermis-cKO | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (proliferation ↓, apoptosis ↑ of keratinocyte) | Pro | 19 | |
| Epidermis-cKO | 4OHT-inducible SOS-F Tg | Cutaneous papilloma | Attenuated (keratinocyte differentiation ↑) | Pro | 19 | |
| ASK2 (MAP3K6) | KO | Induced with DMBA + TPA | Cutaneous papilloma | Enhanced (apoptosis of keratinocyte ↓ [cooperating with ASK1]) | Anti | 13 |
| TAK1 (MAP3K7) | Liver parenchymal-cKO | Spontaneous | HCC | Enhanced (hepatic inflammation, liver fibrosis, cholestasis, ductopenia, hepatocyte apoptosis and necrosis) | Anti | 55 |
| Hepatocyte-cKO | Spontaneous | HCC | Enhanced (dysregulated hepatic inflammation, hepatocyte injury,liver fibrosis, hepatocyte death, compensatory proliferation) | Anti | 53,54 | |
| Myeloid-cKO | Spontaneous | Leukemia | Enhanced (immaturation of myeloid cells, genomic instability, splenomegaly) | Anti | 65 | |
| MEK1 (MAP2K1) | Keratinocyte-Tg (constitutively active mutant) | Spontaneous | Cutaneous papilloma | Enhanced (hyperplasia ↑) | Pro | 15 |
| Keratinocyte-cKO | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (mechanism unknown) | Pro | 14 | |
| MKK7 (MAP2K7) | Epidermis-cKO | K-rasG12D Tg | Lung adenocarcinoma | Enhanced (p53 stability ↓, cellular senescence ↓, cell proliferation ↑) | Anti | 26 |
| MEC-cKO | NeuT Tg | Mammary carcinoma | Enhanced (p53 stability ↓, p53-mediated responses to genotoxic stresses ↓) | Anti | 26 | |
| ERK1 (MAPK3) | KO | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (keratinocyte proliferation ↓) | Pro | 7 |
| p38γ (MAPK12) | KO | Induced with AOM + DSS | Colon adenocarcinoma | Attenuated (expression of pro-inflammatory cytokine and chemokine ↓, recruitment of macrophage and neutrophil ↓, proliferation ↓, apoptosis ↑ of IEC) | Pro | 42 |
| p38δ (MAPK13) | KO | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (keratinocyte proliferation ↓) | Pro | 8 |
| KO | K-rasG12D Tg | Lung adenocarcinoma | Attenuated (mechanism unknown) | Pro | 8 |
For B-Raf, see the referenced review.67 Anti, antitumorigenic role of MAPK pathway; AOM, azoxymethane; cKO, conditional knockout; DMBA, 7,12-dimethylbenz(a)anthracene; DOX, doxycycline; DSS, dextran sodium sulfate; IEC, intestinal epithelial cell; MEC, mammary epithelial cell; NSCLC, non-small cell lung cancer; Pro, pro-tumorigenic role of MAPK pathway; Tg, transgenic. Upright arrows indicate the enhancement of the phenomena and down arrows indicate the attenuation of the phenomena.
Table 2.
Mitogen-activated protein kinase pathway components reported to have both tumorigenic and antitumorigenic functions
| Molecule | Gene manipulation of MAPK pathway components | Tumorigenesis model | Tumor type | Phenotype and concomitant phenomena | Function | References |
|---|---|---|---|---|---|---|
| ASK1 (MAP3K5) | KO | Induced with AOM + DSS | Colon adenocarcinoma | Enhanced (cytotoxicity and apoptosis of macrophage ↓, inflammation ↑) | Anti | 43 |
| KO | Induced with DEN | HCC | Enhanced (death receptor-mediated apoptosis ↓, DNA damage responses ↓) | Anti | 52 | |
| KO | Induced with MNU | Gastric carcinoma | Attenuated (cell proliferation ↓, cell cycle progression ↓) | Pro | 57,58 | |
| KO | Induced with DMBA + TPA | Cutaneous papilloma | Comparable in overall (inflammatory responses ↓) | Pro | 13 | |
| KO | Induced with DMBA + TPA | Cutaneous papilloma | Comparable in overall (apoptosis of keratinocyte ↓ [cooperating with ASK2]) | Anti | 13 | |
| Cot/Tpl2 (MAP3K8) | KO | Induced with urethane | Lung adenoma | Enhanced (hyperproliferation, invasion ↑, cytologic atypia of LEC) | Anti | 32 |
| KO | Apcmin/+ | Intestinal adenoma | Enhanced (intestinal inflammation ↑, Tregs ↓) | Anti | 38 | |
| Lymphocyte-Tg (constitutively active mutant) | Spontaneous | T-cell lymphoma | Enhanced (mechanism unknown) | Pro | 63 | |
| KO | TCR2Ctg/tg Tg | T-cell lymphoma | Enhanced (T cell proliferation ↑) | Anti | 64 | |
| KO | Induced with DMBA + TPA | Cutaneous papilloma | Enhanced (keratinocyte hyperproliferation ↑ triggered by inflammatory cytokines) | Anti | 6 | |
| MEK4/MKK4 (MAP2K4) | Bronchial epithelium-cKO | K-rasG12D Tg | Lung adenocarcinoma | Enhanced (mechanism unknown) | Anti | 25 |
| Keratinocyte-cKO (TAM-inducible) | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (keratinocyte proliferation ↓) | Pro | 9 | |
| JNK1 (MAPK8) | KO | Induced with DEN | HCC | Attenuated (cell death ↓, compensatory cell proliferation ↓, neovascularization ↓) | Pro | 46,47 |
| KO | Induced with DMBA + TPA | Cutaneous papilloma | Enhanced (mechanism unknown) | Anti | 16 | |
| KO | Induced with DMBA + UVA | Cutaneous papilloma | Enhanced (apoptosis ↓) | Anti | 17 | |
| KO | Induced with MNU | Gastric carcinoma | Attenuated (ROS-dependent apoptosis ↓, compensatory cell proliferation ↓) | Pro | 56 | |
| IEC-Tg (constitutively active mutant) | Induced with AOM + DSS | Colon adenocarcinoma | Enhanced (proliferation of progenitor cell ↑) | Pro | 44 | |
| KO | Spontaneous | Intestinal adenoma | Enhanced (mechanism unknown) | Anti | 45 | |
| KO | Trp53+/− | Mammary gland tumor | Enhanced (mechanism unknown) | Anti | 33 | |
| JNK2 (MAPK9) | KO | Induced with DMBA + TPA | Cutaneous papilloma | Attenuated (cell proliferation ↓, vascularization ↓) | Pro | 18 |
| KO | Induced with DMBA + UVA | Cutaneous papilloma | Attenuated (mechanism unknown) | Pro | 17 | |
| KO | Trp53+/− | Mammary gland tumor | Enhanced (mechanism unknown) | Anti | 33 | |
| KO | PyVmT Tg | Mammary carcinoma | Enhanced (dysregulated DNA damage responses, aneuploidy ↑) | Anti | 34 | |
| KO | Apc1638+/−, “Western-style” diet | Intestinal adenoma | Enhanced (inflammation ↑, abberant β-catenin expression) | Anti | 39 | |
| p38α (MAPK14) | KO (4OHT-inducible) | K-rasG12V Tg | NSCLC | Enhanced (maturation ↓, hyperproliferation ↑ of lung epithelium) | Anti | 24 |
| Liver-cKO | Induced with DEN + Pb | HCC | Enhanced (hepatocyte proliferation ↑) | Anti | 51 | |
| Hepatocyte-cKO | Induced with DEN | HCC | Enhanced (ROS production ↑, liver damage ↑, compensatory cell proliferation ↑) | Anti | 49 | |
| Hepatocyte-cKO | Liver (TAA) | HCC | Enhanced (liver fibrogenesis ↑) | Anti | 50 | |
| IEC-cKO | Colon (DSS) | Colon adenocarcinoma | Enhanced (mechanism unknown) | Anti | 41 | |
| IEC-cKO | Colon (AOM + DSS) | Colon adenocarcinoma | Enhanced (inflammatory cell infiltration ↑, IEC apoptosis ↑, compensatory hyperproliferation) | Anti | 40,41 | |
| IEC-cKO (4-OHT-inducible) | Colon (AOM + DSS) | Colon adenocarcinoma | Attenuated (cell proliferation ↓, apoptosis ↑ [tumor maintenance]) | Pro | 41 | |
| Keratinocyte-Tg (dominant negative mutant) | Induced with SUV | Cutaneous papilloma | Attenuated (inflammation ↓, epidermal thicking ↓) | Pro | 23 | |
| Keratinocyte-Tg (dominant negative mutant) | Induced with UVB | Cutaneous papilloma | Attenuated (chronic hyperproliferation ↓) | Pro | 22 |
Many MAPK pathway components seem to have both pro-tumorigenic and antitumorigenic functions, depending on the context. Anti, antitumorigenic function of MAPK pathway; AOM, azoxymethane; cKO, conditional knockout; DEN, diethylnitrosamine; DMBA, 7,12-dimethylbenz(a)anthracene; DSS, dextran sodium sulfate; HCC, hepatocellular carcinoma; IEC, intestinal epithelial cell; LEC, lung epithelial cell; MNU, N-methyl-N-nitrosourea; NSCLC, non-small cell lung carcinoma; 4-OHT, 4-hydroxy-tamoxifen; Pro, pro-tumorigenic function of MAPK pathway; ROS, reactive oxygen species; TAA, thioacetamide; TAM, tamoxifen; Tg, transgenic; Treg, regulatory T cell.
Table 3.
Reports regarding the combined effects of multiple MAPK pathway components in tumorigenesis
| Molecule | Gene manipulation of MAPK pathway components | Tumorigenesis model | Tumor type | Phenotype and concomitant phenomena | Role | References |
|---|---|---|---|---|---|---|
| MEK1 | Mek1 cKO; Mek2 KO (adenovirus with Cre-inducible) | K-rasG12V Tg | NSCLC | Attenuated (mechanism unknown) | Pro | 27 |
| MEK2 (MAP2K2) | ||||||
| MKK4 | Pancreas Mkk4 cKO; Mkk7 cKO | K-rasG12D Tg | Pancreatic ductal adenocarcinoma | Enhanced (trans-differentiation of acinar cell into duct-like cell ↓) | Anti | 62 |
| MKK7 | ||||||
| JNK1 | Jnk1+/−; Jnk2−/− | K-rasG12D Tg | Lung adenocarcinoma | Enhanced (mechanism unknown) | Anti | 26 |
| JNK2 | ||||||
| JNK1 | Hepatocyte-Jnk1 cKO; Jnk2 KO | Induced with DEN | HCC | Enhanced (release of inflammatory cytokines ↑, hepatocyte death ↑, compensatory proliferation ↑) | Anti | 48 |
| JNK2 | ||||||
| JNK1 | Hepatocyte and non-parenchymal-Jnk1 cKO; Jnk2 KO | Induced with DEN | HCC | Attenuated (release of inflammatory cytokines ↓, hepatocyte death ↓, compensatory proliferation ↓) | Pro | 48 |
| JNK2 | ||||||
| p38δ | p38δ KO; p38γ KO | Induced with AOM + DSS | Colon adenocarcinoma | Attenuated (expression of pro-inflammatory cytokine and chemokine ↓, recruitment of macrophage and neutrophil ↓, proliferation ↓, apoptosis ↑ of IEC) | Pro | 42 |
| p38γ | ||||||
| ERK1 | Erk1 KO; Erk2 cKO (adenovirus with Cre-inducible) | K-rasG12V Tg | NSCLC | Attenuated (mechanism unknown) | Pro | 27 |
| ERK2 (MAPK1/2) |
Manipulating multiple MAPK pathway components allows us to examine the combined effects on tumor progression and various biological responses. Anti, antitumorigenic role of MAPK pathway; AOM, azoxymethane; cKO, conditional knockout; DEN, diethylnitrosamine; DSS, dextran sodium sulfate; HCC, hepatocellular carcinoma; IEC, intestinal epithelial cell; NSCLC, non-small cell lung cancer; Pro, pro-tumorigenic role of MAPK pathway; Tg, transgenic.
Meanwhile, from the clinical standpoint, integrative and personalized medicine would be the most effective mainstream practice in cancer therapy. Therefore, drugs that target MAPK pathway components should be selected depending on the origins of tumors and the degree of tumor progression. Prospective research to decipher the precise roles of MAPK pathway components not only in a tumor cell-intrinsic but also in a tumor cell-extrinsic fashion is essential for conquering this, at present, incurable disease.
Acknowledgments
We thank all the members of the Laboratory of Cell Signaling. This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japanese Society for the Promotion of Sciences and the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT), the advanced research for medical products Mining Program of the National Institute of Biomedical Innovation, the “Understanding of molecular and environmental bases for brain health” conducted under the Strategic Research Program for Brain Sciences by MEXT, Ono Medical Research Foundation, Terumo Life Science Foundation, Suzuken Memorial Foundation, and Astellas Foundation for Research on Metabolic Disorders.
Glossary
- AOM
azoxymethane
- ASK
Apoptosis Signal-regulating Kinase
- cKO
conditional knockout
- DEN
diethylnitrosamine
- DMBA
7,12-dimethylbenz(a)anthracene
- DOX
doxycycline
- DSS
dextran sodium sulfate
- GC
gastric cancer
- GEMM
genetically engineered mouse model
- HCC
hepatocellular carcinoma
- IEC
intestinal epithelial cell
- IL
interleukin
- KO
knockout
- MAP2K
MAPK kinase
- MAP3K
MAPK kinase kinase
- 4-OHT
4-hydroxy-tamoxifen
- PanIN
pancreatic intraepithelial neoplasia
- RIP1
rat insulin promoter 1
- Tg
transgenic
- TNF
tumor necrosis factor
Disclosure statement
The authors have no conflict of interest.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–80. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Murakami S, Noguchi T, Takeda K, Ichijo H. Stress signaling in cancer. Cancer Sci. 2007;98:1521–7. doi: 10.1111/j.1349-7006.2007.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCicco-Skinner KL, Trovato EL, Simmons JK, Lepage PK, Wiest JS. Loss of tumor progression locus 2 (tpl2) enhances tumorigenesis and inflammation in two-stage skin carcinogenesis. Oncogene. 2011;30:389–97. doi: 10.1038/onc.2010.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourcier C, Jacquel A, Hess J, et al. p44 Mitogen-activated protein kinase (extracellular signal-regulated kinase 1)–dependent signaling contributes to epithelial skin carcinogenesis. Cancer Res. 2006;66:2700–7. doi: 10.1158/0008-5472.CAN-05-3129. [DOI] [PubMed] [Google Scholar]
- Schindler EM, Hindes A, Gribben EL, et al. p38δ mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res. 2009;69:4648–55. doi: 10.1158/0008-5472.CAN-08-4455. [DOI] [PubMed] [Google Scholar]
- Finegan KG, Tournier C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010;70:5797–806. doi: 10.1158/0008-5472.CAN-09-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–8. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol. 2008;48:199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- Iriyama T, Takeda K, Nakamura H, et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J. 2009;28:843–53. doi: 10.1038/emboj.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl FA, Dumesic PA, Barragan DI, Harada K, Charron J, Khavari PA. Selective role for Mek1 but not Mek2 in the induction of epidermal neoplasia. Cancer Res. 2009;69:3772–8. doi: 10.1158/0008-5472.CAN-08-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feith DJ, Bol DK, Carboni JM, et al. Induction of ornithine decarboxylase activity is a necessary step for mitogen-activated protein kinase kinase-induced skin tumorigenesis. Cancer Res. 2005;65:572–8. [PubMed] [Google Scholar]
- She Q-B, Chen N, Bode AM, Flavell RA, Dong Z. Deficiency of c-Jun-NH2-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002;62:1343–8. [PubMed] [Google Scholar]
- Choi HS, Bode AM, Shim J-H, Lee S-Y, Dong Z. c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer. Mol Cell Biol. 2009;29:2168–80. doi: 10.1128/MCB.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Nomura M, She Q-B, et al. Suppression of skin tumorigenesis in c-Jun NH2-terminal kinase-2-deficient mice. Cancer Res. 2001;61:3908–12. [PubMed] [Google Scholar]
- Ehrenreiter K, Kern F, Velamoor V, et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16:149–60. doi: 10.1016/j.ccr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Dickinson SE, Olson ER, Zhang J, et al. p38 MAP kinase plays a functional role in UVB-induced mouse skin carcinogenesis. Mol Carcinog. 2011;50:469–78. doi: 10.1002/mc.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Yu D, Cho Y-Y, et al. Sunlight UV-induced skin cancer relies upon activation of the p38α signaling pathway. Cancer Res. 2013;73:2181–8. doi: 10.1158/0008-5472.CAN-12-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, Tenbaum S, Perdiguero E, et al. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet. 2007;39:750–8. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- Ahn Y-H, Yang Y, Gibbons DL, et al. Map2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor γ2 expression. Mol Cell Biol. 2011;31:4270–85. doi: 10.1128/MCB.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek D, Kotsinas A, Meixner A, et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet. 2011;43:212–9. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- Blasco RB, Francoz S, Santamaría D, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–63. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-RasG12D. Cancer Discov. 2011;1:128–36. doi: 10.1158/2159-8290.CD-10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff E, Fedorov LM, Siefken R, Walter AO, Papadopoulos T, Rapp UR. Lung-targeted expression of the c-Raf-1 kinase in transgenic mice exposes a novel oncogenic character of the wild-type protein. Cell Growth Differ. 2000;11:185–90. [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wang Z, Perera SA, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67:4933–9. doi: 10.1158/0008-5472.CAN-06-4592. [DOI] [PubMed] [Google Scholar]
- Gkirtzimanaki K, Gkouskou KK, Oleksiewicz U, et al. TPL2 kinase is a suppressor of lung carcinogenesis. Proc Natl Acad Sci USA. 2013;110:E1470–9. doi: 10.1073/pnas.1215938110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellurale C, Weston CR, Reilly J, et al. Role of JNK in a Trp53-dependent mouse model of breast cancer. PLoS ONE. 2010;5:e12469. doi: 10.1371/journal.pone.0012469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, O’Neal JF, Ebelt ND, et al. Jnk2 effects on tumor development, genetic instability and replicative stress in an oncogene-driven mouse mammary tumor model. PLoS ONE. 2010;5:e10443. doi: 10.1371/journal.pone.0010443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- Bertheau P, Lehmann-Che J, Varna M, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22(Suppl 2):S27–9. doi: 10.1016/j.breast.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Näthke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer. 2006;6:967–74. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- Serebrennikova OB, Tsatsanis C, Mao C, et al. Tpl2 ablation promotes intestinal inflammation and tumorigenesis in Apcmin mice by inhibiting IL-10 secretion and regulatory T-cell generation. Proc Natl Acad Sci USA. 2012;109:E1082–91. doi: 10.1073/pnas.1115098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Pohl NM, Yin Z, Yang W. Loss of JNK2 increases intestinal tumor susceptibility in Apc1638+/− mice with dietary modulation. Carcinogenesis. 2011;32:584–8. doi: 10.1093/carcin/bgq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeman D, Schneider JE, Liu J, et al. Deletion of p38-alpha mitogen-activated protein kinase within the intestinal epithelium promotes colon tumorigenesis. Surgery. 2012;152:286–93. doi: 10.1016/j.surg.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta J, del Barco Barrantes I, Igea A, et al. Dual function of p38α MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell. 2014;25:484–500. doi: 10.1016/j.ccr.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Reino Pd, Alsina-Beauchamp D, Escós A, et al. Pro-oncogenic role of alternative p38 mitogen-activated protein kinases p38γ and p38δ linking inflammation and cancer in colitis-associated colon cancer. Cancer Res. 2014;74:6150–60. doi: 10.1158/0008-5472.CAN-14-0870. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Hirata Y, Nakagawa H, et al. Apoptosis signal-regulating kinase 1 regulates colitis and colitis-associated tumorigenesis by the innate immune responses. Gastroenterology. 2010;138:1055–67. doi: 10.1053/j.gastro.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Sancho R, Nateri AS, de Vinuesa AG, et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843–54. doi: 10.1038/emboj.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Yin Z, Song Z, et al. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am J Pathol. 2007;171:297–303. doi: 10.2353/ajpath.2007.061036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci USA. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–53. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25:634–45. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, He G, Matsuzawa A, et al. Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Kudo M, Umemura A, et al. p38α inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 2013;73:215–24. doi: 10.1158/0008-5472.CAN-12-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Bakiri L, Mairhorfer A, et al. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–9. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Hirata Y, Takeda K, et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology. 2011;54:185–95. doi: 10.1002/hep.24357. [DOI] [PubMed] [Google Scholar]
- Inokuchi S, Aoyama T, Miura K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci USA. 2010;107:844–9. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Inokuchi S, Roh YS, et al. Transforming growth factor–β signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte-specific deletion of TAK1. Gastroenterology. 2013;144:1042–54. doi: 10.1053/j.gastro.2013.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettermann K, Vucur M, Haybaeck J, et al. TAK1 suppresses a NEMO-dependent but NF-κB-independent pathway to liver cancer. Cancer Cell. 2010;17:481–96. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Shibata W, Maeda S, Hikiba Y, et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008;68:5031–9. doi: 10.1158/0008-5472.CAN-07-6332. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Hirata Y, Nakagawa H, et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc Natl Acad Sci USA. 2011;108:780–5. doi: 10.1073/pnas.1011418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Hirata Y, Sakitani K, et al. Apoptosis signal-regulating kinase-1 inhibitor as a potent therapeutic drug for the treatment of gastric cancer. Cancer Sci. 2012;103:2181–5. doi: 10.1111/cas.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol. 2009;183:8099–109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- Sobczak I, Galabova-Kovacs G, Sadzak I, Kren A, Christofori G, Baccarini M. B-Raf is required for ERK activation and tumor progression in a mouse model of pancreatic β-cell carcinogenesis. Oncogene. 2008;27:4779–87. doi: 10.1038/onc.2008.128. [DOI] [PubMed] [Google Scholar]
- Davies CC, Harvey E, McMahon RFT, et al. Impaired JNK signaling cooperates with KrasG12D expression to accelerate pancreatic ductal adenocarcinoma. Cancer Res. 2014;74:3344–56. doi: 10.1158/0008-5472.CAN-13-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceci JD, Patriotis CP, Tsatsanis C, et al. Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev. 1997;11:688–700. doi: 10.1101/gad.11.6.688. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C, Vaporidi K, Zacharioudaki V, et al. Tpl2 and ERK transduce antiproliferative T cell receptor signals and inhibit transformation of chronically stimulated T cells. Proc Natl Acad Sci USA. 2008;105:2987–92. doi: 10.1073/pnas.0708381104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamothe B, Lai Y, Hur L, et al. Deletion of TAK1 in the myeloid lineage results in the spontaneous development of myelomonocytic leukemia in mice. PLoS ONE. 2012;7:e51228. doi: 10.1371/journal.pone.0051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer K, Giblett S, Green S, et al. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 2005;65:11493–500. doi: 10.1158/0008-5472.CAN-05-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard C, Carragher L, Aldridge V, et al. Mouse models for BRAF-induced cancers. Biochem Soc Trans. 2007;35:1329–1333. doi: 10.1042/BST0351329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf JA, Ma X, Smith EP, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005;65:4238–45. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- Jeong JH, Wang Z, Guimaraes AS, et al. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS ONE. 2008;3:e3949. doi: 10.1371/journal.pone.0003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Cuevas BD, Winter-Vann AM, Johnson NL, Johnson GL. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene. 2006;25:4998–5010. doi: 10.1038/sj.onc.1209507. [DOI] [PubMed] [Google Scholar]
- Goel VK, Ibrahim N, Jiang G, et al. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene. 2009;28:2289–98. doi: 10.1038/onc.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–23. doi: 10.1093/carcin/bgh261. [DOI] [PubMed] [Google Scholar]
- Gao Y, Tao J, Li MO, et al. JNK1 is essential for CD8+ T cell-mediated tumor immune surveillance. J Immunol. 2005;175:5783–9. doi: 10.4049/jimmunol.175.9.5783. [DOI] [PubMed] [Google Scholar]
- Nasrazadani A, Van Den Berg CL. c-Jun N-terminal kinase 2 regulates multiple receptor tyrosine kinase pathways in mouse mammary tumor growth and metastasis. Genes Cancer. 2011;2:31–45. doi: 10.1177/1947601911400901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay LJ, Felding-Habermann B. Platelets alter tumor cell attributes to propel metastasis: programming in transit. Cancer Cell. 2011;20:553–4. doi: 10.1016/j.ccr.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Reymond N, Borda dÁB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–70. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Amano S, Furuya M, et al. Involvement of p38α mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem. 2006;281:36767–75. doi: 10.1074/jbc.M604371200. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]