

Abstract
Steady-state hematopoiesis responds to extracellular stimuli to meet changing demands and also to pathologically altered intracellular signaling. Granulocyte production increases following infection or in response to cytokine stimulation, and activation of the CCAAT/enhancer-binding protein β (C/EBPβ) transcription factor is required for such stress-induced granulopoiesis, whereas C/EBPα plays a critical role in maintaining steady-state granulopoiesis. Different roles of these C/EBP transcription factors in different modes of hematopoiesis are evolutionally conserved from zebrafish to humans. In addition to reactions against infections, C/EBPβ is responsible for cancer-driven myelopoiesis, which promotes cancer progression, at least in part, by abrogating the immune response in the cancer microenvironment. The BCR–ABL fusion protein activates emergency-specific pathway of granulopoiesis by upregulating C/EBPβ. This in turn causes chronic phase chronic myeloid leukemia, which is characterized by myeloid expansion. The C/EBPβ transcription factor also plays a role in other hematological malignancies of both myeloid and lymphoid lineage origin. Thus, elucidation of the upstream and downstream networks surrounding C/EBPβ will lead to the development of novel therapeutic strategies for diseases mediated by non-steady-state hematopoiesis.
Keywords: Cancer, C/EBPβ, emergency, hematological malignancy, steady-state
Transcription Factor CCAAT/Enhancer Binding Protein β
CCAAT/Enhancer Binding Protein β (C/EBPβ) belongs to the C/EBP leucine zipper domain-containing family of transcription factors (Fig.1).1,2 This intronless gene product binds to certain genomic regulatory regions either as a homodimer or as a heterodimer with other molecules, including other members of the C/EBP family. In addition to direct DNA binding, C/EBPβ cooperates with the switch/sucrose non-fermentable complex to regulate gene expression through chromatin remodeling.3 It induces or represses the expression of target genes and, ultimately, regulates the proliferation, differentiation, metabolism, and survival of many different cell types.1
Fig 1.
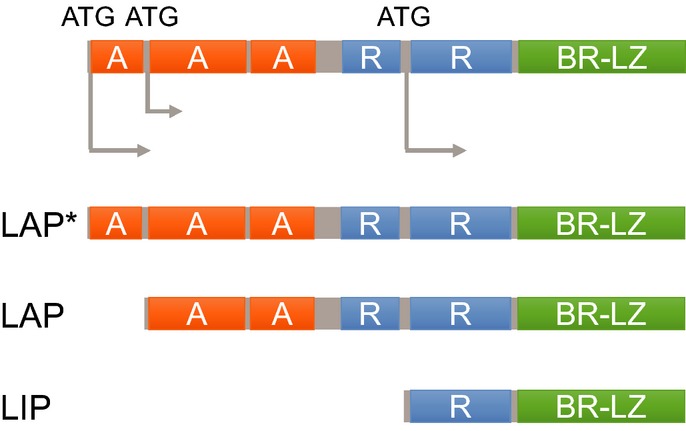
Schematic illustration of the C/EBPβ transcription factor and its isoforms. A, transactivating domain; BR-LZ, basic region–leucine zipper domain; LAP$, liver-enriched activating protein$; LAP, liver-enriched activating protein; LIP, liver-enriched inhibitory protein; R, repression domain.
The expression and function of C/EBPβ are regulated in a complex way during transcription, translation, post-translational modification, and protein–protein interactions.4–8 Notably, alternative translation through the use of different initiation codons generates three different isoforms of C/EBPβ: liver-enriched activating protein$ (LAP$ or full-length), liver-enriched activating protein (LAP), and liver-enriched inhibitory protein (LIP) (Fig.1).6 Both LAP$ and LAP are transcriptional activators, whereas LIP (which is the shortest isoform and lacks transactivation domains but retains DNA binding and dimerization domains) acts as a repressor or a dominant negative inhibitor of other C/EBP family transcription factors.9 The ratio of these isoforms is regulated by different signaling events and has a significant impact on the overall function of C/EBPβ.10,11
Within the hematopoietic system, C/EBPβ is expressed at high levels by monocytes and macrophages, and regulates genes involved in immune and inflammatory responses.12–16 In addition, we found that C/EBPβ plays a crucial role in hematopoiesis, especially under stress conditions.17–19 Here, we discuss the role of this transcription factor in non-steady-state hematopoiesis, including the emergency response to infection and cancer, and in hematological malignancies.
Modes of Hematopoiesis
Hematopoiesis is a continuous process that supplies an organism with all blood cells over its lifetime. To avoid either an excess or lack of any specific type of blood cell, hematopoiesis must be tightly regulated according to demand. During steady-state conditions, the constant production of mature blood cells is maintained by fine-tuning the proliferation and differentiation of hematopoietic precursors in both a cell-intrinsic and a cell-extrinsic manner. By contrast, in emergency situations such as infection or bleeding, large numbers of functionally mature cells are required. These increased demands must be met by an immediate increase in the production or release of specific cell types (Fig.2).20 These physiological non-steady-state responses are triggered by external stimuli and are resolved when the activating signals cease. In addition to responses to infection or bleeding, hematopoietic stress can be elicited by various kinds of pro-inflammatory disease, including cancer and autoimmune diseases.21–24 At the molecular level, steady-state-specific regulatory mechanisms are thought to be modulated in response to external stimuli. The shift from steady-state to emergency hematopoiesis and vice versa is a continuous process, and the extent of the shift is dependent on the type, strength, and duration of the stimuli and/or signals.20,25 It is difficult to determine clear boundaries between steady-state and emergency hematopoiesis, partly because the shift between these modes may be an extension of the fine-tuning of steady-state hematopoiesis mediated by co-operation between steady-state-specific and emergency-specific signals. Recent studies report the involvement of inflammatory signals in both the development and ageing of hematopoiesis.26–28 Therefore, it is necessary to identify the emergency-specific signals if we are to fully understand the mechanisms that fine-tune hematopoiesis in general.
Fig 2.
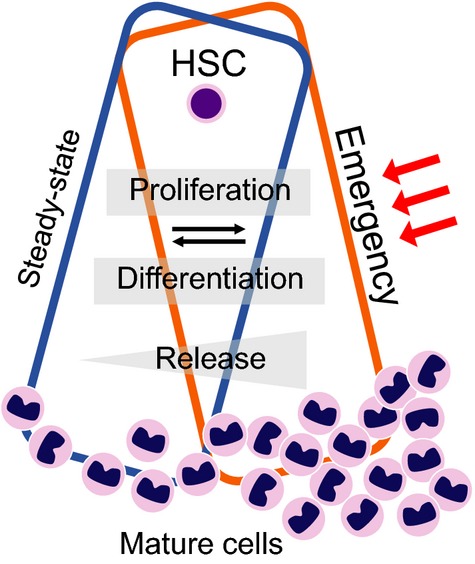
Modes of hematopoiesis. The hematopoietic system in the bone marrow supplies mature blood cells on demand. Under both steady-state and emergency conditions, overlapping and distinct signals ensure the adequate production and release of mature cells. Red arrows indicate extracellular stimuli, including infection and cancer. HSC, hematopoietic stem cells.
Emergency Granulopoiesis
Neutrophilic granulocytes (granulocytes) are recruited to the frontline of infection, where they expel their granular contents to fight microbes.20,25 In the bone marrow, hematopoietic stem cells give rise to mature granulocytes through successive intermediates, such as common myeloid progenitors and granulocyte–macrophage progenitors.29 Granulocytes have an extremely short half-life; therefore, they must be produced continuously in the bone marrow, stored, and supplied to the periphery. As is the cases with other hematopoietic lineages, either an excess or a lack of granulocytes is harmful to the host; therefore, granulopoiesis must be tightly regulated according to demand. It is well known that C/EBPα plays critical roles in granulopoiesis. In Cebpa-deficient mice, transition from common myeloid progenitors to granulocyte–macrophage progenitors is completely abrogated and no granulocytes are present under steady-state conditions.17,30,31 Overexpression of C/EBPα represses the proliferation of leukemic cells and induces their differentiation into granulocytes.17,32 Collectively, these findings suggest that C/EBPα is the master regulator of steady-state granulopoiesis.
While searching for the regulatory mechanisms involved in emergency granulopoiesis, we found that granulopoiesis can be induced by cytokines in the absence of C/EBPα. This suggests the existence of a C/EBPα-independent pathway of granulopoiesis under emergency conditions.17 Interestingly, all members of the C/EBP family, except C/EBPβ, were downregulated in response to cytokine stimulation. Cytokine- or infection-induced enhancement of granulopoiesis is impaired in Cebpb knockout mice, and the C/EBPα-independent pathway of granulopoiesis is significantly attenuated by inhibiting C/EBPβ.17 By contrast, C/EBPβ is not necessary for steady-state granulopoiesis. These results clearly suggest that C/EBPβ is required for stress-induced granulopoiesis; indeed, this requirement has been verified in other mouse models and in a zebrafish model.33–35 Both C/EBPβ and C/EBPα share many common target molecules, including genes associated with granulocytic differentiation.36 By contrast, they show a differing ability to regulate the cell cycle. C/EBPα strongly inhibits the cell cycle through direct or indirect interactions with cell cycle regulators,37–39 whereas C/EBPβ has a less inhibitory effect.17,40 These differences might be the reason for the selective requirement of C/EBPα and C/EBPβ for steady-state and emergency granulopoiesis, respectively. As the transition from steady-state to emergency granulopoiesis (or vice versa) is a continuous process, C/EBPα or C/EBPβ might collaborate with each other to ensure an adequate supply of granulocytes by fine-tuning the proliferation and differentiation of granulocyte precursors (Fig.3). Furthermore, we also found that CEBPβ is required by early granulocyte precursors under emergency conditions; we are currently investigating the role of CEBPβ in regulating hematopoietic stem cells.18
Fig 3.
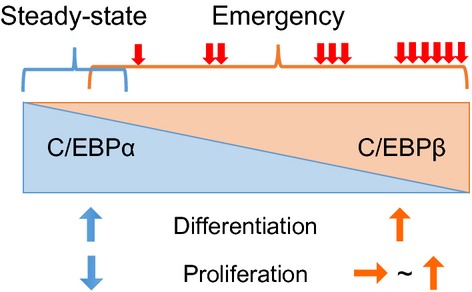
Role of C/EBP transcription factors in steady-state and emergency granulopoiesis. Activation of the C/EBPβ transcription factor is required to generate increased numbers of granulocytes under emergency conditions, such as severe infection or cytokine exposure, whereas C/EBPα plays a critical role under steady-state conditions. C/EBPβ and C/EBPα share many common target molecules, including genes associated with granulocytic differentiation. By contrast, they show a differing ability to regulate the cell cycle. C/EBPα strongly inhibits the cell cycle, whereas C/EBPβ has a less inhibitory effect. Red arrows indicate extracellular stimuli that activate C/EBPβ.
Role of C/EBPs in the Pathophysiology of Severe Congenital Neutropenia
Severe congenital neutropenia (SCN) is an inherited condition characterized by severe neutropenia in the peripheral blood (<500/μL) and by arrest of myeloid precursor maturation at the promyelocyte/myelocyte stage in the bone marrow, resulting in increased vulnerability to bacterial and fungal infections.41,42 The majority of patients with SCN respond to treatment with recombinant granulocyte-colony stimulating factor, which increases the neutrophil count and reduces both the frequency and severity of infections. Patients with SCN harbor mutations in diverse genes. These heterogeneous genetic alterations reflect the complex mechanisms governing the homeostasis of neutrophils.42 Establishing induced pluripotent stem cells from SCN cells in combination with an in vitro differentiation system will further our understanding of both the pathogenesis of this disease and the physiological regulation of granulopoiesis.43,44 The majority of SCN patients harbor mutations in ELANE and HAX1 (approximately 60% and 10%, respectively).45 Recently, the lymphoid enhancer-binding factor 1 (LEF-1) transcription factor was identified as a common factor responsible for defective granulopoiesis in SCN patients with mutations in ELANE (ELA2) or HAX1.46 LEF-1 regulates C/EBPα during granulopoiesis. Both the expression and function of LEF-1 and C/EBPα are severely reduced in myeloid precursors in SCN patients with ELANE or HAX1 mutations, and the reduction in C/EBPα (the master regulator of steady-state granulopoiesis) might be a critical mechanism underlying neutropenia in SCN.46,47 Maturation arrest can be overcome by treatment with granulocyte-colony stimulating factor, presumably because it activates the C/EBPβ-mediated pathway of granulopoiesis, which is thought to be intact in SCN patients.48 The new insights into the pathophysiology of SCN suggest that the different roles of C/EBPα and C/EBPβ during granulopoiesis may also be true in humans.
Cancer-associated Myelopoiesis
Cancer progression, including tumor growth, invasion, and metastasis, cannot be achieved by tumor cells alone; it requires the appropriate microenvironment.49,50 Accumulating evidence suggests that myeloid cells are major components of the cancer microenvironment.21–23 Indeed, there is a strong association between increased numbers of macrophages or neutrophils in cancer tissues and poor patient survival.51,52 Thus, these myeloid cells can be good candidate therapeutic targets.
Tumor cells, or other stromal cells, in the microenvironment produce a variety of growth factors and chemokines, which then recruit myeloid cells from the bone marrow or reservoir tissues.21–23 Therefore, the mode of hematopoiesis is altered in the presence of cancer, and hematopoietic systems release a variety of myeloid cells into the cancer microenvironment. Such cells include monocytes, macrophages (tumor-associated macrophages), dendritic cells, neutrophils (tumor-associated neutrophils), and eosinophils. Recent studies by ourselves and others identified fibrocytes as important constituents of the cancer microenvironment.53–55 In such microenvironments, myeloid cells support cancer progression by secreting growth factors and promoting angiogenesis and/or tissue remodeling. In addition, it is widely accepted that a special subset of myeloid cells, called myeloid-derived suppressor cells (MDSCs), are induced by tumor-induced factors and are responsible for immune dysfunction.22,56 MDSCs in mice are classified as either monocytic or granulocytic based on their surface expression of Ly6C and Ly6G, respectively. Upregulated in the bone marrow of tumor-bearing hosts, C/EBPβ regulates the expression of enzymes such as arginase and inducible nitric oxide synthase, both of which are required for the lymphocyte-inhibitory activities of MDSCs (Fig.4a).57 Accordingly, in tumor-bearing mice, both the emergence and the immunosuppressive function of MDSCs are severely abrogated in the absence of C/EBPβ, resulting in attenuated tumor spread.57 A similar relationship between C/EBPβ and cancer-driven myelopoiesis is observed in humans.58 These findings suggest that C/EBPβ plays a critically important role in cancer-induced inflammation; thus, C/EBPβ may be a therapeutic target for regulating the cancer microenvironment. Further studies should examine the roles of C/EBPβ in generating or regulating the function of MDSCs in other diseases.
Fig 4.

Involvement of C/EBPβ in non-infectious modes of hematopoiesis. (a) C/EBPβ is upregulated in the bone marrow of tumor-bearing hosts. C/EBPβ regulates the differentiation of myeloid-derived myeloid suppressor cells (MDSCs) and the expression of enzymes such as arginase and inducible nitric oxide synthase (iNOS), both of which are required for the lymphocyte-inhibitory activities of MDSCs. (b) In chronic phase chronic myeloid leukemia (CML), C/EBPβ is activated by signal transducer and activator of transcription 5 (STAT5), which is located downstream of BCR–ABL. C/EBPβ is involved in BCR–ABL-mediated myeloid expansion and leukemic stem cell exhaustion in chronic phase CML. (c) Acute promyelocytic leukemia (APL) is characterized by a promyelocytic leukemia-retinoic acid receptor α(PML-RARa)-mediated differentiation block at the promyelocyte stage. During the processes of differentiation-inducing therapy using all trans retinoic acid (ATRA), C/EBPβ is upregulated in the presence of PML-RARα and increases the number of neutrophils derived from APL cells by promoting their proliferation and differentiation.
Role of CEBPβ in Chronic Myeloid Leukemia
Chronic phase chronic myeloid leukemia (CP-CML) is characterized by a massive expansion of myeloid cells.59 In sharp contrast to acute myeloid leukemia (AML) with leukemic hiatus, both myeloid progenitors and mature granulocytes accumulate in the bone marrow, peripheral blood, and spleen in CP-CML. The myeloid expansion in CP-CML is attributed to the BCR–ABL fusion protein, which arises from a translocation between chromosomes 9 and 22.59 The leukocytosis observed in patients with infections, severe burns, or cancer is sometimes referred to as a “leukemoid” reaction because of the marked increase in the number of myeloid cells with a “left shift” in the shape of the nucleus. The resemblance between leukemoid reactions and CP-CML prompted us to examine whether BCR–ABL might use the emergency-specific pathway of granulopoiesis. Therefore, we investigated the role of C/EBPβ in CP-CML (Fig.4b). BCR–ABL upregulates C/EBPβ, at least in part, by activating signal transducer and activator of transcription 5.19 Myeloid differentiation and proliferation (induced by BCR–ABL) are significantly impaired in Cebpb-deficient bone marrow cells both in vitro and in vivo.19 Interestingly, higher numbers of Cebpb-deficient leukemic stem cells were maintained after serial transplantation than wild-type leukemic stem cells in this mouse model.19 These results suggest that C/EBPβ is involved in BCR–ABL-mediated myeloid expansion and leukemic stem cell exhaustion in CP-CML. Consistent with our observations, C/EBPβ is markedly upregulated in a pluripotent hematopoietic cell line transduced with BCR–ABL.60 By contrast, downregulation of C/EBPβ is associated with progression of CML toward a blast crisis.61 Changes in the BCR–ABL-mediated regulation of C/EBPβ during the progression of CML may be a consequence of genetic or epigenetic changes. Isoforms of C/EBPβ involved in the pathogenesis of CML remain to be identified. Further identification of the molecular mechanisms underlying the regulation of C/EBPβ and C/EBPβ-mediated leukemic stem cell exhaustion might lead to novel therapeutic strategies for eradicating CML stem cells.
Role of CEBPβ in other Hematological Malignancies
Hematological malignancies are the consequence of dysregulated differentiation and/or proliferation; therefore, they can be regarded as a form of pathologically induced non-steady-state hematopoiesis. Because C/EBPα promotes neutrophilic differentiation and inhibits the cell cycle, many cases of AML are associated with recurrent mutations in, or dysregulation of, C/EBPα.62–64 By contrast, no recurrent mutations in C/EBPβ have been identified in AML,65 possibly reflecting the fact that this transcription factor is required for emergency-specific responses. However, C/EBPβ plays a role in the pathogenesis of many hematological malignancies. In AML, LIP (the shortest isoform of C/EBPβ) collaborates with a proto-oncogene, Evi1, to induce leukemia in a mouse bone marrow transplantation model.66 The same isoform is induced by signaling downstream of internal tandem duplication of fms-like tyrosine kinase 3, thereby supporting the proliferation of blasts.67 These findings suggest that regulating the amount or the ratio of C/EBPβ isoforms might be a common pathway that is abrogated during the development of AML.
Acute promyelocytic leukemia (APL) is a subtype of AML characterized by a promyelocytic leukemia-retinoic acid receptor α(PML-RARa)-mediated differentiation block at the promyelocyte stage, which occurs (at least in part) through an impairment in C/EBPα function.68 This block is reversed by all-trans retinoic acid (ATRA), which is used as frontline therapy for APL.69 After the start of ATRA treatment, mature neutrophil-like cells originate from leukemic promyelocytes and their numbers increase in the bone marrow and peripheral blood of responder APL cases. During this process of differentiation-inducing therapy, C/EBPβ is upregulated in the presence of PML-RARα and increases the number of neutrophils derived from APL cells by promoting their proliferation and differentiation (Fig.4c).70 In other words, C/EBPβ is an ATRA-dependent PML-RARα target gene in APL cells.
It is clear that C/EBPβ regulates not only myeloid hematopoiesis, but also bone marrow B lymphopoiesis, in both a cell-intrinsic and cell-extrinsic manner.71,72 One study examined the contribution of C/EBPβ to the development of lymphoid neoplasias in cases with acute B-cell precursor leukemia and identified recurrent translocations in C/EBPβ, which resulted in the upregulation of C/EBPβ.73
Anaplastic large cell lymphoma (ALCL) is a subset of non-Hodgkin’s lymphoma characterized by unique cell morphology and expression of CD30.74 In ALCL cells, the anaplastic lymphoma kinase (ALK) gene is frequently fused to the nucleophosmin (NPM) gene, and the resulting ALK activity is the central driver for the survival of ALCL cells. Recently, C/EBPβ was identified as a downstream target of ALK-mediated signaling.74 C/EBPβ is upregulated in the presence of activated ALK through signal transducer and activator of transcription 374,75 or by post-transcriptional regulation,76 whereupon it contributes to the transformation and survival of ALCL cells.77
The pathogenesis of multiple myeloma remains unclear and, at present, this plasma cell disorder is incurable. A recent report shows that C/EBPβ is overexpressed in myeloma cells and is involved in regulating several transcription factors, including IRF4, XBP1, and BLIMP1, all of which are critical for the proliferation and survival of myeloma cells.78 Inhibiting C/EBPβ translation in myeloma cells using immunomodulatory derivatives of thalidomide has been proposed as a novel therapeutic strategy for multiple myeloma.79
Conclusions
The expression and/or function of C/EBPβ are upregulated in the hematopoietic system in response to various kinds of cell-extrinsic stress, including infections and cancer. This upregulation increases the supply of myeloid cells. Dysregulation of C/EBPβ is observed in several hematological malignancies, resulting in the maintenance or progression of disease. Although the roles of C/EBPβ in hematopoiesis have not been fully elucidated, it appears to play a key role in non-steady-state hematopoiesis, including hematological malignancies, and hematopoiesis in host with cancers in addition to hematopoietic responses against infections (Fig.5). Even though direct targeting of this transcription factor might be technically difficult, identifying the upstream and downstream networks involving C/EBPβ will lead to a better understanding of the pathogenesis and pathophysiology of diseases mediated by non-steady-state hematopoiesis.
Fig 5.
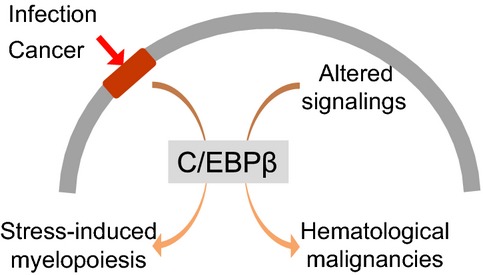
Role of C/EBPβ and non-steady-state hematopoiesis. Different types of cell-extrinsic stress, including infections and cancer, activate C/EBPβ to increase the supply of functionally mature myeloid cells or myeloid-derived suppressor cells. Dysregulation of C/EBPβ is observed in some hematological malignancies, resulting in maintenance or progression of disease.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to H.H., A.Y., and T.M.), a grant from the Project for Development of Innovative Research on Cancer Therapeutics from MEXT (to H.H. and T.M.), a Grant-in-Aid from the Ministry of Health, Labour and Welfare in Japan (to T.M.), the National Cancer Center Research and Development Fund (to T.M.), the Takeda Science Foundation (to H.H.), a research grant from the Princess Takamatsu Cancer Research Fund (to T.M.), and the Kobayashi Foundation for Cancer Research (to T.M.).
Disclosure Statement
The authors have no conflict of interest.
References
- Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–75. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17:318–24. doi: 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Kowenz-Leutz E, Leutz A. A C/EBP beta isoform recruits the SWI/SNF complex to activate myeloid genes. Mol Cell. 1999;4:735–43. doi: 10.1016/s1097-2765(00)80384-6. [DOI] [PubMed] [Google Scholar]
- Ruffell D, Mourkioti F, Gambardella A, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci USA. 2009;106:17475–80. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Kamio N, Huang G, et al. Cyclic AMP responsive element binding proteins are involved in ‘emergency’ granulopoiesis through the upregulation of CCAAT/enhancer binding protein beta. PLoS One. 2013;8:e54862. doi: 10.1371/journal.pone.0054862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–79. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- Trautwein C, Caelles C, van der Geer P, Hunter T, Karin M, Chojkier M. Transactivation by NF-IL6/LAP is enhanced by phosphorylation of its activation domain. Nature. 1993;364:544–7. doi: 10.1038/364544a0. [DOI] [PubMed] [Google Scholar]
- Bradley MN, Zhou L, Smale ST. C/EBPbeta regulation in lipopolysaccharide-stimulated macrophages. Mol Cell Biol. 2003;23:4841–58. doi: 10.1128/MCB.23.14.4841-4858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descombes P, Chojkier M, Lichtsteiner S, Falvey E, Schibler U. LAP, a novel member of the C/EBP gene family, encodes a liver-enriched transcriptional activator protein. Genes Dev. 1990;4:1541–51. doi: 10.1101/gad.4.9.1541. [DOI] [PubMed] [Google Scholar]
- An MR, Hsieh CC, Reisner PD, et al. Evidence for posttranscriptional regulation of C/EBPalpha and C/EBPbeta isoform expression during the lipopolysaccharide-mediated acute-phase response. Mol Cell Biol. 1996;16:2295–306. doi: 10.1128/mcb.16.5.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilova B, Kowenz-Leutz E, Scheller M, Leutz A. Lymphoid to myeloid cell trans-differentiation is determined by C/EBPbeta structure and post-translational modifications. PLoS One. 2013;8:e65169. doi: 10.1371/journal.pone.0065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuka S, Akira S, Nishio Y, et al. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood. 1992;79:460–6. [PubMed] [Google Scholar]
- Tanaka T, Akira S, Yoshida K, et al. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–61. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- Screpanti I, Romani L, Musiani P, et al. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J. 1995;14:1932–41. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Pietsch D, Panterodt T, Brand K. Regulation of C/EBPbeta and resulting functions in cells of the monocytic lineage. Cell Signal. 2012;24:1287–96. doi: 10.1016/j.cellsig.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Cain DW, O’Koren EG, Kan MJ, et al. Identification of a tissue-specific, C/EBPbeta-dependent pathway of differentiation for murine peritoneal macrophages. J Immunol. 2013;191:4665–75. doi: 10.4049/jimmunol.1300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Zhang P, Dayaram T, et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–9. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- Satake S, Hirai H, Hayashi Y, et al. C/EBPbeta is involved in the amplification of early granulocyte precursors during candidemia-induced “emergency” granulopoiesis. J Immunol. 2012;189:4546–55. doi: 10.4049/jimmunol.1103007. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Hirai H, Kamio N, et al. C/EBPbeta promotes BCR-ABL-mediated myeloid expansion and leukemic stem cell exhaustion. Leukemia. 2013;27:619–28. doi: 10.1038/leu.2012.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–14. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Etzrodt M, Newton A, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. 2012;109:2491–6. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii W, Ashihara E, Hirai H, et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J Immunol. 2013;191:1073–81. doi: 10.4049/jimmunol.1203535. [DOI] [PubMed] [Google Scholar]
- Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–11. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- Li Y, Esain V, Teng L, et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014;28:2597–612. doi: 10.1101/gad.253302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Zhang C, Wang L, et al. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood. 2015;125:1098–106. doi: 10.1182/blood-2014-09-601542. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Iwasaki-Arai J, Iwasaki H, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21:853–63. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Saitoh T, O’Kelly J, Akira S, Gombart AF, Koeffler HP. Impaired response to GM-CSF and G-CSF, and enhanced apoptosis in C/EBPbeta-deficient hematopoietic cells. Blood. 2008;111:2999–3004. doi: 10.1182/blood-2007-04-087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–71. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CJ, Flores MV, Oehlers SH, et al. Infection-responsive expansion of the hematopoietic stem and progenitor cell compartment in zebrafish is dependent upon inducible nitric oxide. Cell Stem Cell. 2012;10:198–209. doi: 10.1016/j.stem.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Jones LC, Lin ML, Chen SS, et al. Expression of C/EBPbeta from the C/ebpalpha gene locus is sufficient for normal hematopoiesis in vivo. Blood. 2002;99:2032–6. doi: 10.1182/blood.v99.6.2032. [DOI] [PubMed] [Google Scholar]
- Porse BT, Pedersen TA, Xu XF, et al. E2F repression by C/EBP alpha is required for adipogenesis and granulopoiesis in vivo. Cell. 2001;107:247–58. doi: 10.1016/s0092-8674(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Wang HM, Iakova P, Wilde M, et al. C/EBP alpha arrests cell proliferation through direct inhibition of cdk2 and cdk4. Mol Cell. 2001;8:817–28. doi: 10.1016/s1097-2765(01)00366-5. [DOI] [PubMed] [Google Scholar]
- Johansen LM, Iwama A, Lodie TA, et al. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol. 2001;21:3789–806. doi: 10.1128/MCB.21.11.3789-3806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian T, Johnson PF. Stop and go: anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle. 2006;5:953–7. doi: 10.4161/cc.5.9.2733. [DOI] [PubMed] [Google Scholar]
- Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011;6:26. doi: 10.1186/1750-1172-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C. Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol. 2011;29:399–413. doi: 10.1146/annurev-immunol-030409-101259. [DOI] [PubMed] [Google Scholar]
- Hiramoto T, Ebihara Y, Mizoguchi Y, et al. Wnt3a stimulates maturation of impaired neutrophils developed from severe congenital neutropenia patient-derived pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110:3023–8. doi: 10.1073/pnas.1217039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima T, Watanabe K, Niwa A, et al. Genetic correction of HAX1 in induced pluripotent stem cells from a patient with severe congenital neutropenia improves defective granulopoiesis. Haematologica. 2014;99:19–27. doi: 10.3324/haematol.2013.083873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler C, Germeshausen M, Klein C, Welte K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol. 2009;144:459–67. doi: 10.1111/j.1365-2141.2008.07425.x. [DOI] [PubMed] [Google Scholar]
- Skokowa J, Cario G, Uenalan M, et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med. 2006;12:1191–7. doi: 10.1038/nm1474. [DOI] [PubMed] [Google Scholar]
- Skokowa J, Klimiankou M, Klimenkova O, et al. Interactions among HCLS1, HAX1 and LEF-1 proteins are essential for G-CSF-triggered granulopoiesis. Nat Med. 2012;18:1550–9. doi: 10.1038/nm.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skokowa J, Lan D, Thakur BK, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 2009;15:151–8. doi: 10.1038/nm.1913. [DOI] [PubMed] [Google Scholar]
- Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 2010;29:243–8. doi: 10.1007/s10555-010-9227-2. [DOI] [PubMed] [Google Scholar]
- Taketo MM. Roles of stromal microenvironment in colon cancer progression. J Biochem. 2012;151:477–81. doi: 10.1093/jb/mvs035. [DOI] [PubMed] [Google Scholar]
- Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol. 2009;27:4709–17. doi: 10.1200/JCO.2008.18.9498. [DOI] [PubMed] [Google Scholar]
- Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–85. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Maric I, DiPrima MJ, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013;122:1105–13. doi: 10.1182/blood-2012-08-449413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deventer HW, Palmieri DA, Wu QP, McCook EC, Serody JS. Circulating fibrocytes prepare the lung for cancer metastasis by recruiting Ly-6C+ monocytes via CCL2. J Immunol. 2013;190:4861–7. doi: 10.4049/jimmunol.1202857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Fujishita T, Kurimoto K, et al. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin Exp Metastasis. 2014;31:977–89. doi: 10.1007/s10585-014-9684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo I, Bosio E, Solito S, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Lechner MG, Megiel C, Russell SM, et al. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J Transl Med. 2011;9:90. doi: 10.1186/1479-5876-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330–40. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- Minami Y, Stuart SA, Ikawa T, et al. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc Natl Acad Sci USA. 2008;105:17967–72. doi: 10.1073/pnas.0808303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerzoni C, Bardini M, Mariani SA, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood. 2006;107:4080–9. doi: 10.1182/blood-2005-08-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C. C/EBP alpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–28. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AD. C/EBPalpha in normal and malignant myelopoiesis. Int J Hematol. 2015;111:33041. doi: 10.1007/s12185-015-1764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegesna V, Takeuchi S, Hofmann WK, et al. C/EBP-beta, C/EBP-delta, PU.1, AML1 genes: mutational analysis in 381 samples of hematopoietic and solid malignancies. Leuk Res. 2002;26:451–7. doi: 10.1016/s0145-2126(01)00150-3. [DOI] [PubMed] [Google Scholar]
- Watanabe-Okochi N, Yoshimi A, Sato T, et al. The shortest isoform of C/EBPbeta, liver inhibitory protein (LIP), collaborates with Evi1 to induce AML in a mouse BMT model. Blood. 2013;121:4142–55. doi: 10.1182/blood-2011-07-368654. [DOI] [PubMed] [Google Scholar]
- Haas SC, Huber R, Gutsch R, et al. ITD- and FL-induced FLT3 signal transduction leads to increased C/EBPbeta-LIP expression and LIP/LAP ratio by different signalling modules. Br J Haematol. 2010;148:777–90. doi: 10.1111/j.1365-2141.2009.08012.x. [DOI] [PubMed] [Google Scholar]
- Guibal FC, Alberich-Jorda M, Hirai H, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. 2009;114:5415–25. doi: 10.1182/blood-2008-10-182071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhu H, Hu J, et al. Progress in the treatment of acute promyelocytic leukemia: optimization and obstruction. Int J Hematol. 2014;100:38–50. doi: 10.1007/s12185-014-1603-1. [DOI] [PubMed] [Google Scholar]
- Duprez E, Wagner K, Koch H, Tenen DG. C/EBPbeta: a major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003;22:5806–16. doi: 10.1093/emboj/cdg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu W, Ambrosino C, et al. Impaired generation of bone marrow B lymphocytes in mice deficient in C/EBPbeta. Blood. 1997;90:156–64. [PubMed] [Google Scholar]
- Yoshioka S, Miura Y, Yao H, et al. CCAAT/enhancer-binding protein beta expressed by bone marrow mesenchymal stromal cells regulates early B-cell lymphopoiesis. Stem Cells. 2014;32:730–40. doi: 10.1002/stem.1555. [DOI] [PubMed] [Google Scholar]
- Akasaka T, Balasas T, Russell LJ, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) Blood. 2007;109:3451–61. doi: 10.1182/blood-2006-08-041012. [DOI] [PubMed] [Google Scholar]
- Piva R, Pellegrino E, Mattioli M, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116:3171–82. doi: 10.1172/JCI29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasov N, Bonzheim I, Rudelius M, et al. C/EBPbeta expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica. 2010;95:760–7. doi: 10.3324/haematol.2009.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergalet J, Fawal M, Lopez C, et al. HuR-mediated control of C/EBPbeta mRNA stability and translation in ALK-positive anaplastic large cell lymphomas. Mol Cancer Res. 2011;9:485–96. doi: 10.1158/1541-7786.MCR-10-0351. [DOI] [PubMed] [Google Scholar]
- Bonzheim I, Irmler M, Klier-Richter M, et al. Identification of C/EBPbeta target genes in ALK+ anaplastic large cell lymphoma (ALCL) by gene expression profiling and chromatin immunoprecipitation. PLoS One. 2013;8:e64544. doi: 10.1371/journal.pone.0064544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Janz M, Galson DL, et al. C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood. 2009;114:3890–8. doi: 10.1182/blood-2009-01-201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Pal R, Monaghan SA, et al. IMiD immunomodulatory compounds block C/EBP{beta} translation through eIF4E down-regulation resulting in inhibition of MM. Blood. 2011;117:5157–65. doi: 10.1182/blood-2010-10-314278. [DOI] [PMC free article] [PubMed] [Google Scholar]