

Abstract
Osteosarcoma (OS) is the most frequent primary solid malignant tumor of bone. Its prognosis remains poor in the substantial proportion of patients who do not respond to chemotherapy and novel therapeutic options are therefore needed. We previously established a mouse model that mimics the aggressive behavior of human OS. Enzyme-linked immunosorbent assay-based screening of such mouse tumor lysates identified platelet-derived growth factor–BB (PDGF-BB) as an abundant soluble factor, the gene for which was expressed dominantly in surrounding non-malignant cells of the tumor, whereas that for the cognate receptor (PDGF receptor β) was highly expressed in OS cells. Platelet-derived growth factor-BB induced activation of both MEK–ERK and phosphatidylinositol 3-kinase–protein kinase B signaling pathways and promoted survival in OS cells deprived of serum, and these effects were blocked by the PDGF receptor inhibitor imatinib. However, these actions of PDGF-BB and imatinib were mostly masked in the presence of serum. Whereas imatinib alone did not manifest an antitumor effect in mice harboring OS tumors, combined treatment with imatinib and adriamycin exerted a synergistic antiproliferative effect on OS cells in vivo. These results suggest that treatment of OS with imatinib is effective only when cell survival is dependent on PDGF signaling or when imatinib is combined with another therapeutic intervention that renders the tumor cells susceptible to imatinib action, such as by inducing cellular stress.
Keywords: Animal model, drug resistance, imatinib, osteosarcoma, platelet-derived growth factor (PDGF) signaling
Osteosarcoma (OS), the most common primary malignant solid tumor of bone, is distinguished by its aggressive local growth at the primary site and systemic hematogenous dissemination at an early stage of the disease.1 Although chemotherapy regimens have improved the survival rate, 20–30% of patients are still refractory to the conventional treatment.2 Novel molecularly targeted therapies of more efficacy and less toxicity are thus needed.3,4 The development of such approaches has been hindered, however, by a lack of definitive insight into the molecular drivers of the aggressive behavior of OS.
We previously established a mouse OS model by c-MYC overexpression in bone marrow stromal cells from Ink4a/Arf knockout mice.5 Inoculating these AXT cells into syngeneic C57BL/6 mice results in lethal osteoblastic tumors accompanying osteoid formation and rapid systemic dissemination including metastasis to the lung, both of which reflect the pathology of human high-grade osteosarcoma. Not only cell intrinsic events, such as regulation of PPARγ,5 Imp3,6 and Twist27 gene expressions, but also environmental factors were found to contribute to the malignant phenotype.8–10
The tumor microenvironment is an important source of factors for tumor progression. Neighboring stromal cells and inflammatory cells provide cancer cells with soluble agents such as growth factors, cytokines, and chemokines that confer invasive and malignant properties.11,12 Characterization of the mechanisms by which the growth of OS is regulated by its microenvironment would embrace some possibility to provide a basis for novel treatments. To identify candidate growth-promoting molecules, we screened for soluble factors using our model system. There we found platelet-derived growth factor (PDGF)-BB as a factor whose concentration in a tumor lysate was greater than that in serum.
Platelet-derived growth factor signaling has been considered a potential target in cancer therapy for its ability to evoke tumorigenic cellular responses.13 Dysregulation of such signaling has been detected in various malignancies, including glioma, gastrointestinal stromal tumor (GIST), and dermatofibrosarcoma protuberans. Imatinib is a tyrosine kinase inhibitor targeting the BCR–ABL fusion protein as well as c-Kit and the PDGF receptor (PDGFR). Dermatofibrosarcoma protuberans is driven by constitutive expression of PDGFB, and imatinib has been approved as a first-line therapy for this tumor.14
In OS, although PDGF signaling has been implicated in its malignancy and inhibition of such signaling has been proven effective in some studies,15–19 imatinib has not yet achieved marked success in clinical treatment.20,21 This controversy has been left unsolved and it remains unclear how PDGF signaling contributes to OS pathogenesis or whether its attenuation is a rational treatment strategy. We have now examined the role of PDGF signaling in OS cells as well as the efficacy and validity of PDGFR-targeted therapy for this malignancy using our newly established mouse model.
Materials and Methods
Cell culture
AXT cells were established as previously described.5,6 Human OS cell lines SAOS2, SJSA1, and U2OS were obtained from ATCC (Manassas, VA, USA) and the human CML cell line K562 was from Japanese Collection of Research Bioresources (Osaka, Japan). AXT and other OS cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM) (Invitrogen, Carlsbad, CA, USA) and K562 cells were maintained in RPMI-1640 (Invitrogen), both of which were supplemented with FBS. Recombinant murine PDGF-BB was obtained from PeproTech (Rocky Hill, NJ, USA) and imatinib was from Cayman Chemical (Ann Arbor, MI, USA).
Quantitative analysis of PDGF-BB
Tumors and adjacent host tissue were suspended in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) and disrupted with a Biomasher (Nippi, Tokyo, Japan). The concentration of PDGF-BB in the tumor lysate was quantified with Bio-Plex Pro Mouse Cytokine 9-plex Assay G2 (Bio-Rad, Hercules, CA, USA).8
Cell proliferation assay
Proliferation of OS or CML cells in 96-well tissue culture plates was measured in triplicate with the use of a Cell Titer Glo cell proliferation assay kit (Promega, Madison, WI, USA).
Immunoblot analysis
Immunoblot analysis was carried out according to standard procedures. Primary antibodies to phosphorylated or total forms of ERK1/2, protein kinase B (Akt), and glycogen synthase kinase 3β were obtained from Cell Signaling Technology and those to α-tubulin and β-actin were from Sigma-Aldrich (St. Louis, MO, USA). For detection of PDGFRβ phosphorylation, cell lysates were subjected to immunoprecipitation with anti-PDGFRβ antibodies (Epitomics, Burlingame, CA, USA), and the immunoprecipitates were subjected to immunoblot analysis with antibodies to phospho-PDGFRβ (Epitomics).
Cell migration assay
Transwell membrane filter inserts with a pore size of 8 μm (BD Biosciences, San Jose, CA, USA) were placed in 24-well plates containing IMDM supplemented with 0.5% FBS. Cells were seeded inside the inserts in serum-free IMDM with or without PDGF-BB (200 ng/mL). After culture for 4 h, cells were fixed and stained as previously described.8 Stained cells were counted microscopically and values for four randomly chosen high-magnification (×400) fields of each membrane were summed. Each assay was carried out in triplicate.
Tumor xenograft model
Animal care and procedures were carried out in accordance with the guidelines of Keio University (Tokyo, Japan). AXT cells (1 × 106) were injected s.c. into syngeneic 7-week-old female C57BL/6 mice (SLC, Shizuoka, Japan). Tumors were isolated 1 month after cell injection for extraction of total RNA. For evaluation of the effect of combined treatment with imatinib and adriamycin (ADR), mice were injected s.c. and bilaterally with AXT cells (5 × 105) on day 0. The mice were then randomly assigned to four groups for treatment with imatinib (100 mg/kg) by mouth on days 10–15 and 18–23, with ADR (6 mg/kg) injected once daily i.v. on days 13 and 15, or with neither or both. Tumors and lungs were isolated for analysis on day 24 after euthanasia.
Reverse transcription and real-time PCR analysis
Total RNA extraction, reverse transcription, and real-time PCR analysis was carried out as previously described.8 The sequences of the primers (sense and antisense, respectively) were 5′-GTGAGACAGTAGTGACCCCT-3′ and 5′-TTCTCACCGTCCGAATGGTC-3′ for Pdgfb, 5′-GTCTACTCGCAACATGTCTG-3′ and 5′-GAAGGAGAGCTGGACCTCAT-3′ for Pdgfrb, and 5′-CAACCGTGAAAAGATGACCC-3′ and 5′-TACGACCAGAGGCATACAG-3′ for Actb.
Immunohistochemistry
Immunohistochemical analysis was carried out by standard methods. Sections were stained with antibodies to Ki67 (Abcam, Cambridge, UK), GFP (Santa Cruz Biotechnology, Dallas, TX, USA), or PDGFRβ (Epitomics). Immune complexes were detected with the use of Histofine (Nichirei Bioscience, Tokyo, Japan) and ImmPACT DAB (Vector Laboratories, Burlingame, CA, USA). For quantification of Ki67 positivity, positively stained cells and the total number of cells was evaluated in four high-magnification (×400) fields selected from the regions of maximal staining and the values were summed to calculate the ratio of the stain-positive cells to the total cell count. To evaluate the therapeutic efficacy in lung metastasis, the ratio of positively stained pixel area for GFP to the whole pixel area of the lung section was quantified using BZ-II analyzer software (Keyence, Osaka, Japan). A lung tissue section was randomly obtained from each lobe.
Human OS tissue array
An array of human OS specimens was obtained from Folio Biosciences (Powell, OH, USA) and was subjected to immunohistochemical staining for PDGFRβ. Comprehensive evaluation of the staining intensity and positivity was carried out as described previously.22
Measurement of intracellular reactive oxygen species
Intracellular reactive oxygen species (ROS) levels in AXT cells were quantified in triplicate with the use of CellROX Deep Red Reagent (Invitrogen), and the fluorescence was detected by flow cytometry.
Statistical analysis
Quantitative data are presented as means ± SD (unless indicated otherwise) and were analyzed with Student’s t-test. A P-value of <0.05 was considered statistically significant. $P < 0.05, $$P < 0.005.
Results
Platelet-derived growth factor-BB and PDGFRβ are highly expressed in OS tumors
We previously established a mouse OS model based on highly tumorigenic AXT cells that can be distinguished by their expression of GFP and c-MYC.5,6 To gain insight into soluble factors that promote OS pathogenesis, we used ELISA-based screening of AXT-derived tumor tissue lysates for such factors.8 We found PDGF-BB to be highly expressed in tumors, with its concentration in tumor lysates (63.16 ± 35.2 pg/mL, n = 5) being higher than that in serum (17.56 ± 4.91 pg/mL, n = 3), suggesting that PDGF-BB was not supplied by the circulation but was generated within the tumor tissue. The abundance of Pdgfb mRNA was significantly higher in AXT-derived tumors than in AXT cell cultures (Fig.1a). In addition, sorting of tumor and surrounding stromal cells from tumors revealed that GFP-negative non-malignant cells expressed Pdgfb at a higher level than did GFP-positive OS cells (Fig.1a). Immunohistochemical analysis showed that the PDGF-BB receptor, PDGFRβ, was also highly expressed in OS tumors (Fig.1b). In contrast to the expression pattern of Pdgfb, the abundance of Pdgfrb mRNA was significantly greater in tumor cells than in stromal cells of AXT tumors (Fig.1c). These results suggested that PDGF-BB secreted from stromal cells might interact with OS cells via PDGFRβ and thereby influence tumor behavior in vivo.
Fig 1.
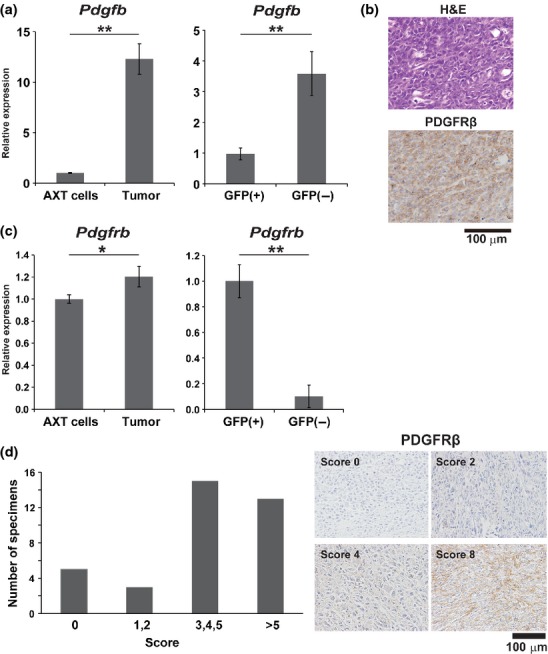
Expression of platelet-derived growth factor receptor β (PDGFRβ) in AXT cells and osteosarcoma (OS) tissue. (a) RT and quantitative PCR analysis of Pdgfb expression in AXT cells and s.c. AXT cell-derived OS tumors (left) as well as in fractionated GFP(+) OS cells and GFP(−) stromal cells from AXT tumors (right). (b) Representative histology of s.c. AXT tumors as revealed by H&E staining (top) and immunohistochemical staining for PDGFRβ (lower). (c) RT and quantitative PCR analysis of Pdgfrb expression as in (a). (d) Immunohistochemical staining for PDGFRβ in human OS samples (n = 36). Quantitative analysis was done according to the Allred scoring system.20 Representative images are shown. $P < 0.05; $$P < 0.005.
Immunohistochemical analysis of a tissue array containing human OS specimens revealed that 31 of 36 cases (86.1%) were positive for PDGFRβ (Fig.1d). Evaluation of the expression level on the basis of the Allred score,22 a combined scoring system for staining positivity and intensity (0 for negative and 8 for maximal), revealed that 28 out of 36 (77.8%) showed a moderate or high expression level (score of 3 to 8). This frequent detection of high PDGFRβ expression in human OS is consistent with previous reports.15–17
Platelet-derived growth factor-BB promotes OS cell survival in serum-free culture
To clarify whether PDGFRβ is functional in OS cells, we examined the effect of PDGF-BB on OS cell proliferation and migration. Platelet-derived growth factor-BB supported the growth of AXT cells in serum-free culture but did not affect cell proliferation in the presence of serum (Fig.2a); PDGF-BB also tended to promote cell migration under low-serum conditions, although this effect did not achieve statistical significance (Fig.2b).
Fig 2.
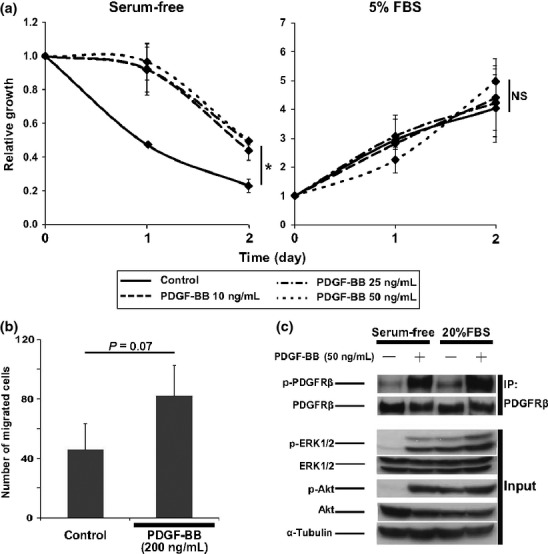
Effects of platelet-derived growth factor-BB (PDGF-BB) on AXT osteosarcoma cell proliferation and migration. (a) Cell proliferation assay with AXT cells in the presence of the indicated concentrations of PDGF-BB in the absence (left) or presence (right) of FBS. Data are expressed relative to the value for time 0. $P < 0.05. NS, not significant. (b) Migration assay with AXT cells in the absence or presence of PDGF-BB. (c) Immunoblot analysis of AXT cells deprived of serum for 2 h and then stimulated with PDGF-BB for 15 min in serum-free or FBS medium. PDGF receptor β (PDGFRβ) was immunoprecipitated (IP) from cell lysates before immunoblot analysis.
To investigate the intracellular molecular events induced by PDGF-BB, we examined mediators of PDGFRβ signaling by immunoblot analysis. The MEK–ERK and phosphatidylinositol 3-kinase–Akt pathways were previously shown to be activated by PDGF.23 We found that PDGF-BB induced marked phosphorylation of PDGFRβ in AXT cells maintained in both the absence and presence of serum (Fig.2c). Serum deprivation resulted in the inactivation of ERK and Akt in AXT cells, and PDGF-BB induced reactivation of both molecules in the serum-deprived cells. However, PDGF-BB had little effect on ERK or Akt phosphorylation on top of that induced by 20% serum (Fig.2c), possibly accounting for the limited effect of PDGF-BB on OS cell growth in serum-supplemented culture (Fig.2a).
Imatinib inhibits the effects of PDGF-BB in AXT cells in vitro
Imatinib is a molecularly targeted drug used in the clinical setting for the treatment of several neoplastic disorders including CML and GIST.14,23,24 It serves as a competitive inhibitor of BCR–ABL (in CML), c-Kit (in GIST), and PDGFR.25 In the treatment of dermatofibrosarcoma protuberans, which is caused by unregulated expression of PDGFB, imatinib is given as a therapeutic inhibitor of PDGFR.14,26,27 Given that we found that PDGFRβ is expressed at a high frequency in OS (Fig.1) and that PDGF-BB activates survival and proliferative signals in OS tumor cells (Fig.2), we next investigated whether imatinib attenuate OS cell growth.
Imatinib blocked the supportive effect of PDGF-BB on AXT cell growth under serum-free conditions, whereas it had no effect on cell proliferation in serum-containing medium regardless of the presence of PDGF-BB (Fig.3a). In contrast, imatinib inhibited the growth of K562 cells (imatinib-sensitive human CML cells harboring the BCR–ABL fusion gene) even in the presence of 10% serum (Fig.3b). Importantly, imatinib successfully blocked activation of PDGFRβ in AXT cells induced by PDGF-BB both in serum-free and in serum-containing culture (Fig.3c). In the absence of serum, imatinib completely inhibited the activation of ERK and Akt induced by PDGF-BB in AXT cells (Fig.3d). The additional activation caused by PDGF-BB was also cancelled by imatinib, whereas the activating signal from serum was scarcely affected. Consistent with a previous report,19 AXT cell viability under the presence of serum was significantly affected with higher doses of imatinib (Fig. S1a). Nonetheless, even 10 μM imatinib could not suppress the intracellular signal activation of ERK and Akt induced by serum (Fig. S1b). This observation suggests that the growth suppression by higher concentration of imatinib does not rely on the signal inhibition by way of PDGFR blockade.
Fig 3.
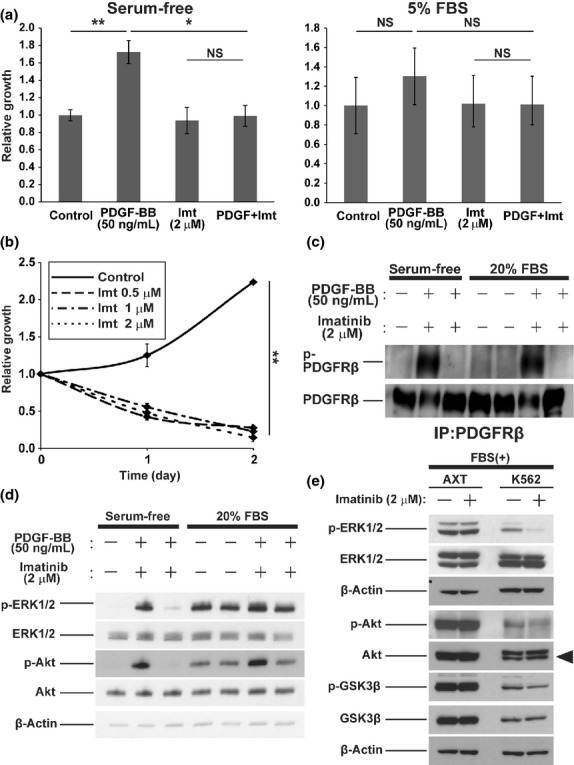
Inhibitory effects of imatinib (Imt) in AXT and K562 cells. (a) Cell proliferation assay with AXT cells incubated for 2 days in the absence (left) or presence (right) of FBS and with or without platelet-derived growth factor-BB (PDGF-BB) or imatinib. Data are expressed relative to the control value. (b) Cell proliferation assay with K562 cells incubated in the presence of FBS and the indicated concentrations of imatinib. Data are expressed relative to the value for time 0. (c) Phosphorylation of PDGF receptor β (PDGFRβ) was evaluated by immunoprecipitation (IP) and immunoblot analysis. AXT cells were cultured without serum for 2 h in the absence or presence of imatinib. Then the medium was replaced with those containing PDGF-BB, imatinib, and/or FBS as indicated. Cells were cultured for 15 min prior to lysate collection. (d) Immunoblot analysis of whole cell lysates from AXT cells as in (c). (e) Immunoblot analysis of the indicated molecules in AXT and K562 cells deprived of serum for 2 h in the absence or presence of imatinib and then stimulated with FBS for 15 min (in the continued absence or presence of imatinib). The arrowhead shows the total form of protein kinase B (Akt). $P < 0.05; $$P < 0.005. GSK3β, glycogen synthase kinase 3β.
On the contrary, in K562 cells, imatinib attenuated the activation of ERK in the presence of serum (Fig.3e). Consistent with previous observations,28 imatinib also inhibited the activation of Akt in these cells, albeit to a lesser extent than it did that of ERK (Fig.3e). Phosphorylation of the Akt substrate glycogen synthase kinase 3β was also inhibited by imatinib and all these changes were not observed in AXT cells. These results thus showed that imatinib was able to block intracellular signaling triggered by PDGFRβ ligation in OS cells, whereas it had little effect on such signaling events triggered by serum. They also revealed that AXT cells differ from K562 cells in terms of their sensitivity to imatinib, even though both cell types express receptor tyrosine kinases with tumor-promoting capacity that is targetable by this drug.
Combined treatment with ADR and imatinib has a synergistic cytotoxic effect on OS cells in vitro
Whereas PDGF-BB induced intracellular signal activation and supported OS cell growth in a manner sensitive to imatinib, these effects were masked in the presence of serum. On the basis of these findings, there seems to be two options to enhance the antitumor activity of imatinib against OS: blocking growth-promoting signals from other pathways that can compensate the influence of PDGFR inhibition, or administering imatinib treatment under cellular stresses induced by serum deprivation. We examined the latter possibility.
Serum deprivation induces the intracellular accumulation of ROS and the consequent oxidative stress is responsible in part for the resulting cell death.29,30 Indeed, serum deprivation triggered an increase in the intracellular ROS level in AXT cells (Fig.4a), which raised the hypothesis that elevated oxidative stress might improve OS cell sensitivity toward imatinib treatment.
Fig 4.

Enhanced sensitivity of osteosarcoma cells to imatinib (Imt) by reactive oxygen species (ROS). (a) Left, flow cytometric histogram of ROS levels in AXT cells in the absence or presence of FBS for 6 h. Right, quantification of ROS level (n = 3). (b) Cell proliferation assay with AXT cells incubated for 24 h in medium containing FBS in the absence or presence of adriamycin (ADR) or N-acetyl cysteine (NAC) as indicated. Data are expressed relative to the value for non-treated cells. NS, not significant. (c) Cell proliferation assay with AXT cells incubated for 24 h in medium containing FBS in the absence or presence of ADR or the indicated concentrations of imatinib. (d–f) Cell proliferation assay carried out as in (c) with human osteosarcoma cell lines. $P < 0.05; $$P < 0.005.
Given that chemotherapeutic drugs also elicit intracellular ROS accumulation,31,32 we examined whether the cytotoxicity of ADR, a drug used in standard chemotherapy for OS, in AXT cells is related to ROS burden. The proliferation of AXT cells in vitro was suppressed by ADR treatment, which was counteracted by the antioxidant N-acetyl cysteine (Fig.4b), suggesting that this effect of ADR is mediated, at least in part, by the induction of oxidative stress. Although imatinib alone did not affect the growth of AXT cells in the presence of serum, it significantly enhanced the inhibitory effect of ADR on cell growth and this enhancement was apparent at an imatinib concentration as low as 0.01 μM (Fig.4c). Similar synergistic effects of imatinib and ADR were observed in three different human OS cell lines (Fig.4d–f).
Combined treatment with ADR and imatinib synergistically inhibits OS cell proliferation in vivo
We next examined the effect of combined treatment with imatinib and ADR on growth of OS tumors formed by AXT cells in mice. Treatment with imatinib or ADR alone at the current dose did not achieve significant reduction in tumor weight (Fig.5a). However, notably, combined treatment of imatinib and ADR showed significant antitumor effects (Fig.5a). Ki67-positive cells in tumor specimens were significantly reduced by single treatment with ADR and, notably, the effect was further enhanced by imatinib combined with ADR, albeit without statistical significance. Imatinib alone did not show such an inhibitory effect (Fig.5b). Moreover, evaluation of the therapeutic efficacy on lung metastatic lesions, which was based on GFP-positive areas using immunohistochemical staining of lung lobes, revealed the consistent tendency with the primary lesions (Fig.5c). Therefore, consistent with in vitro analyses (Fig.4c–f), the combination of imatinib and ADR showed synergistic antitumor effects even in vivo.
Fig 5.

Antitumor activity of imatinib (Imt) in vivo. (a) Weight of AXT cell-derived s.c. tumors in mice treated with adriamycin (ADR), imatinib, or both. (b) Proportion of Ki67+ cells determined by immunohistochemical (IHC) analysis of tumors from mice treated as in (a). Left, representative images of Ki67 staining. Right, quantification of Ki67 positivity. (c) Quantitative analysis for lung metastatic lesions in mice treated as in (a). Left, representative images of lung metastatic lesions. Right, quantitative data of metastatic area presented as box-and-whisker plots. $P < 0.05. NS, not significant.
Discussion
The pathology of OS has recently been found to be influenced by soluble factors provided by tumor-surrounding cells.4,8–10 With the use of our newly established immunocompetent OS model, we found that PDGF-BB was present in tumor lysates at a concentration higher than that in serum. We also found that PDGFRβ was highly expressed in our OS cells of the mouse model as well as in human OS specimens. We therefore examined the possible contribution of PDGFRβ signaling to OS pathogenesis as well as its potential as a therapeutic target.
Although PDGF-BB clearly induced the intracellular signal activation through PDGFRβ in OS cells and promotes their cellular growth, these effects could be mostly masked in the presence of serum. Accordingly, successful PDGFR signal inhibition by imatinib did not bring about successful results regarding antitumor activity in the presence of serum.
It is clear that targeting PDGFR would not be sufficient to control the growth of OS, even in tumor cells that frequently express PDGFR. This is because the PDGF signaling pathway does not appear to be the main driver of OS cell fate, especially in the presence of other growth-promoting factors such as those in serum. They may also shed light on the unfavorable results obtained in a clinical trial of imatinib in patients with OS,20,21 considering the fact that OS is mostly equipped with rich vascularity, thus with plenty of growth signal supply. The application of molecularly targeted therapy to OS would therefore be expected to be effective only if OS cells are “addicted to” the target molecule for their survival, as in the case of imatinib therapy for CML, which is crucially dependent on the driver oncogene BCR–ABL.33
Nevertheless, we found serum deprivation optimized the effect of imatinib on OS cells and that combination of imatinib and ADR achieved the synergistic antiproliferative effect in OS cells in vitro and even therapeutic efficiency in vivo. Furthermore, our observations suggest that oxidative stress induced by ADR, similar to that induced by serum deprivation, might have some contribution to alter cellular status toward such that imatinib could exert its cytotoxic effect. Collectively, these findings imply that cellular dependency to the PDGFR pathway is relatively dynamic and can be shifted in a therapeutically desirable direction by exogenous stimuli such as serum deprivation or ADR treatment that, at least in part, trigger the intracellular accumulation of ROS. Identification of conditions capable of rendering OS cells sensitive to imatinib cytotoxicity or of OS subtypes with intrinsic sensitivity to this agent should facilitate the application of imatinib as a novel therapeutic alternative for OS.
Acknowledgments
We thank I. Ishimatsu for technical assistance as well as K. Arai and M. Sato for secretarial assistance. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (T.S. and H.S.), MEXT-Supported Program for the Strategic Research Foundation at Private Universities, Takeda Science Foundation (T.S.), and a grant from the National Institute of Biomedical Innovation of Japan (H.S.).
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Anti-osteosarcoma tumor effects of imatinib at higher doses.
References
- Fletcher CDM, Unni KK, Mertens F. Osteogenic Tumours: WHO Classification Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. pp. 259–85. [Google Scholar]
- Gorlick R. Current concepts on the molecular biology of osteosarcoma. Cancer Treat Res. 2009;152:467–78. doi: 10.1007/978-1-4419-0284-9_27. [DOI] [PubMed] [Google Scholar]
- Posthuma DJ, Witlox MA, Kaspers GJL, Van Royen BJ. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clin Exp Metastasis. 2011;28:493–503. doi: 10.1007/s10585-011-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botter SM, Nerri D, Fuchs B. Recent advances in osteosarcoma. Curr Opin Pharmacol. 2014;16:15–23. doi: 10.1016/j.coph.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Ishikawa T, Sugihara E, et al. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010;29:5687–99. doi: 10.1038/onc.2010.312. [DOI] [PubMed] [Google Scholar]
- Ueki A, Shimizu T, Masuda K, et al. Up-regulation of Imp3 confers in vivo tumorigenicity on murine osteosarcoma cells. PLoS One. 2012;7(11):e50621. doi: 10.1371/journal.pone.0050621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Shimizu T, Ueki A, et al. Twist2 functions as a tumor suppressor in murine osteosarcoma cells. Cancer Sci. 2013;104:880–8. doi: 10.1111/cas.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Ishikawa T, Iwai S, et al. Fibroblast growth factor-2 is an important factor that maintains cellular immaturity and contributes to aggressiveness of osteosarcoma. Mol Cancer Res. 2012;10(3):454–68. doi: 10.1158/1541-7786.MCR-11-0347. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Sugihara E, Yamaguchi IS, et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014;74:6531–41. doi: 10.1158/0008-5472.CAN-14-0914. [DOI] [PubMed] [Google Scholar]
- Mori T, Sato Y, Miyamoto K, et al. TNFα promotes osteosarcoma progression by maintaining tumor cells in an undifferentiated state. Oncogene. 2014;33:4236–41. doi: 10.1038/onc.2013.545. [DOI] [PubMed] [Google Scholar]
- McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010;28:4022–8. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- Seruga B, Zhang H, Bernatein LJ, et al. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer. 2008;8:887–99. doi: 10.1038/nrc2507. [DOI] [PubMed] [Google Scholar]
- Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19(8):460–73. doi: 10.1016/j.molmed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Duffaud F, Le Cesne A. Imatinib in the treatment of solid tumours. Target Oncol. 2009;4(1):45–6. doi: 10.1007/s11523-008-0101-x. [DOI] [PubMed] [Google Scholar]
- Abdeen A, Chou AJ, Healey JH, et al. Correlation between clinical outcome and growth factor pathway expression in osteogenic sarcoma. Cancer. 2009;115:5243–50. doi: 10.1002/cncr.24562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Piperdi S, Rosenblum J, et al. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer. 2008;112:2119–29. doi: 10.1002/cncr.23437. [DOI] [PubMed] [Google Scholar]
- Kim MY, Lin J, Piperdi S, et al. Cell surface receptor expression patterns in osteosarcoma. Cancer. 2012;118:740–9. doi: 10.1002/cncr.26339. [DOI] [PubMed] [Google Scholar]
- McGary EC, Weber K, Mills L, et al. Inhibition of platelet-derived growth factor-mediated proliferation of osteosarcoma cells by the novel tyrosine kinase inhibitor STI571. Clin Cancer Res. 2002;8:3584–91. [PubMed] [Google Scholar]
- Gobin B, Moriceau G, Ory B, et al. Imatinib mesylate exerts anti-proliferative effects on osteosarcoma cells and inhibits the tumour growth in immunocompetent murine models. PLoS One. 2014;9(3):e90795. doi: 10.1371/journal.pone.0090795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura H, Fujiwara Y, Ando M, et al. Multicenter phase II trial assessing effectiveness of imatinib mesylate on relapsed or refractory KIT-positive or PDGFR-positive sarcoma. J Orthop Sci. 2010;15:654–60. doi: 10.1007/s00776-010-1506-9. [DOI] [PubMed] [Google Scholar]
- Bond M, Bernstein ML, Pappo A, et al. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumours: a Children’s Oncology Group study. Pediatr Blood Cancer. 2008;50:254–8. doi: 10.1002/pbc.21132. [DOI] [PubMed] [Google Scholar]
- Allred DC, Harvey JM, Clark GM, et al. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–68. [PubMed] [Google Scholar]
- Östman A, Heldin CH. PDGF receptors as targets in tumor treatment. Adv Cancer Res. 2007;97:247–74. doi: 10.1016/S0065-230X(06)97011-0. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Griffith DJ, Druker BJ, et al. Inhibition of c-kit receptor tyrosine kinase by STI571, selective tyrosine kinase inhibitor. Blood. 2000;96:925–32. [PubMed] [Google Scholar]
- Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–45. [PubMed] [Google Scholar]
- Handolias D, McArthur GA. Imatinib as effective therapy for dermatofibrosarcoma protuberans: proof of concept of the autocrine hypothesis for cancer. Future Oncol. 2008;4:211–7. doi: 10.2217/14796694.4.2.211. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Shimizu A, O’Brien KP, et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001;61:5778–83. [PubMed] [Google Scholar]
- Jacquel A, Herrant M, Legros L, et al. Imatinib induces mitochondria-dependent apoptosis of the Bcr-Abl-positive K562 cell line and its differentiation toward the erythroid lineage. FASEB J. 2003;17:2160–2. doi: 10.1096/fj.03-0322. [DOI] [PubMed] [Google Scholar]
- Satoh T, Sakai N, Enokido Y, et al. Survival factor-insensitive generation of reactive oxygen species induced by serum deprivation in neuronal cells. Brain Res. 1996;733(1):9–14. doi: 10.1016/0006-8993(96)00527-6. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim JJ, Kim TW, et al. Serum deprivation-induced reactive oxygen species production is mediated by Romo1. Apoptosis. 2010;15:204–18. doi: 10.1007/s10495-009-0411-1. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Margreiter R, Amberger A, et al. Changes in Mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim Biophys Acta. 2011;1813:1144–52. doi: 10.1016/j.bbamcr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Bátkai S, et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2009;296:1466–83. doi: 10.1152/ajpheart.00795.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambacorti-Passerini C, le Coutre P, Mologni L, et al. Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells Mol Dis. 1997;23:380–94. doi: 10.1006/bcmd.1997.0155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Anti-osteosarcoma tumor effects of imatinib at higher doses.