

Abstract
The Fanconi anemia (FA) pathway plays a key role in interstrand crosslink (ICL) repair and maintenance of the genomic stability, while inhibition of this pathway may sensitize cancer cells to DNA ICL agents and ionizing radiation (IR). The active FA core complex acts as an E3 ligase to monoubiquitinate FANCD2, which is a functional readout of an activated FA pathway. In the present study, we aimed to identify FANCD2-targeting agents, and found that the natural compound celastrol induced degradation of FANCD2 through the ubiquitin–proteasome pathway. We demonstrated that celastrol downregulated the basal and DNA damaging agent-induced monoubiquitination of FANCD2, followed by proteolytic degradation of the substrate. Furthermore, celastrol treatment abrogated the G2 checkpoint induced by IR, and enhanced the ICL agent-induced DNA damage and inhibitory effects on lung cancer cells through depletion of FANCD2. These results indicate that celastrol is a FANCD2 inhibitor that could interfere with the monoubiquitination and protein stability of FANCD2, providing a novel opportunity to develop FA pathway inhibitor and combinational therapy for malignant neoplasms.
Keywords: Celastrol, DNA damaging agents, FANCD2, lung cancer, proteasomal degradation
Lung cancer is the leading cause of cancer-related mortality which in 2012 caused 1.59 million deaths worldwide. The 5-year overall survival rate of lung cancer is only 15% for all stages combined.1,2 This malignant neoplasm can be divided into two major forms: non-small cell lung cancer (NSCLC) which accounts for approximately 85% of lung cancer and is comprised of three major histological subtypes (squamous cell carcinoma, adenocarcinoma and large-cell carcinoma), and small cell lung cancer (SCLC) which accounts for 15% of lung cancer.3 Current treatment includes surgery, platinum doublet therapy, radiation therapy and targeted therapy, determined by histologic subtype and stage of the disease. For decades, cisplatin has been widely used in treating NSCLCs,4 mainly via a mode of action involving the generation of DNA lesions followed by the activation of the DNA damage response and the induction of mitochondrial apoptosis.5 However, cisplatin treatment often results in the development of chemoresistance, leading to therapeutic failure.5 Therefore, strategies to overcome drug resistance are desirable to tame this deadly disease.
Cisplatin is able to crosslink with the purine bases on the DNA, interfere with DNA repair mechanisms, cause DNA damage and trigger programmed cell death of cancer cells.6 The bioreductive drug mitomycin C (MMC) primarily causes interstrand crosslinks (ICL),7 while ionizing radiation (IR) (e.g. X-rays) induces DNA double strand breaks as well as single strand breaks. These processes recruit DNA repair signaling complex including Fanconi anemia (FA) proteins to the DNA lesion spots.8 The FA pathway is involved in homologous recombination, nucleotide excision repair and translesion synthesis to repair the DNA lesions and ensure the appropriate DNA replication process.9,10 Fifteen FA proteins have been identified, and eight of them (FA complementation group A, B, C, E, F, G, L and M) form an FA core complex in the DNA lesion spots, which is responsible for the monoubiquitination of the central complex of the FA pathway: FANCD2 and FANCI.11,12 The E3 ubiquitin ligase FANCL and the E2 protein UBE2T mediate monoubiquitination of FANCD2 and FANCI at Lys561 and Lys523, respectively. Another E3 ligase, RAD18, has also been implicated in efficient monoubiquitination of FANCD2 and FANCI.13–16 FA component FAN1 acts downstream of the monoubiquitinated FANCD2/FANCI complex and facilitates the repair of DNA ICL.17,18 Proteasome is involved in many aspects of DNA damage response and controls monoubiquitination of FANCD2, foci formation of 53BP1, phosphorylated ataxia telangiectasia mutated (ATM), NBS1, BRCA1, FANCD2 and RAD51, and proteasome inhibitors may sensitize tumor cells to DNA-damaging agents through the inhibition of these processes.19,20 Recent studies show that some natural agents, such as curcumin21,22 and phenylbutyrate,23 interfere with the FA/BRCA pathway and inhibit monoubiquitination of FANCD2, leading to sensitization of tumor cells to cisplatin. Depletion of FAN1 also leads to dysfunction of the FA pathway and sensitizes cancer cells to DNA ICL agents.18 However, agents inhibiting the FA pathway through decreasing protein stability of FANCD2 have not been reported, and the therapeutic potential of FANCD2-targeting agents warrants further investigation.
In this study, we aim to identify a novel FANCD2-targeting agent and investigate its combinational effect with ICL agents. We found that a natural compound celastrol, which potentiated the inhibitory effect of cisplatin on lung cancer cells in vitro and in vivo via inhibition of the CIP2A–Akt pathway,24 promoted FANCD2 degradation through the ubiquitin–proteasome pathway, inhibited basal and DNA damage-induced FANCD2 monoubiquitination, and enhanced ICL agent-induced inhibitory effects on lung cancer cells, providing a novel opportunity to develop the FA pathway inhibitor and combinational therapy for malignant neoplasms.
Materials and Methods
Reagents
Celastrol was purchased from Calbiochem (San Diego, CA, USA) and Pie & Pie Technologies (Shenzhen, Guangdong, China). Proteasome inhibitor PS341 was obtained from Millennium Pharmaceuticals (Cambridge, MA, USA) and MG132 was obtained from Calbiochem. Cycloheximide (CHX) was obtained from Beyotime Institute of Biotechnology (Haimen, Jiangsu, China). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Amresco (Solon, OH, USA). Cisplatin (CDDP), hydroxyurea (HU) and MMC were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture, cytotoxicity and cell cycle assays
The NSCLC cell lines NCI-H1975 and A549, liver cancer cell line HepG2 and mammary adenocarcinoma cell line MCF7 were obtained from the American Tissue Culture Collection (ATCC [Manassas, VA, USA]). A549, HepG2 and MCF7 cells were cultured in DMEM containing 10% fetal bovine serum (FBS; Gibco/BRL, Grand Island, NY, USA), and NCI-H1975 cells were cultured in RPMI 1640 supplemented with 10% FBS.
The cells were treated with cisplatin, HU or MMC at indicated concentrations for the indicated time points. The viability of cells was determined by the MTT assay. Briefly, exponentially growing cells (1 × 104 in 180 μL) were plated in 96-well microplates, and 20 μL of 10× drug was added to each well. Cells were incubated with or without celastrol for 44 h, followed by co-incubation with 100 μg MTT for 4 h. The microplates were centrifuged, supernatants were removed, and MTT formazan crystals were resolubilized by adding 150 μL DMSO to each well. Microplates were then agitated on a plate shaker for 5 min, and absorbance was measured using a multiplate reader at the wavelength of 570 nm.25 For cell cycle analysis, cells were synchronized to G1/S boundary by a double-thymidine block,26 and then treated with celastrol for indicated time points. Cells were harvested, fixed with 70% cold ethanol, incubated with RNase, and stained with propidium iodide.27 Cell cycle distribution was analyzed by flow cytometry (BD FACS, Callbur, NJ, USA) and CellQuest software (BD).
Transfection
siRNAs were transfected into the cells with the Lipofectamine 2000 (Invitrogen, Frederick, MD, USA). Plasmids containing FANCD2 (kindly provided by Professor Jun Huang, Zhejiang University of China) were transfected into A549 cells according to the optimized protocol for A549 cell line developed by Lonza in a Lonza Nucleofector II (Allendale, NJ, USA).
Immunofluorescence microscopy
Cells were seeded on the 18 × 18 mm cover slides with 1% gelatin in six-well cell culture plates for 24 h. After co-incubation with drugs at indicated concentrations and time points, cells were washed with PBS twice and fixed with 4% formaldehyde for 15 min at room temperature. The cover slides were rinsed three times with PBS containing 100 mM glycine and permeabilized with 0.3% Triton X-100/PBS for 20 min at room temperature. Cells were blocked with 5% BSA/PBS for 30 min at room temperature and then incubated with primary antibody overnight at 4°C, washed with 0.05% tween-20/PBS three times, followed by incubation with secondary antibody (fluorescein-conjugated AffiniPure goat anti-mouse IgG [H+L], 1:200) for 2 h at room temperature, and washed with 0.05% tween-20/PBS three times. Nuclei were stained with DAPI (Sigma-Aldrich), washed, and observed under a confocal microscope LSM 510 Meta (Zeiss, Oberkochen, Germany).
Immunoblotting
Cell pellets were lysed in RIPA buffer containing 50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 1% deoxycholate, 1% Triton X-100, 1 mM DTT, 1 mM NaF, 1 mM sodium vanadate, and protease inhibitors cocktail (Sigma-Aldrich). Cells were lysed on ice for 15 min in RIPA buffer, lysates were centrifuged, and the supernatant was dissolved with 5× sample loading buffer and boiled for 5 min. Protein extracts were quantitated and loaded on 8–12% sodium dodecyl sulfate polyacrylamide gel, electrophoresed and transferred to a nitrocellulose membrane (Whatman International, Maidstone, Kent, UK). The membrane was incubated with primary antibody, washed and incubated with HRP-conjugated secondary antibody (Thermo Fisher Scientific, Rockford, IL, USA). Detection was performed by using a SignalFire™ ECL Reagent (Cell Signaling, Beverly, MA, USA). The antibodies used were anti-FANCD2 (Santa Cruz Biotechnology, catalog # SC-20022, Santa Cruz, CA, USA), anti-γH2AX (Abcam, catalog # ab2893, Cambridge, MA, USA), anti-PARP (Cell Signaling, catalog # 9542) and anti-β-actin (Sigma-Aldrich) antibodies.
RNA preparation and RT-PCR
The total RNA of the cells was extracted using the TRIZOL Reagent (Invitrogen, Carlsbad, CA, USA) and the phenol–chloroform extraction method according to the manufacturer’s instruction. Total RNA (2 μg) was annealed with Oligo (dT) primers at 65°C for 5 min. The cDNA was synthesized using a First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD, USA). Primers for FANCD2 detection were as follows: forward primer 5′-CCTTCCCTGTGAACTCCT-3′, and reverse primer 5′-CCCCAGACTATGTCCCTC-3′.
Immunoprecipitation
The cells (5 × 106) were seeded in 10-cm dishes and treated with biotin or biotinylated-celastrol (Bio-Cel),24 washed five times with PBS, scraped and lysed in IP buffer. Cell lysates were incubated at 4°C on a rotator for 1 h, and pellets were discarded by centrifugation. The supernatants were incubated with 25 μL streptavidin agarose resin (Thermo Fisher Scientific) in 4°C overnight. The streptavidin agarose resin were washed with lysis buffer five times, then resuspended in 25 μL 2× SDS loading buffer and denatured in 99°C for 6 min. Proteins were resolved on 10% SDS-PAGE gels and immunoblotted as described.24
Statistical analysis
All statistical analyses were conducted using GraphPad Prism 5 and SPSS software 12.0 for Windows (Chicago, IL, USA). Statistically significant differences were determined by Student’s t-test. P-values < 0.05 were considered statistically significant. All experiments were repeated at least three times and the data are presented as the mean ± SD unless noted otherwise.
Results
FANCD2 is rapidly downregulated in cells treated with celastrol
We tested the effects of several compounds on the expression of FANCD2, and found that celastrol induced reduction of FANCD2 in a dose- and time-dependent manner. Treatment of A549 cells with celastrol at 5 μM for 6–18 h drastically downregulated FANCD2 at protein level (Fig.1a). Similarly, treatment with celastrol at 1–5 μM for 6 h reduced the expression of FANCD2 in A549 cells (Fig.1a). These observations were confirmed in H1975 cells treated with celastrol (Fig.1b). Celastrol also reduced FANCD2 at protein level in hepatocarcinoma HepG2 cells and breast cancer MCF7 cells (Fig.1c).
Fig 1.
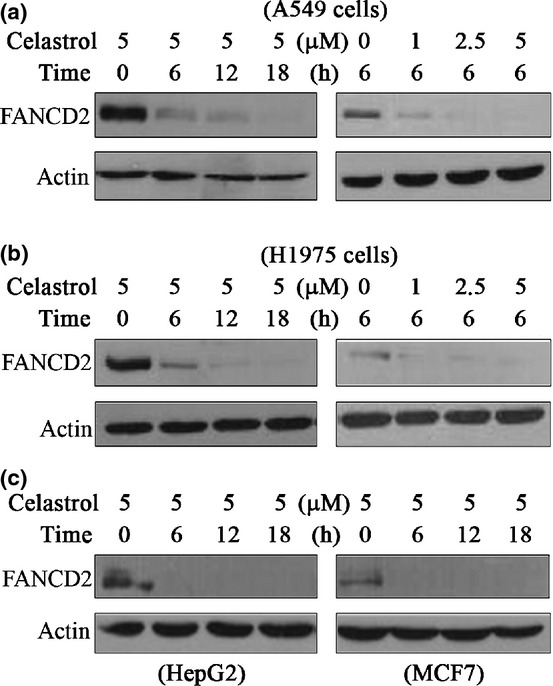
Celastrol induces degradation of FANCD2. (a, b) A549 cells and H1975 cells were treated with celastrol at indicated time points and indicated concentrations, lysed, and Western blotting was performed using anti-FANCD2 and anti-Actin antibodies. (c) Western blot analysis of FANCD2 expression in HepG2 and MCF-7 cells treated with celastrol.
FANCD2 is degraded by proteasome
We investigated the underlying mechanism of celastrol-induced downregulation of FANCD2. By RT-PCR assay, we showed that treatment with celastrol at 5 μM for 6 h did not perturb FANCD2 expression at mRNA level in A549 cells (Fig.2a). Protein synthesis inhibitor CHX was employed to detect the protein stability of FANCD2. We found that in A549 cells treated with CHX (50 μg/mL) alone, the expression of FANCD2 was not decreased within 6 h (Fig.2b). Interestingly, treatment with CHX in combination with celastrol dramatically decreased FANCD2 at protein level within 4 h (Fig.2b), indicating that celastrol impaired the protein stability of FANCD2 by triggering its degradation.
Fig 2.
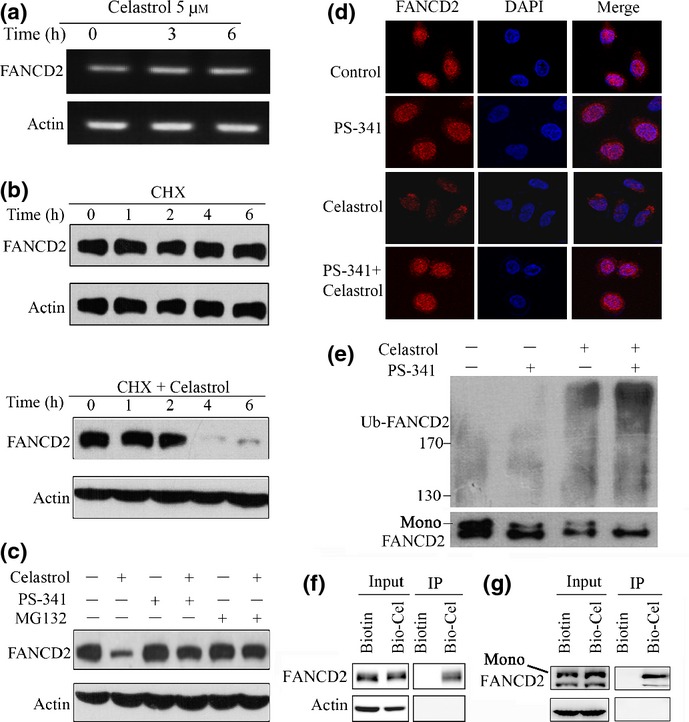
Celastrol triggers FANCD2 degradation through the ubiquitin–proteasome pathway. (a) RT-PCR assays for detecting FANCD2 RNA level in A549 cells upon celastrol. (b) Effects of cycloheximide (CHX) (50 μg/mL) alone or in combination with celastrol (5 μM) on FANCD2 expression, evaluated by Western blotting in A549 cells. (c) A549 cells were pre-incubated with MG132 (10 μM) or PS341 (100 nM) for 1 h, followed by celastrol (5 μM) treatment for 6 h. Cell lysates were subjected to Western blotting using anti-FANCD2 antibody. (d) A549 cells were pretreated with PS341 (100 nM) for 1 h, followed by celastrol (5 μM) incubation for 6 h. The cells were then analyzed by immunofluorescence assay labeling with anti-FANCD2 antibody and DAPI. (e) A549 cells were pretreated with or without PS341 (100 nM) for 1 h, followed by celastrol incubation for 2 h. Cell lysates were subjected to immunoprecipitation with FANCD2 antibody, followed by Western blotting using antibodies against FANCD2 and ubiquitin. (f, g) A549 cells were treated with Biotin or Bio-Cel at 50 μM for 4 h, lysed, and the cell lysates were subjected to immunoprecipitation using streptavidin agarose and Western blotting using indicated antibodies. Mono, monoubiquitinated FANCD2.
The ubiquitin–proteasome system is responsible for the degradation of most endogenous proteins.28 To investigate the role that proteasome plays in celastrol-induced FANCD2 degradation, proteasome inhibitors bortezomib (PS341)29 and MG13230 were used to treat A549 cells in the absence or presence of celastrol. We found that while PS341 and MG132 did not cause proteolysis of FANCD2, they markedly inhibited celastrol-triggered FANCD2 degradation (Fig.2c). These results were confirmed by immunofluorescence staining with anti-FANCD2 antibody in cells co-incubated with PS341/MG132 with or without celastrol (Fig.2d). It has been reported that proteasomes play an important role in monoubiquitination of FANCD2, and MG132 could decrease monoubiquitination of FANCD2.19 We showed that while PS341 and celastrol decreased monoubiquitination and increased polyubiquitination of FANCD2, combined use of both agents enhanced these effects (Fig.2e), indicating that celastrol interfered with monoubiquitination and polyubiquitination of FANCD2.
We then tested whether celastrol had the ability to bind FANCD2 using a biotinylated celastrol,24 and found that in A549 cells treated with Bio-Cel, FANCD2 was pulled down by streptavidin agarose, indicating that celastrol could directly target FANCD2 (Fig.2f). We further showed that Bio-Cel could bind both FANCD2 and its monoubiquitinated derivative (Fig.2g), suggesting that celastrol binding could reduce the stability of ubiquitinated as well as unmodified FANCD2.
Celastrol inhibits DNA damage-induced Fanconi anemia pathway activation
We examined the effect of celastrol on monoubiquitinated FANCD2 in NSCLC cells, and found that compared to the unmodified FANCD2, the monoubiquitinated FANCD2 was decreased more rapidly upon celastrol treatment at 5 μM for 1–4 h (Fig.3a), suggesting that celastrol decreased monoubiquitination of FANCD2 and then reduced protein stability. Furthermore, while cisplatin, HU, MMC and IR induced monoubiquitination of FANCD2 in A549 cells, celastrol treatment (at 5 μM for 4 h) markedly attenuated this effect (Fig.3b). By immunofluorescence assay, we observed that cisplatin induced marked increase in FANCD2 in the nucleus (Fig.3c), suggesting formation of nuclear foci in A549 cells. This phenomenon was attenuated by celastrol (Fig.3c).
Fig 3.

Celastrol downregulates monoubiquitinated FANCD2 and attenuates DNA-damaging agent-induced FA pathway activation. (a) A549 and H1975 cells were treated with celastrol at 5 μM for indicated time points, lysed, and Western blotting was performed using anti-FANCD2 antibody. (b) FANCD2 monoubiquitination induced by different DNA-damaging agents was inhibited by celastrol. A549 cells were pre-incubated with cisplatin (CDDP; 20 μM/L), mitomycin C (MMC) (0.4 μg/mL) or hydroxyurea (HU; 1 mmol/L) for 24 h or subjected to ionizing radiation (IR) (15 Gy), then incubated with celastrol (5 μM) for additional 4 h. The cells were lysed and Western blot was performed. (c) A549 cells were pretreated with cisplatin (20 μM/L) for 24 h, followed by celastrol (5 μM) incubation for additional 4 h. The cells were subjected to immunofluorescence assay by labeling with anti-FANCD2 antibody and DAPI. (d) A549 cells were irradiated by X-ray (15 Gy), then incubated immediately with 5 μM celastrol at indicated time points; whole-cells extracts were used for detection of FANCD2 and γ-H2AX. (e) A549 cells were transfected with siRNA against negative control (siRNA-NC) or FANCD2 (siFANCD2), irradiated by X-ray (15 Gy), then incubated immediately with 5 μM celastrol at indicated time points. The whole-cells extracts were used for detection of FANCD2 and γ-H2AX. (f) A549 cells were transfected with pDONR201-vector or pDONR201-FANCD2, irradiated by X-ray (15 Gy), then incubated immediately with 5 μM celastrol at indicated time points, and the whole-cells extracts were used for detection of FANCD2 and γ-H2AX. (g) A549 cells were irradiated by X-ray (15 Gy), incubated immediately with 5 μM celastrol for an additional 4 or 8 h. Cell cycle distribution was then determined. (h) A549 cells were transfected with siRNA-NC or si-FANCD2, irradiated by X-ray (15 Gy), then incubated immediately with 5 μM celastrol for additional 8 h. Cell cycle distribution was then determined. (i) A549 cells were transfected with pDONR201-vector or pDONR201-FANCD2, irradiated by X-ray (15 Gy), and then incubated immediately with 5 μM celastrol for an additional 8 h. Cell cycle distribution was determined.
Ionizing radiation exposure could induce rapid phosphorylation of histone H2AX at Ser 139 (γ-H2AX) at the DNA damage site until the DNA lesions are properly repaired.31,32 In A549 cells irradiated with 15 Gy X-ray, the monoubiquitinated FANCD2 was increased in 0–8 h and the γ-H2AX was upregulated in 2 h but then returned to the basic level (Fig.3d). In irradiated cells treated with celastrol, the expression of monoubiquitinated and unmodified FANCD2 was downregulated, and the γ-H2AX remained high in 8 h (Fig.3d). In siRNA-mediated FANCD2 silencing A549 cells, IR increased γ-H2AX expression compared to the scrambled siRNA-treated group (Fig.3e, left panel). Combined use of FANCD2 siRNA and celastrol led to enhanced reduction of FANCD2 and an increase of γ-H2AX (Fig.3e, right panel). In A549 cells transfected with pDONR201-FANCD2, overexpressed FANCD2 suppressed the increase of γ-H2AX by IR alone or in combination with celastrol (Fig.3f). These results suggested that celastrol suppressed DNA repair through inhibition of monoubiquitination of FANCD2.
The cell cycle G2 checkpoint could be activated upon DNA damage to prevent aberrant damaged chromosome segregation in mitosis,33,34 and the FA pathway is involved in the DNA damage-induced G2 checkpoint.35 We showed that in A549 cells exposed to IR, cells in G2/M phase significantly increased in 8 h after irradiation (Fig.3g). However, celastrol significantly reduced G2/M cells induced by IR (Fig.3g). Celastrol treatment and FANCD2-siRNA transfection inhibited the IR-induced G2/M checkpoint in A549 cells, and combined treatment of celastrol and FANCD2 siRNA enhanced this effect (Fig.3h). In contrast, transfection of pDONR201-FANCD2 into A549 cells inhibited the effects of celastrol (Fig.3i). These results indicated that celastrol could interfere with normal DNA repair function and disrupt the DNA damage-induced activation of the cell cycle checkpoint.
Celastrol enhances interstrand crosslink agent-induced DNA damage and cell death
We examined the combined effects of celastrol and DNA damage agents in A549 cells by MTT assay, and found that while cisplatin (5–20 μM) and MMC (0.2–0.8 μg/mL) inhibited cell proliferation, celastrol significantly increased the inhibition rate of the ICL agents (Fig.4a). Furthermore, while cisplatin and MMC upregulated the monoubiquitinated FANCD2, celastrol downregulated monoubiquitinated and unmodified FANCD2 (Fig.4b). Cisplatin and MMC increased the expression of γ-H2AX, and celastrol treatment further upregulated γ-H2AX (Fig.4b). The poly (ADP-ribose) polymerases (PARP)36 was cleaved in cisplatin-treated and MMC-treated cells, and celastrol enhanced this observation (Fig.4b), indicating that celastrol potentiated the apoptotic effect of cisplatin and MMC. In FANCD2-knockdown A549 cells, FANCD2-silencing enhanced the effects of celastrol on CDDP/MMC sensitization (Fig.4c) and γ-H2AX upregulation as well as PARP cleavage (Fig.4d). In FANCD2-transfected A549 cells, FANCD2 overexpression attenuated cell proliferation inhibition caused by celastrol or combined use of celastrol and CDDP/MMC (Fig.4e), and suppressed γ-H2AX upregulation and PARP cleavage (Fig.4f).
Fig 4.

Celastrol enhances inhibitory effects of DNA-damaging agents on lung cancer cells. (a) A549 cells were pretreated with cisplatin (5–20 μM) or mitomycin C (MMC) (0.2–0.8 μg/mL) at indicated concentrations for 24 h, followed by celastrol (5 μM) incubation for an additional 12 h, and cell viability was detected by MTT assay. (b) A549 cells pretreated with cisplatin or MMC at indicated concentrations for 20 h, followed by celastrol incubation for an additional 4 h. The whole-cell extracts were used for detection of FANCD2, γ-H2AX and PARP by Western blot. (c, d) A549 cells were transfected with siRNA-NC or si-FANCD2, treated with cisplatin or MMC for 24 h, followed by celastrol treatment for an additional 12 h. The cells were analyzed by MTT assay (c) or lysed for Western blot using indicated antibodies (d). (e, f) A549 cells were transfected with pDONR201-vector or pDONR201-FANCD2, treated with cisplatin or MMC for 24 h, followed by celastrol treatment for an additional 12 h. The cells were analyzed by MTT assay (e) or lysed for Western blot using the indicated antibodies (f).
Discussion
Increasing evidence shows that the FA proteins have critical roles in DNA damage response. Defects in the FA pathway often lead to autosomal recessive disorder and spontaneous chromosomal instability.37 Abnormalities in the FA pathway occur in subsets of diverse human cancers, including lung cancer.38,39 The hypersensitivity of FA pathway-deficient cells to DNA ICL agents renders these genes attractive targets for a genotype-based, individualized anticancer therapy.40,39 Here we reported for the first time that celastrol promotes FANCD2 degradation in cancer cells through the ubiquitin–proteasome pathway, and synergizes with DNA ICL agents and IR-induced apoptosis, exhibiting therapeutic potentials through FA pathway inhibition in lung cancer.
FANCD2 is phosphorylated by ATM at serine 222 upon DNA damage by IR, and this phosphorylation event is required for the activation of the S-phase checkpoint.41 However, FANCD2 is monoubiquitinated at a specific lysine upon MMC-induced DNA damage, and the monoubiquitinated FANCD2 is activated and recruited to the BRCA1 foci.12 A ubiquitin ligase PHF9 which contains a RING-like domain (PHD domain) is required for FANCD2 ubiquitination in vivo.15 Mutations in the conserved lysine residue of FANCD2 abrogate DNA damage-induced monoubiquitination, translocation of FANCD2 to the BRCA1 foci, and the ability of FANCD2 to rescue the DNA repair defects of FA mutant cell lines.12 We found that the basal monoubiquitination level of FANCD2 was significantly abrogated upon celastrol treatment within 4 h, which corresponds to the fact that further celastrol incubation markedly inhibited monoubiquitination of FANCD2 upon DNA-damaging agents (Fig.3), indicating that celastrol could first disrupt the monoubquitination process of FANCD2, then promote FANCD2 polyubiquitination and degradation. Different forms of ubiquitin chains determine the fate of the ubiquitinated proteins. In addition, lysine 63-linked monoubiquitination could regulate the protein function in DNA repair and gene expression, and Lys 48-linked polyubiquitination plays an important role in protein degradation.42 Intriguingly, celastrol has effects on both FANCD2 monoubiquitination and polyubiquitination (Figs2 and 3), indicating that celastrol plays a dual role in FANCD2 ubiquitination modulation and DNA repair inhibition. In line with this, relatively long-term exposure to celastrol could deplete the total protein level of FANCD2 and induce persistent DNA damage, which suggests the role of celastrol in FA pathway inhibition and sensitizing NSCLC cells to therapeutic DNA-damaging drug-induced apoptosis.
The proteasome function is responsible for monoubiquitination of FANCD2, and the proteasome inhibitor is able to attenuate monoubiquitination of FANCD2.19 Celastrol is reported to be able to inhibit proteasome function in prostate cancer cells,43 but in lung cancer this proteasome inhibiting activity is very weak.24 We found that while celastrol and PS341 alone reduced monoubiquitination of FANCD2, combined use of both agents resulted in a synergistic reduction in the monoubiquitinated FANCD2. While PS341 did not trigger proteolysis of FANCD2, it prevented celastrol-induced degradation of FANCD2 (Fig.2), demonstrating that this degradation is proteasome-dependent and celastrol is not a proteasome inhibitor, at least in lung cancer cells. Celastrol could bind FANCD2 directly, and possibly contribute to the inhibition of monoubiquitination and degradation of FANCD2, but the detailed mechanisms, including the binding sites, conformation changes and the potential E3 ligase involved, warrant further investigation.
Recent studies have demonstrated that elevated DNA repair capacity has a significant relationship with the inefficiency of cisplatin in lung cancer treatment, and lower DNA repair capacity may enhance therapeutic efficacy with cisplatin-based chemotherapy.42,44,45 We found that celastrol incubation after chemo drug exposure caused persistent DNA damage and lung cancer cell apoptosis (Fig.4), suggesting that celastrol might induce a reduction of DNA repair capacity. Celastrol is extracted from a Chinese medicinal herb Tripterygium wilfordii Hook. F., which has been used for centuries in treating inflammatory diseases. It can inhibit topoisomerase II, NF-κB, Hsp90 and AKT/Mammalian target of rapamycin pathway.46 We recently showed that celastrol inhibited cell proliferation and induced apoptosis of NSCLC cells, and potentiated the inhibitory effect of cisplatin on lung cancer cells in vitro and in vivo via inhibition of the cancerous inhibitor of phosphatase PP2A (CIP2A).24 These results indicate that celastrol may target multiple oncogenic pathways to enhance the efficacy of cisplatin and, hence, serves as an efficient chemo-sensitizer that has therapeutic potentials in lung cancer.
Acknowledgments
This work was supported by the National Natural Science Funds for Distinguished Young Scholars (81425025), the National Key Program for Basic Research (2012CB910800), and the National Natural Science Foundation of China (81171925, 81201537).
Disclosure Statement
The authors have no conflict of interest to declare.
References
- World Health Organization. GLOBOCAN 2012. GLOBOCAN 2013. [Cited 24 Dec 2014.] Available from URL: http://globocaniarcfr/Pages/fact_sheets_canceraspx.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno L, Favaretto A, Rosell R. Platinum drugs and DNA repair mechanisms in lung cancer. Anticancer Res. 2014;34:493–501. [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharm. 2014;740:364–78. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato M, Phillips RM. Tailoring targeted therapy to individual patients: lessons to be learnt from the development of mitomycin C. Cancer Genomics Proteomics. 2007;4:175–86. [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. How the Fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SN, Bindra RS. Targeting the DNA damage response for cancer therapy. DNA Repair. 2009;8:1153–65. doi: 10.1016/j.dnarep.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2009;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–62. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Longerich S, San Filippo J, Liu D, Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem. 2009;284:23182–6. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–77. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood. 2011;117:5078–87. doi: 10.1182/blood-2010-10-311761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ghosal G, Yuan J, Chen J, Huang J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. 2010;329:693–6. doi: 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- Kratz K, Schopf B, Kaden S, et al. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 Sensitizes cells to interstrand crosslinking agents. Cell. 2010;142:77–88. doi: 10.1016/j.cell.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and Fanconi anemia pathway activation. Cancer Res. 2007;67:7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- Aghajanian C, Dizon DS, Sabbatini P, Raizer JJ, Dupont J, Spriggs DR. Phase I trial of bortezomib and carboplatin in recurrent ovarian or primary peritoneal cancer. J Clin Oncol. 2005;23:5943–9. doi: 10.1200/JCO.2005.16.006. [DOI] [PubMed] [Google Scholar]
- Chirnomas D, Taniguchi T, de la Vega M, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–61. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- Chen P, Li J, Jiang HG, Lan T, Chen YC. Curcumin reverses cisplatin resistance in cisplatin-resistant lung caner cells by inhibiting FA/BRCA pathway. Tumor Biol. 2014 doi: 10.1007/s13277-014-2996-4. http://dx.doi.org/10.1007/s13277-014-2996-4. [DOI] [PubMed] [Google Scholar]
- Burkitt K, Ljungman M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol Cancer. 2008;7:24. doi: 10.1186/1476-4598-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Ma L, Zs Wen, et al. Cancerous inhibitor of PP2A (CIP2A) is targeted by natural compound celastrol for degradation in non-small-cell lung cancer. Carcinogenesis. 2014;35:905–14. doi: 10.1093/carcin/bgt395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, Mitchell JB, DeGraff WG, et al. Chemosensitivity testing of human lung cancer cell lines using the MTT assay. Br J Cancer. 1988;57:540–7. doi: 10.1038/bjc.1988.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobey RA, Petersen DF, Anderson EC, Puck TT. Life cycle analysis of mammalian cells. 3. The inhibition of division in Chinese hamster cells by puromycin and actinomycin. Biophys J. 1966;6:567–81. doi: 10.1016/s0006-3495(66)86678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–93. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Adams J, Palombella VJ, Sausville EA, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–22. [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–71. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Mah LJ, El-Osta A, Karagiannis TC. [gamma]H2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–86. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. In: Bjergbæk L, editor. DNA Repair Protocols. vol. 920 edn. New York: Humana Press; 2012. pp. 613–26. [DOI] [PubMed] [Google Scholar]
- Beamish H, Lavin MF. Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int J Radiat Biol. 1994;65:175–84. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- Wang X, Khadpe J, Hu B, Iliakis G, Wang Y. An overactivated ATR/CHK1 pathway is responsible for the prolonged G2 accumulation in irradiated AT cells. J Biol Chem. 2003;278:30869–74. doi: 10.1074/jbc.M301876200. [DOI] [PubMed] [Google Scholar]
- Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet. 2001;2:446–59. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–85. [PubMed] [Google Scholar]
- D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- Duan W, Gao L, Aguila B, Kalvala A, Otterson GA, Villalona MA. Fanconi anemia repair pathway dysfunction, a potential therapeutic target in lung cancer. Front Oncol. 2014;19:368. doi: 10.3389/fonc.2014.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia//BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2003;23:1000–4. doi: 10.1038/sj.onc.1207256. [DOI] [PubMed] [Google Scholar]
- Gallmeier E, Kern SE. Targeting Fanconi Anemia/BRCA2 Pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res. 2007;13:4–10. doi: 10.1158/1078-0432.CCR-06-1637. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, et al. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–72. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- Sadowski M, Suryadinata R, Tan AR, Roesley SNA, Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–42. doi: 10.1002/iub.589. [DOI] [PubMed] [Google Scholar]
- Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine”, is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–65. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24:123–34. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick CJ, Jackson D, Diffley JFX. Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem. 2004;279:20067–75. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Ma L, Zhou GB. The main anticancer bullets of the Chinese medicinal herb, Thunder God Vine. Molecules. 2011;16:5283–97. doi: 10.3390/molecules16065283. [DOI] [PMC free article] [PubMed] [Google Scholar]