

Abstract
Telomeres are considered anti-cancer targets, as telomere maintenance above a minimum length is necessary for cancer growth. Telomerase abrogation in cancer-prone mouse models, however, only decreased tumor growth after several mouse generations when telomeres reach a critically short length, and this effect was lost upon p53 mutation. Here, we address whether induction of telomere uncapping by inhibition of the TRF1 shelterin protein can effectively block cancer growth independently of telomere length. We show that genetic Trf1 ablation impairs the growth of p53-null K-RasG12V-induced lung carcinomas and increases mouse survival independently of telomere length. This is accompanied by induction of telomeric DNA damage, apoptosis, decreased proliferation, and G2 arrest. Long-term whole-body Trf1 deletion in adult mice did not impact on mouse survival and viability, although some mice showed a moderately decreased cellularity in bone marrow and blood. Importantly, inhibition of TRF1 binding to telomeres by small molecules blocks the growth of already established lung carcinomas without affecting mouse survival or tissue function. Thus, induction of acute telomere uncapping emerges as a potential new therapeutic target for lung cancer.
Keywords: cancer, drug development, shelterin, telomeres, TRF1
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide (Siegel et al, 2012). Adenocarcinoma, a subtype of non-small-cell lung cancer (NSCLC), is the single most common form of lung cancer, comprising 29% of cases (Herbst et al, 2008). The overall 5-year survival rate is of only 15%, due to the long-term ineffectiveness of current therapies and late stage at time of diagnosis (Siegel et al, 2012).
Activating mutations in the K-Ras proto-oncogene are found in 30% of human NSCLC (Rodenhuis et al, 1988). Mutations in the p53 tumor suppressor gene are also common in NSCLC, affecting 50% of the cases (Chiba et al, 1990). Several lung cancer mouse models have been generated that recapitulate human NSCLC by using K-Ras-mutated alleles (Johnson et al, 2001; Guerra et al, 2003). In particular, the lox-stop-lox-K-RasG12V knock-in mouse model, in which endogenous expression of the K-RasG12V oncogene is induced upon Cre recombinase expression, has allowed the study of early stages of lung tumorigenesis (Guerra et al, 2003). Combination of K-RasG12D expression with p53 deficiency recapitulates late-stage lung cancers, including occurrence of invasion, stromal desmoplasia, and metastasis (Jackson et al, 2005). The lox-stop-lox-K-RasG12V mouse model has been instrumental to test novel therapeutic strategies against lung cancer, such as c-Raf, Cdk4, EGF receptor, and Notch (Puyol et al, 2010; Blasco et al, 2011; Maraver et al, 2012). However, to date all therapeutic targets tested have failed to impair the growth of K-RasG12D-induced lung tumors in the context of p53 deficiency (Navas et al, 2012).
Telomeres are specialized heterochromatin structures at the ends of chromosomes composed of tandem TTA (Perera et al, 2008) GGG repeats bound by a protein complex, known as shelterin, that protects chromosome ends from degradation and DNA repair activities (Blasco, 2007; Palm & de Lange, 2008). The shelterin complex is composed of six core proteins, including the telomeric repeat binding factor 1 or TRF1 (de Lange, 2005).
During each cell division cycle, telomeres shorten owing to the incomplete replication of chromosome ends by conventional DNA polymerases, the so-called end-replication problem (Watson, 1972; Olovnikov, 1973). Telomerase can elongate telomeres de novo, thus compensating for telomere shortening in those cells where it is expressed, such as pluripotent and adult stem cells, as well as the majority of late-stage human cancers (Shay & Bacchetti, 1997; Deng & Chang, 2007; Shay & Wright, 2010). Telomerase is composed of a catalytic subunit or TERT and of an RNA component or Terc which is used as template for the de novo synthesis of telomeric repeats (Greider & Blackburn, 1985).
Telomeres are usually shorter in tumor cells compared to the healthy surrounding tissue (de Lange et al, 1990; Shay & Wright, 2010). To maintain a minimum functional telomere length, 80–90% of human tumors reactivate telomerase (Kim et al, 1994; Shay & Bacchetti, 1997; Joseph et al, 2010), and the remaining activate an alternative mechanism to maintain telomeres, known as ALT, based on recombination between telomeric sequences (Bryan et al, 1997). Supporting a role of telomerase deregulation in human cancer, single nucleotide polymorphisms in the locus of human hTERT are associated with various malignancies, including glioma, lung cancer, urinary bladder cancer, melanoma, and breast cancer, among others (McKay et al, 2008; Wang et al, 2008; Rafnar et al, 2009; Shete et al, 2009; Petersen et al, 2010; Melin et al, 2012; Mocellin et al, 2012; Bojesen et al, 2013; Garcia-Closas et al, 2013; Horn et al, 2013; Huang et al, 2013). These findings lead to the development of telomerase-based therapeutic strategies for cancer treatment, some of which are currently tested in clinical trials (Buseman et al, 2012).
Telomerase abrogation in the context of mouse models, however, has only shown anti-tumorigenic activity after several mouse generations of telomerase-deficient Terc−/− mice, when telomeres reach a critically short length, and this anti-tumorigenic effect is abrogated in the absence of p53 (Chin et al, 1999; Greenberg et al, 1999). Similarly, in the context of the K-RasG12V lung carcinogenesis mouse model, telomerase deficiency decreased tumor growth only after five mouse generations, and this effect was lost upon p53 abrogation (Perera et al, 2008).
To circumvent these potential shortcomings of telomerase inhibition, here we set out to address the therapeutic effect of acute telomere uncapping owing to Trf1 abrogation (Martinez et al, 2009) in the K-RasG12V lung cancer model (Guerra et al, 2003). TRF1 is an essential component of shelterin and, in addition, it is enriched in adult stem cells and pluripotent stem cells, suggesting that its inhibition may also target cancer stem cells (Boue et al, 2010; Schneider et al, 2013). In this regard, we have previously shown that TRF1 deletion in stratified epithelia could promote cancer development when in a p53-deficient background (Martinez et al, 2009). However, in contrast to POT1, another shelterin component, TRF1, has not been found mutated in human cancer (Ramsay et al, 2013; Robles-Espinoza et al, 2014; Shi et al, 2014; Bainbridge et al, 2015). Indeed, TRF1 has been reported to be overexpressed in several tumor types (Matsutani et al, 2001; Ohyashiki et al, 2001; Fujimoto et al, 2003; Oh et al, 2005; Bellon et al, 2006). Thus, here we set out to address whether Trf1 deletion in the context of oncogenic K-Ras-induced lung cancer mouse model would act as a tumor suppressor or as an oncogene.
We found that Trf1 genetic ablation effectively reduces the size and malignancy of p53-null K-RasG12V lung carcinomas and increases mouse survival. This tumor-suppressive effect of Trf1 deficiency occurs already at the first mouse generation and is independent of telomere length. Furthermore, long-term conditional whole-body Trf1 deletion in adult mice does not affect mouse viability and survival. Moreover, we show that chemical inhibition of TRF1 can be achieved in vivo by using small molecules, which effectively impair the growth of already established lung adenocarcinomas without affecting mouse and tissue viability. Thus, acute telomere uncapping owing to TRF1 inhibition represents a novel potent therapeutic strategy for K-Ras-induced lung cancer.
Results
Trf1 deficiency impairs immortalization of MEFs expressing the K-RasG12V oncogene even in a p53-deficient background
To assess the effect of Trf1 abrogation in the context of lung cancer induced by expression of the K-RasG12V oncogene, we crossed K-Ras+/LSLG12Vgeo mice (designated from now on as K-Ras+/G12V) (Guerra et al, 2003) to a strain carrying a floxable allele of Trf1 (Trf1lox/lox) either wild-type or deficient for p53 (p53−/−) (Martinez et al, 2009) (Fig.1A). First, we isolated primary (passage 2) mouse embryonic fibroblasts (MEFs) and transduced them with a retrovirus encoding the Cre recombinase (pBabe-Cre). This allowed the expression of the resident K-RasG12V oncoprotein simultaneously with the deletion of exon 1 of the Trf1lox allele (Fig1A). A p53-null background was used to allow for the growth of Trf1-deleted cells, which otherwise show severe proliferative defects (Supplementary Fig S1A) (Martinez et al, 2009; Thanasoula et al, 2010). While Trf1+/+ K-Ras+/G12V p53−/− MEFs showed a 16-fold increase in cell number at day 7 post-plating, Trf1Δ/Δ K-Ras+/G12V p53−/− MEFs only increased their population by fourfold in the same time (Supplementary Fig S1A), indicating a severe growth impairment of Trf1-deficient K-RasG12V-expressing cells compared to the Trf1+/+ K-Ras+/G12V p53−/− controls.
Figure 1.
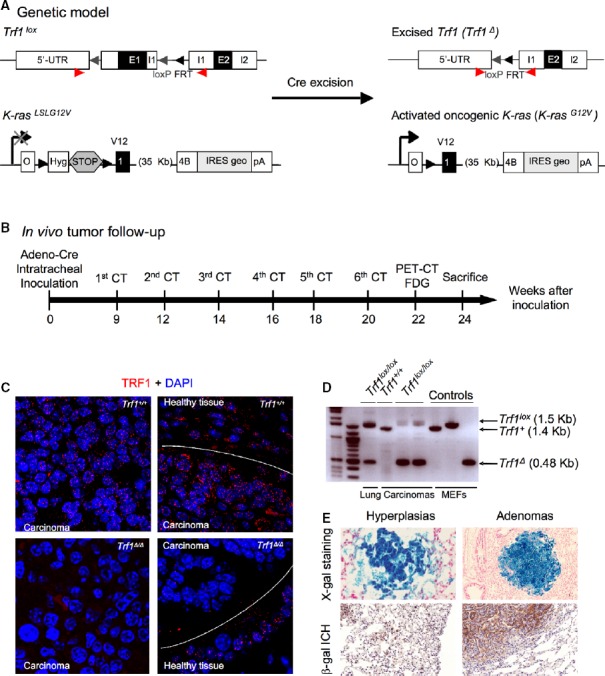
- Genetic model. Trf1lox and K-RasLSLGV12 alleles are depicted before and after Cre-mediated excision.
- In vivo imaging schedule. Eight- to ten-week-old mice were intratracheally infected with adeno-Cre, mice were analyzed every 2 weeks by computerized tomography (CT), and 22 weeks post-infection, a positron emission tomography (PET) was performed. Mice were sacrificed 24 weeks post-infection for further histological analysis.
- TRF1 immunofluorescence of the lungs. Notice the absence and presence of TRF1 signal in the carcinomas and surrounding healthy tissue of Trf1Δ/Δ mice, respectively.
- Analysis of Trf1 excision by PCR. Notice the completed excision in carcinomas of Trf1lox/lox lungs.
- Detection of β-galactosidase activity in the lungs as a surrogate marker of oncogenic K-RasG12V expression.
To address how Trf1 ablation impairs the growth of K-RasG12V-expressing MEFs, we first analyzed cellular senescence by the β-galactosidase senescence-associated activity. Trf1Δ/Δ K-Ras+/G12V p53+/+ MEFs presented 1.9-fold higher percentage of senescent cells 7 days post-plating compared to Trf1+/+ K-Ras+/G12V p53+/+ MEFs (Supplementary Fig S1B) (Martinez et al, 2009). Of note, p53 deficiency did not abolish Trf1 deficiency-mediated senescence. Indeed, Trf1Δ/Δ K-Ras+/G12V p53−/− MEFs showed a 21-fold higher percentage of senescent cells compared to Trf1+/+ K-Ras+/G12V p53−/− MEFs 7 days post-plating (Supplementary Fig S1B), most likely reflecting an additive effect of K-Ras oncogene-induced senescence and Trf1 deficiency-induced senescence (Serrano et al, 1997; Martinez et al, 2009). No significant differences in apoptosis were detected between genotypes by caspase-3 activation (Supplementary Fig S1C). Thus, Trf1 abrogation in MEFs expressing mutant K-Ras leads to higher numbers of senescent cells even in the absence of p53.
Next, we addressed the effect of Trf1 abrogation in immortalization of MEFs. To this end, we performed a colony formation assay, which reflects on the clonogenic capacity of individual cells. p53-proficient MEFs did not form any colonies in agreement with the fact that p53 wild-type MEFs do not spontaneously immortalize (Supplementary Fig S1D and E) (Harvey & Levine, 1991; Parrinello et al, 2003). In contrast, p53-deficient MEFs were able to form immortalized colonies, although Trf1Δ/Δ K-Ras+/G12V p53−/− MEFs formed fewer and smaller colonies than Trf1+/+ K-Ras+/G12V p53−/− MEFs. In summary, Trf1 deficiency limits both proliferation and cellular immortalization of K-Ras-expressing cells in vitro even in the absence of p53.
Trf1 deficiency impairs K-RasG12V-mediated lung cancer and increases mouse survival even in the absence of p53
We next set out to determine the impact of Trf1 deficiency in vivo in the K-Ras-induced lung carcinogenesis model. To this end, 8-week-old Trf1lox/lox K-Ras+/G12V p53+/+ and Trf1lox/lox K-Ras+/G12V p53−/− mice, as well as their respective Trf1 wild-type controls, were infected by intratracheal instillation with replication-defective adenoviruses encoding the Cre recombinase (adeno-Cre) (Materials and Methods). This strategy allowed the expression of the resident K-RasG12V oncoprotein simultaneously with the ablation of the Trf1lox allele in the infected lung cells (Fig1A). Nine weeks after viral inoculation, tumor growth was measured by using computed tomography (CT) every second week until 24th week post-infection when the experiment was concluded. At 22 weeks post-infection, positron emission tomography (PET) was performed to monitor tumor malignancy (Fig1B). At 24th week post-infection, the animals were sacrificed to carry a full histopathological analysis of the lungs, and to confirm K-RasG12V expression and Trf1 deletion in the lesions (Fig1B). Trf1 deletion was monitored in all tumors by TRF1 immunofluorescence and by PCR (Fig1C and D). K-RasG12V expression in tumors was confirmed by detecting the expression of its beta-galactosidase (β-geo) reporter (Fig1E).
In vivo tumor follow-up by CT scan showed that in a p53-proficient background, Trf1-deleted mice showed a delayed onset of the first CT scan-detectable lesions from 9 weeks in Trf1 wild-type lungs to 12 weeks in the Trf1-deleted ones (Fig2A). However, after this initial lag, both genotypes showed a similar growth of the tumor lesions by CT scan (Fig2A). Post-mortem lung analysis revealed that the number of tumors per mouse was higher in Trf1 wild-type than in Trf1lox/lox mice although tumors were histologically identical (Fig2B). Importantly, immunofluorescence analysis of Trf1 expression showed that all tumors in Trf1-deleted lungs were escapers and had normal Trf1 expression (Fig2C). Thus, Trf1 is essential for K-RasG12V-induced lung tumor development in a p53-proficient background as no tumors lacking Trf1 expression were found (Fig2C).
Figure 2.
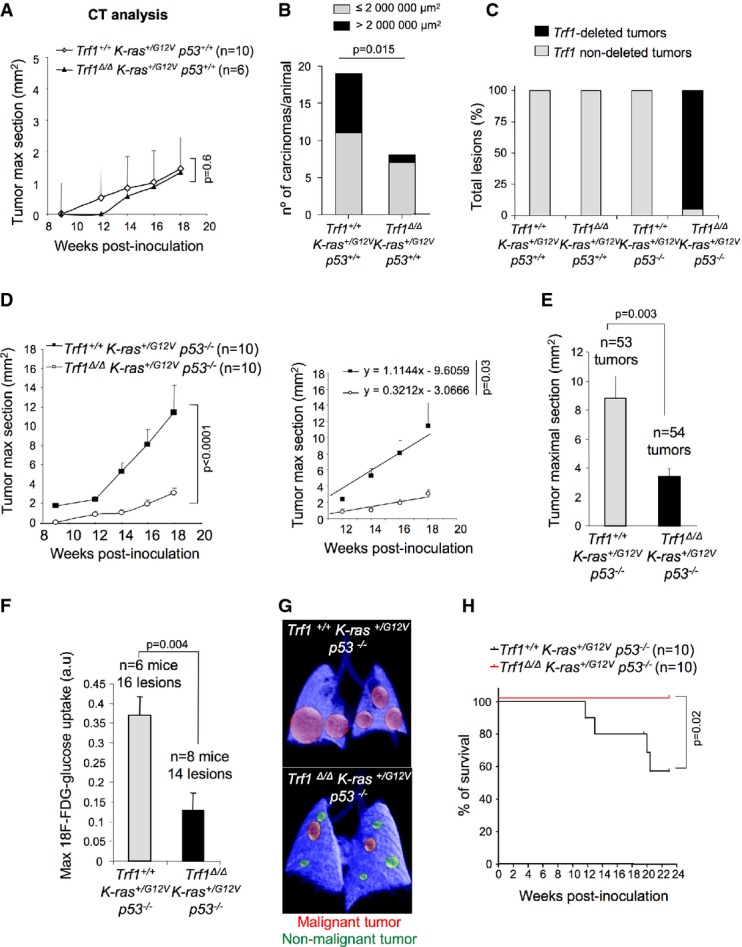
- Tumor growth curve of Trf1+/+ K-Ras+/G12V p53+/+ and Trf1Δ/Δ K-Ras+/G12V p53+/+ measured by computed tomography (CT).
- Quantification of the number and size of Trf1+/+ K-Ras+/G12V p53+/+ and Trf1Δ/Δ K-Ras+/G12V p53+/+ carcinomas at death point.
- Percentage of tumors that have deleted Trf1 quantified by TRF1 immunofluorescence after mice had been sacrificed. Post-mortem analysis of Trf1 deletion in each tumor revealed that none of the Trf1lox/lox K-Ras+/G12V p53+/+ ones had excised Trf1.
- Tumor growth curve and tumor growth slope of Trf1+/+ K-Ras+/G12V p53−/− and Trf1Δ/Δ K-Ras+/G12V p53−/− measured by CT.
- Tumor maximal section of Trf1+/+ K-Ras+/G12V p53−/− and Trf1Δ/Δ K-Ras+/G12V p53−/− lungs measured by histological analysis before death point by CT.
- Maximum 18F-FDG-glucose uptake by Trf1+/+ K-Ras+/G12V p53−/− and Trf1Δ/Δ K-Ras+/G12V p53−/− tumors 22 weeks after infection by positron emission tomography (PET).
- Representative PET-CT image of Trf1+/+ K-Ras+/G12V p53−/− and Trf1Δ/Δ K-Ras+/G12V p53−/− lungs.
- Survival curve of Trf1+/+ K-Ras+/G12V p53−/− and Trf1Δ/Δ K-Ras+/G12V p53−/− mice.
Data information: Error bars represent standard error. t-test, chi-squared (B) test, or log-rank (Mantel–Cox; H) test was used to assess statistical significance. The number of mice and the number of tumors are indicated in each case.
Next, we studied the impact of Trf1 deletion in K-RasG12V-induced lung tumors in a p53-deficient background, a situation that resembles many human lung tumors. CT analysis revealed that Trf1Δ/Δ K-Ras+/G12V p53−/− tumors grew markedly slower and reached a smaller size at their end point than Trf1+/+ K-Ras+/G12V p53−/− tumors (Fig2D and E). PET analysis at 22 weeks post-infection revealed that Trf1Δ/Δ K-Ras+/G12V p53−/− tumors showed less metabolic activity than Trf1+/+ K-Ras+/G12V p53−/− tumors indicating a lower grade of malignancy (Fig2F and G). Notably, in agreement with the lower malignancy, Trf1lox/lox K-Ras+/G12V p53−/− mouse survival was significantly higher than that of Trf1+/+ K-Ras+/G12V p53−/− mice (Fig2H). Of note, in this setting only 5% of the Trf1lox/lox p53−/− tumors were found to be escapers (Fig2C).
By blind histopathological analysis of the lungs, we confirmed that Trf1Δ/Δ K-Ras+/G12V p53−/− lungs developed less carcinomas than Trf1+/+ K-Ras+/G12V p53−/− lungs (Supplementary Fig S2A–C). In addition, the malignant lesions in Trf1Δ/Δ K-Ras+/G12V p53−/− lungs were smaller compared to Trf1+/+ K-Ras+/G12V p53−/− lungs (Supplementary Fig. S2A–C). In summary, Trf1 deletion effectively impairs progression to full-blown carcinomas even in the absence of p53.
Trf1 abrogation induces telomeric DNA damage and apoptosis in p53-deficient lung carcinomas
Previous reports have shown that abrogation of Trf1 in different cell types causes a persistent DNA damage response at chromosome ends, which leads to decreased cell viability (Martinez et al, 2009; Sfeir et al, 2009; Beier et al, 2012; Schneider et al, 2013). To address whether the decreased growth and lower malignancy of Trf1-deficient lung tumors were associated with increased DNA damage in the lesions, we quantified γH2AX DNA damage foci and their colocalization to telomeres (the so-called telomere-induced foci or TIFs) in lung carcinoma sections. The percentage of γH2AX-positive cells was significantly higher in Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas compared to Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig3A). Furthermore, co-immunofluorescence staining of γH2AX foci with RAP1, another shelterin component localized at telomeres, showed increased number of DNA damage foci at telomeres in Trf1Δ/Δ K-Ras+/G12V p53−/− than in Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig3B).
Figure 3.
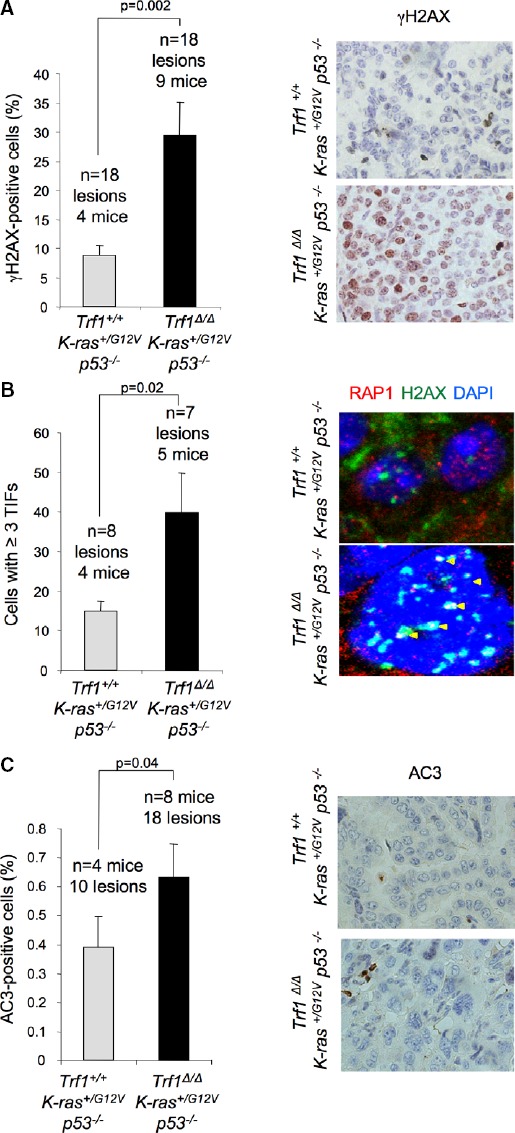
- Percentage of cells showing γH2AX foci in carcinomas of the indicated genotypes (left panel). Representative images of γH2AX immunohistochemistry (right panel).
- Percentage of cells showing 3 or more γH2AX and RAP1 colocalizing foci (TIFs) (left panel). Representative images of γH2AX and RAP1 double immunofluorescence (right panel). Yellow arrowheads: colocalization of γH2AX and RAP1.
- Percentage of active caspase-3 (AC3)-positive cells (left panel). Representative images of AC3 immunohistochemistry (right panel).
Data information: Error bars represent standard error. The number of mice and carcinomas analyzed per genotype is indicated. t-test was used to assess statistical significance.
To address the cellular effects of increased telomere damage in lung carcinomas, we first determined the percentage of apoptotic cells in lung carcinoma lesions. The percentage of carcinoma cells that were positive for the apoptotic marker active caspase-3 (AC3) was higher in Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas compared to Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig3C). Thus, Trf1 deletion in lung carcinomas leads to increased telomeric damage and subsequent induction of apoptosis.
Trf1 deficiency leads lower proliferation and increased G2 arrest and mitotic defects in p53-deficient lung carcinomas
To determine whether Trf1 deletion in the context of K-RasG12V-induced lung cancer also leads to proliferation defects, we performed Ki67 immunohistochemistry directly on lung carcinoma sections. We observed a lower proliferation index (Ki67-positive cells) in Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas compared to Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig4A). To determine the cell cycle phase where Trf1-deleted cells showed defects, we analyzed the staining pattern of phospho-histone H3 (Ser10). A pH3 pan-nuclear staining is a distinctive feature of mitotic cells, whereas pH3 foci are characteristic of G2 cells (Hendzel et al, 1991). We found that the percentage of cells positive for G2-distinctive pH3 foci pattern was significantly increased in Trf1Δ/Δ K-Ras+/G12V p53−/− carcinoma lesions compared to Trf1+/+ K-Ras+/G12V p53−/− lesions (Fig4B), suggestive of increased G2 arrest.
Figure 4.

- Percentage of Ki67-positive cells in the carcinomas of the indicated genotypes (left panel). Representative images of Ki67 immunohistochemistry (right panel).
- Percentage of pH3-positive cells in the carcinomas of the indicated genotypes (left panel). Representative images of pH3 immunohistochemistry (right panel). Red arrowheads: pH3-positive cells.
- Percentage of giant nuclei in the carcinomas of the indicated genotype.
- Percentage of anaphase bridges out of total anaphases in the carcinomas of the indicated genotypes.
- Representative images of giant nuclei, multilobulated nuclei, anaphase bridges, and multipolar mitoses. Red arrowheads indicate the corresponding mitotic aberrations indicated in the image.
Data information: Error bars represent standard error. The number of mice and carcinomas analyzed per genotype is indicated. t-test was used to assess statistical significance.
Persistent telomere damage can result in bypass of mitosis leading to endoreduplication and tetraploidy (Davoli et al, 2010). In line with this, Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas presented an increased number of giant nuclei indicative of endoreduplication, as well as increased anaphase bridges, compared to Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig4C–E). Similarly, Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas presented cells showing bizarre multilobulated nuclei and multipolar mitosis, which were not present in Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Fig4E). This type of aberrant nuclei have been previously related to mitotic catastrophes (Vakifahmetoglu et al, 2008). These observations suggest that lung carcinoma cells require TRF1 for proper completion of mitosis and that Trf1 deletion leads to severe mitotic defects.
Trf1 downregulation in mouse cell lines derived from K-RasG12V p53-deficient lung carcinomas as well as in human lung cancer cell lines impairs cancer growth and metastasis in xenograft models
To validate these results using an independent strategy to inhibit TRF1, as well as to assess the effect of TRF1 abrogation in already established K-Ras-induced lung tumors, we downregulated Trf1 expression by using shRNA technology in three K-RasΔ/G12Vgeo p53−/− mouse cancer cell lines derived from three independent mouse lung carcinoma lesions and assessed the effects on tumor growth using two independent allograft experiments. First, to address the effect of TRF1 inhibition in tumor growth in vivo, we subcutaneously injected 50,000 lung carcinoma cells into immunodeficient mice and followed tumor onset and growth. Trf1 downregulation resulted in a marked delay in tumor onset as well as in a significantly decreased tumor growth (Fig5A–D). Three weeks after injection, mice were sacrificed and the tumors were histologically analyzed. We confirmed that Trf1 downregulation was maintained during in vivo tumor development (Fig5E). Trf1-downregulated tumors showed decreased proliferation and increased DNA damage as well as increased apoptosis compared to controls (Fig5F–H). Again, Trf1-downregulated tumors presented a high proportion of aberrant nuclei and anaphase bridges (Fig5I). Next, to study the effect of TRF1 inhibition in the metastatic potential of established lung cancer cells induced by K-Ras expression, we intravenously injected 150,000 K-RasΔ/LG12Vgeo p53−/− lung cells into immunodeficient mice. Tail vein injection of tumor cells results in lung metastasis (Elkin & Vlodavsky, 2001). Three weeks after injection, the mice were sacrificed and lungs were subjected to full histopathology analysis. Again, we confirmed that Trf1 downregulation was maintained in the generated lung metastasis (Fig5J). Importantly, Trf1 downregulation resulted in smaller lung metastasis (Fig5K and I), coincidental with increased DNA damage, decreased proliferation, and increased apoptosis compared to the controls (Fig5M–O). These results indicate that even a partial decrease in TRF1 levels of approximately 50% in tumors very significantly impairs lung tumor growth and lung metastasis, arguing that putative small molecule inhibitors of TRF1 could be effective.
Figure 5.

- A–C The latency (A), volume (B), and weight (C) of subcutaneous tumors generated by control and Trf1-downregulated K-RasG12V-transformed lung cells in athymic mice.
- D Representative images of the subcutaneous tumors.
- E Trf1 expression levels measured by qPCR in the injected cell line and in the generated subcutaneous tumors.
- F–H Number of Ki67-positive (F), number of γH2AX-positive (G), and number of active caspase-3-positive (H) cells per field in the subcutaneous tumors.
- I Representative images of aberrant giant nuclei and anaphase bridges in the Trf1-downregulated subcutaneous tumors compared to the normal nuclei of control tumors.
- J TRF1 immunofluorescence shows the downregulation of Trf1 in lung tumors of the mice intravenously injected with control and Trf1-downregulated cells.
- K Tumor area measured in the lungs of the mice intravenously injected with control and Trf1-downregulated cells.
- L Representative images of the lungs colonized by control and Trf1-downregulated cells, respectively.
- M–O Number of Ki67-positive (M), number of γH2AX-positive (N), and number of active caspase-3-positive (O) cells per field in the lung tumors.
- P Trf1 expression levels measured by qPCR in the A549 cell line infected either with sh-scrambled or sh-Trf1.
- Q Growth of A549-derived tumors.
Data information: Error bars represent standard error. The number of mice and tumors analyzed per condition is indicated. t-test was used to assess statistical significance.
Next, we tested whether TRF1 depletion had similar effects on human lung cancer cell lines. To this end, we downregulated Trf1 levels by using shRNAs in a K-Ras-mutated human lung carcinoma cell line, the A549 (ATCC n°; CCL-185), a human lung cancer cell line harboring wild-type p53 (Fig5P). We then injected subcutaneously in immunodeficient mice 150,000 cells either infected with sh-scrambled or Trf1-shRNA and followed tumor development. TRF1 downregulation resulted in a markedly delayed tumor onset and growth. Indeed, control cells started to develop tumors 11 days after injection, while the latency of TRF1-depleted cells was of 17 days. Moreover, after 24 days of follow-up, only two out of eight injections with TRF1-downregulated cells were able to generate tumors whose growth were significantly impaired as compared to control cells (Fig5Q and Supplementary Fig S3). Thus, TRF1 downregulation blocks the growth of cell lines derived from already formed lung mouse tumors and has proven efficacy in one human cancer cell line.
Trf1 deficiency impairs lung carcinomas independently of telomere length
To demonstrate that the increased apoptosis and proliferation defects observed in Trf1-deleted lung carcinomas were not due to shorter telomeres compared to the TRF1-proficient tumors, we determined telomere length by telomere quantitative FISH directly on lung carcinoma sections. Indeed, telomeres were longer in the Trf1Δ/Δ K-Ras+/G12V p53−/− carcinomas compared to Trf1+/+ K-Ras+/G12V p53−/− carcinomas (Supplementary Fig S4A–C). As telomere length reflects the proliferative history of a given tissue, the observation that Trf1+/+ K-Ras+/G12V p53−/− carcinomas present shorter telomeres than TRF1-deficient ones is likely to reflect the lower proliferation rate of TRF1-deficient tumors.
Whole-body Trf1 depletion allows normal mouse survival and normal tissue function
A prerequisite that must be fulfilled by any potential anti-cancer target is that its systemic inhibition in healthy tissues does not compromise organism viability. It has been shown that Trf1 deletion is deleterious at early points of development (Karlseder et al, 2003; Martinez et al, 2009). In addition, acute TRF1 removal from bone marrow in 8-week-old mice leads to bone marrow failure (Beier et al, 2012). To validate TRF1 as a therapeutic drug target in lung cancer treatment, we set out to analyze the effects of whole-body TRF1 depletion in the context of adult mice and its impact on long-term mouse viability. To this end, we first generated a new mouse model, Trf1lox/lox hUBC-CreERT2 mice, in which the expression of CreERT2 is transcriptionally controlled by the ubiquitously and constitutively regulated ubiquitin promoter (Ruzankina et al, 2007; Martinez et al, 2009) (Fig6A). Then, twelve-week-old Trf1+/+ hUBC-CreERT2 and Trf1lox/lox hUBC-CreERT2 mice were subjected to a tamoxifen-containing diet for 7 weeks in order to induce Trf1 deletion. After this period of time, a total of eight mice of each genotype were sacrificed to analyze the extent of Trf1 deletion in a panel of different tissues as well as to perform full histopathological analysis. qPCR analysis showed that Trf1 had been successfully deleted from heart, intestine, lung, skin, blood, liver, kidney, bone marrow, brain, and stomach (Fig6B). TRF1 immunofluorescence in skin and small intestine sections confirmed partial depletion of TRF1 protein in these tissues (Fig6C). Despite successful TRF1 depletion after 7 weeks in a tamoxifen-containing diet, neither Trf1+/+ nor Trf1Δ/Δ showed signs of viability loss or decreased survival (Fig6D). Histopathological analysis of the tissues revealed that the Trf1Δ/Δ hUBC-CreERT2 mice showed alterations in rapidly proliferating tissues consistent with the stem cell compartment being affected (Fig6E). TRF1-depleted basal skin, intestinal crypts, and bone marrow progenitors presented anisocytosis (Supplementary Figs S5 and S6). Trf1-deficient basal skin displayed areas with low cellularity and follicular cysts (Fig6E and Supplementary Fig S5A). Trf1Δ/Δ intestinal crypts showed an increased number of mitosis, but the microvilli length was normal (Fig6E and Supplementary Fig S5B). Trf1Δ/Δ bone marrow presented megakaryocytes with reduced cytoplasm (Fig6E and Supplementary Fig S6A). Blood counts showed a small decrease in the number of platelets and lymphocytes (Fig6F). Of note, only one Trf1Δ/Δ hUBC-CreERT2 mouse showed a moderate bone marrow aplasia (Fig6E and Supplementary Fig S6A). Of note, 6 months on tamoxifen diet did not further decrease Trf1 expression as compared to the levels observed after 7 weeks and did not aggravate neither pathologies nor mouse viability (Fig6G and data not shown). Importantly, tamoxifen retrieval from the diet resulted in a rescue of pathologies as well as in a recovery of Trf1 expression levels (Fig6E–G). Indeed, after 3 weeks, placing the mice on standard diet Trf1 levels in blood, intestine, skin, and bone marrow increased 20-, 10-, 30- and 200-fold, respectively, compared to the levels observed in mice on tamoxifen diet. However, recovery of Trf1 expression did not reach wild-type levels (Fig6G). After 3 weeks and 4 months on standard diet, platelet blood counts were also recovered to wild-type levels (Fig6F). Thus, a partial ubiquitous TRF1 downregulation, although resulting in decreased cellularity in some highly proliferative compartments, such as the bone marrow and the blood, is compatible with mouse viability.
Figure 6.

- Trf1lox/lox hUBC-CreERT2 genetic mouse model.
- Trf1 expression levels in the indicated tissues of wild-type and Trf1lox/lox hUBC-CreERT2 mice subjected to a tamoxifen-containing diet for 7 weeks.
- Representative images of TRF1 immunofluorescence and quantification of the percentage of TRF1-positive cells in skin and intestine sections of wild-type and Trf1lox/lox hUBC-CreERT2 mice subjected to a tamoxifen-containing diet for 7 weeks.
- Survival curve of wild-type and Trf1lox/lox hUBC-CreERT2 mice subjected to a tamoxifen-containing diet for 7 weeks.
- Quantification of the histological alterations observed in tamoxifen-treated Trf1lox/lox hUBC-CreERT2 mice and 4 months after tamoxifen retrieval compared to their wild-type counterparts.
- Quantification of blood cell populations in wild-type and Trf1lox/lox hUBC-CreERT2 mice subjected to a tamoxifen-containing diet for 7 weeks and after 3 weeks and 4 months of tamoxifen retrieval.
- Trf1 expression levels in blood, intestine, skin, and bone marrow of Trf1lox/lox hUBC-CreERT2 mice subjected to a tamoxifen-containing diet either for 7 weeks or for 6 months and after 3 weeks tamoxifen retrieval compared to wild-type mice.
Data information: Error bars represent standard error. The number of mice analyzed per genotype is indicated in each case. t-test was used to assess statistical significance. TMX, tamoxifen.
Identification of chemical compounds that disrupt TRF1 binding to telomeres
We next set out to identify chemicals that disrupt TRF1 binding to telomeres. To do so, we designed a cell-based system for high-throughput screening. Induced pluripotent stem (iPS) cells derived from eGFP-TRF1 knock-in (KI) mouse embryo fibroblasts (MEFs) were used (Schneider et al, 2013). The eGFP-TRF1 protein forms fluorescence foci localizing at the telomere that strongly decrease in intensity in control cells expressing a sh-Trf1 or in cells heterozygous for eGFP-TRF1 expression (eGFP-Trf1+/KI) showing that this cellular system is an excellent tool to track TRF1 and telomeres in vivo (Supplementary Fig S7A). Homozygous version (eGFP-Trf1KI/KI)—with strong foci intensity—, heterozygous version (eGFP-Trf1+/KI) and knockdown for TRF1 (sh-Trf1)—with low foci intensity—were used to validate this system (Supplementary Fig S7B). The Z'-factor coefficient, a statistical parameter that in addition to considering the window in the assay also considers the variance around both the high and low signals in the assay, is commonly used to assess the robustness of high-throughput screening (HTS) assays. The Z'-factor was calculated as follows: Z'-factor = 1 − 3 × (sp + sn)/¦mp − mn¦, where m: mean fluorescence intensity and s: standard deviation; n: negative control (sh-Trf1 or eGFP-Trf1+/KI heterozygous, minimum signal) and p: positive control (homozygous eGFP-Trf1KI/KI, maximum signal). A Z'-factor value of 0.75, when comparing homozygous (homo) vs. heterozygous (het) or 0.86 comparing homo vs. sh-Trf1, confirmed the feasibility of this screening system (Supplementary Fig 7C). We carried out a screening campaign using a small collection of 640 compounds selected as representative of the ETP-CNIO library (Materials and Methods). Screening was performed at 8 h and at 12.5 μM final concentration on eGFP-Trf1KI/KI cells. Foci distribution of the homozygous eGFP-Trf1KI/KI (control) or sh-Trf1, were taken as internal controls. Compounds decreasing percentage of high-intensity foci above 25% of the control were selected as “hit” candidates, for further validations. In addition, the number of nuclei was counted. Compounds significantly affecting cell viability were not considered as positive “hits”.
The screening retrieved a number of positive hits belonging to different chemical classes. The identified hits were newly resynthesized and tested confirming the observed activity. The search for analogues within the ETP-CNIO library and their screening identified additional active compounds. Among the different hits, two of them were selected for further biological investigation attending to their primary activity as “TRF1 inhibitors” and additionally to their potential to be used as tool compounds for in vivo validation experiments. The selected hits ETP-47228 and ETP-47037 are included as examples in CNIO international patent applications WO2010119264 and WO2011089400, respectively. The general structures of both compounds are depicted in Supplementary Fig S8A.
We next validated our hits by measuring eGFP-TRF1 fluorescence intensity in e-GFP-Trf1KI/KI iPS cells after treatment with DMSO, ETP-47228, or ETP-47037 for 8 h at 10 μM. iPS cells transduced with a sh-Trf1 were used as a positive control. Both ETP-47228 and ETP-47037 induced a decrease of 31.33 and 27.32%, respectively, of e-GFP-TRF1 levels, as compared to a 57% decrease observed in cells treated with sh-Trf1 (Supplementary Fig S7D).
To address whether these compounds also decrease endogenous TRF1 levels, we treated wild-type iPS cells (eGFP-Trf1KI/KI) with 10 μM ETP-47228 for 8 h and quantified TRF1 levels by immunofluorescence. Cells treated with ETP-47228 contained 25% lower levels of TRF1 signal as compared to control cells treated with DMSO (Supplementary Fig S9A). Interestingly, neither the Trf1 transcriptional levels nor the total TRF1 protein levels were affected by ETP-47228 or ETP-47037 (Supplementary Fig S9B). In analogy to Trf1 genetic ablation, chemical inhibition of TRF1 foci intensity was accompanied by increased γH2AX DNA damage foci and increased telomeric DNA damage foci (TIFs), together with impairment of cellular proliferation with increasing ETP-47228 concentrations (Supplementary Fig S9C–E).
Effective chemical Trf1 inhibition in lung adenocarcinoma-derived cells
Next, we treated lung mouse adenocarcinoma cell lines with 10 μM of both ETP-47228 and ETP-47037 for 24 and 48 h and quantified TRF1 levels compared to DMSO-treated cells. Again, treatment of lung cancer cells with both ETP-47228 and ETP-47037 resulted in decreased TRF1 foci levels, increased γH2AX foci, as well as induction of TIFs (Fig7A–C). Similarly, both compounds affected proliferation from concentrations of approximately 5 μM (Fig7D).
Figure 7.

- Quantification of TRF1 levels by immunofluorescence in lung tumor-derived cell line treated with DMSO, with 10 μM ETP-47228 (24 h), and with 10 μM ETP-47037 (48 h). Representative images are shown to the right.
- Quantification of γH2AX levels by immunofluorescence in lung tumor-derived cell line treated with DMSO, with ETP-47228 (24 h), and with ETP-47037 (48 h). Representative images are shown to the right.
- Quantification of telomere-induced foci (TIFs) by double immunofluorescence with anti-RAP1 and anti-γH2AX antibodies. Representative images are shown to the right. White arrowheads: colocalization of γH2AX and RAP1.
- Effect of different ETP-47228 and ETP-47037 concentrations during 24 h on proliferation in lung tumor-derived cell line relative to the growth of DMSO-treated cells.
- Tumor growth quantification in allograft model ETP-47037 or with ETP-47228.
Data information: The data represent the mean values of two to three independent experiments (A–D). Error bars represent standard errors. t-test was used to assess statistical significance.
Next, we generated allograft mouse models with lung cancer cells pretreated with either DMSO, ETP-47228, or ETP-47037 and followed tumor onset and growth during 12 days. Treatment with both compounds significantly impaired initial tumor development in allograft models (Fig7E).
In vivo treatment with ETP-47037 disrupts TRF1 and effectively impairs lung carcinoma progression
Next, we addressed whether TRF1 chemical inhibitors administered in vivo could inhibit the growth of already established K-RasG12V lung carcinomas lacking p53. To this end, we selected compound ETP-47037 owing to its in vivo pharmacokinetic properties. ETP-47037 pharmacokinetic properties were determined after IV and PO (per os) administration in BALB/c mice at doses of 3.0 and 9 mg/kg of body weight, respectively (Supplementary Fig S8). In particular, ETP-47037 is an orally bioavailable compound with an absorbed fraction F = 29.5%. The half-life after IV administration is 0.5 h, and the same parameter in the oral arm extends up to 5.2 h. ETP-47037 is highly stable in vivo with a total clearance of 0.65 l/h/kg, which means 12% of the hepatic blood flow for mice. The compound is distributed in tissues, as inferred by a volume of distribution of 0.6 l/kg similar to the total body water content (Supplementary Materials and Methods).
Mice with already developed p53-null lung carcinomas were treated by oral gavage during 10 days (8 days in total with a 2-day gap) with 75 mg/kg body weight of ETP-47037. Control mice were similarly treated with vehicle. Previously to the start and at the end of the treatment, mice were subjected to computerized tomography (CT) for quantification of tumor growth area. Strikingly, 10 days of treatment with this compound effectively impaired the progression of already formed lung carcinomas compared to the group treated with vehicle (Fig8A and B). Of note, one of the tumors detected before the treatment was located in a highly inflammatory region, and for this reason, we could not accurately determine its size before the treatment (white arrowhead in Fig8B). TRF1 foci levels were significantly decreased in tumor samples as well as in intestines of mice treated with ETP-47037 compared to the vehicle (Fig8C). Decrease in TRF1 foci signal was accompanied with a significant increase in γH2AX-positive cells in both intestines and lung tumors (Fig8D). In addition, ET-47037-treated tumors showed a significant decrease in proliferating and mitotic cells and increase in the number of G2-arrested cells (Fig9A–D). Importantly, during the treatment, the mice showed a normal viability and did not show any signs of sickness. Histopathological analysis of the intestine did not reveal any apparent lesion although we saw increased aberrant mitotic figures and nuclei characteristic of TRF1 inhibition (Fig9E). In bone marrow, moderate aplasia, necrotic cells, and hemosiderosis were observed. In the skin, multinucleated cells and giant nuclei were detected (Fig9F), a hallmark of TRF1 targeting. Of note, ETP-47037 did not induce changes in telomere length in treated tumors as compared to untreated ones (Fig9G). These findings indicate that TRF1 inhibition can be achieved in vivo using chemical compounds and that there is a therapeutic window for targeting TRF1 in cancer that merits further work.
Figure 8.
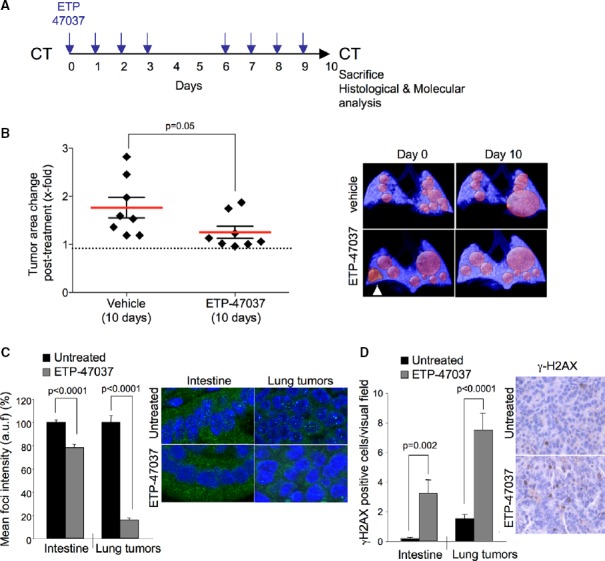
- Schematic representation of the ETP-47037 treatment protocol. Mice with already developed lung carcinomas were subjected to computerized tomography (CT) measurements before the start of the treatment. ETP-47037 was given at a dose of 75 mg/kg body weight by oral gavage 8 days out of the ten that the experiment lasted as indicated. At the end of the treatment, a CT was performed for quantification of tumor area and mice were sacrificed for further histological and molecular analysis. Control mice were similarly treated with vehicle.
- Quantification of tumor growth relative to initial tumor size. Representative CT images are shown to the right. The white arrowhead indicates a tumor within a highly inflammatory region.
- Quantification of TRF1 levels by immunofluorescence in intestine and lung tumors samples of mice treated with vehicle or with ETP-47037 for 10 days. Representative images are shown to the right (n = 4).
- Number of cells showing γH2AX foci in intestine and lung tumors samples of mice treated with vehicle or with ETP-47037 for 10 days. Representative images are shown to the right (n = 4).
Data information: The data represent the mean values obtained from three mice in each group. Error bars represent standard errors. t-test was used to assess statistical significance.
Figure 9.
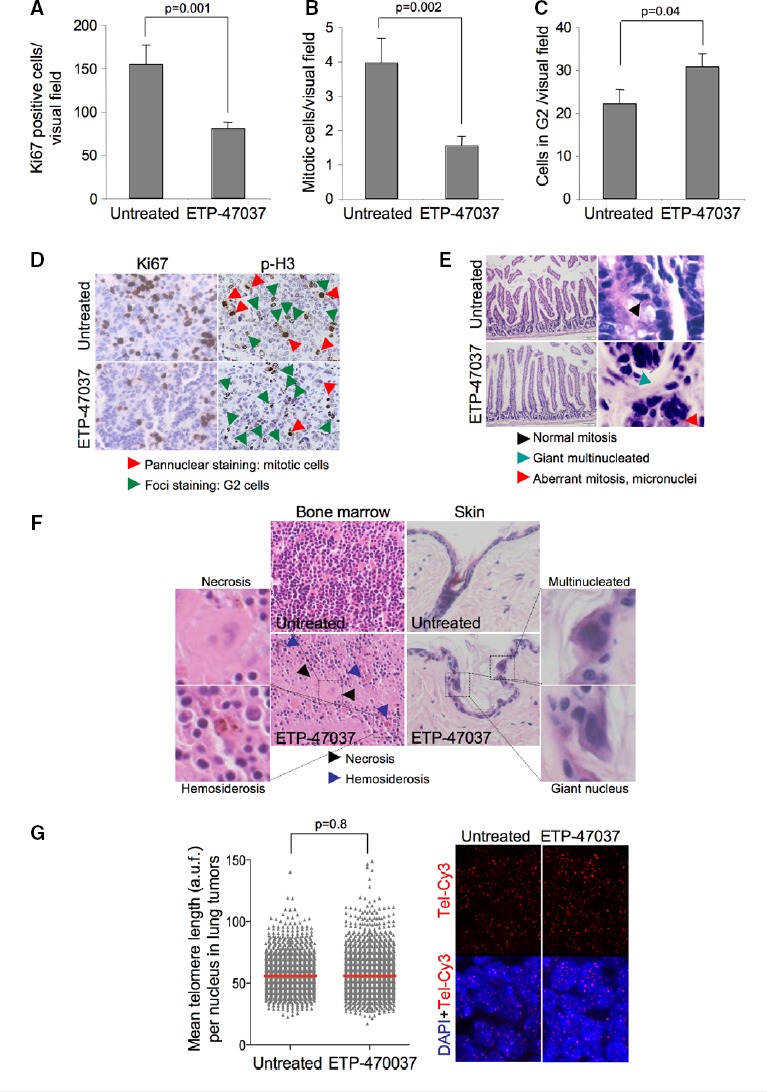
- A–C Quantification of the number of (A) Ki67-, (B) pan-nuclear p-H3 pattern-, and (C) foci p-H3 pattern-positive cells in untreated and ETP-470037-treated lung carcinomas. The data represent the mean values obtained for three mice in each group. Error bars represent standard errors.
- D Representative Ki67 and p-H3 images.
- E Representative H&E images of intestine samples corresponding to untreated and ETP-47037-treated animals. High-magnification images are shown to the right indicating the presence of normal mitosis, giant multinucleated and aberrant mitotic figures.
- F Representative H&E images of bone marrow and skin samples corresponding to untreated and ETP-47037-treated animals. High-magnification images are shown indicating the presence of necrosis, hemosiderosis, multinucleated cells, and giant nuclei. Bone marrow showed moderated aplasia.
- G Telomere length in untreated and ETP-47037-treated lung tumor samples. Representative images are shown to the right.
Data information: t-test was used to assess statistical significance.
Discussion
Aberrant telomerase activation is a common feature of human cancers, where it allows the growth of malignant cells by ensuring maintenance of a minimal functional telomere length that permits cell division (Kim et al, 1994; Hahn et al, 1999; Gonzalez-Suarez et al, 2000). Indeed, mutations in the telomerase gene or its regulatory regions have been found associated with many different types of cancer (McKay et al, 2008; Wang et al, 2008; Rafnar et al, 2009; Shete et al, 2009; Petersen et al, 2010; Melin et al, 2012; Bojesen et al, 2013; Garcia-Closas et al, 2013; Horn et al, 2013; Huang et al, 2013). To date, targeting of telomeres in human cancer has been mainly via targeting telomerase activity, typically through direct small molecule inhibitors of the enzyme activity (Brennan et al, 2010; Joseph et al, 2010), or through immunotherapy-based approaches (Brunsvig et al, 2006; Suso et al, 2011). Telomeric repeats can also form DNA higher order structures known as G-quartets, and molecules that stabilize G-quartets have also been proposed to inhibit telomerase-mediated telomere elongation in cancer (Sun et al, 1997; Shin-ya et al, 2001; Huang et al, 2008). A predicted shortcoming of therapeutic strategies based on telomerase inhibition to treat cancer is that they will be effective only when telomeres shorten below a minimum length. Indeed, telomerase activity is dispensable for transformation of cells with long telomeres (Seger et al, 2002), and studies with telomerase inhibitors indicate that they are effective preferentially in cells with short telomeres (Hahn et al, 1999; Herbert et al, 1999; Wang et al, 2004; Brennan et al, 2010; Wu et al, 2012; reviewed in Buseman et al, 2012). In line with this, telomerase abrogation in the context of cancer-prone mouse models, including the K-Ras+/G12D lung tumorigenesis mouse model, only showed anti-tumorigenic activity after several mouse generations in the absence of telomerase when telomeres reached a critically short length (Chin et al, 1999; Greenberg et al, 1999; Gonzalez-Suarez et al, 2000; Perera et al, 2008). Moreover, these anti-tumorigenic effects of short telomeres owing to telomerase deficiency are abrogated in the absence of p53 (Chin et al, 1999; Greenberg et al, 1999).
In contrast to telomerase inhibition, telomere uncapping has been shown to cause rapid induction of cell death and/or senescence in a manner that is independent of telomerase activity and telomere length (Karlseder et al, 1999; Smogorzewska & de Lange, 2002; Martinez et al, 2009). Owing to the fact that telomere uncapping can be achieved independently of telomere length, it emerges as a more universal way to rapidly impair the growth of dividing cells. Indeed, in our experimental system, Trf1 abrogation results in a dramatic reduction in the number and the size of malignant lung carcinoma lesions, even in the absence of p53, already in the first mouse generation and in the absence of telomere shortening, indicating that Trf1 deficiency severely impairs cancer progression in the context of oncogenic K-Ras. As a consequence of this, all the Trf1Δ/Δ K-Ras+/G12V p53−/− mice survived until the end point of the experiment (24 weeks post-infection), while only 50% of the Trf1+/+ K-Ras+/G12V p53−/− mice survived the same period. These findings indicate that Trf1 deficiency impairs the development of K-Ras-induced lung carcinomas. Of note, this represents the first time that effective impairment of K-Ras+/G12V p53−/− carcinomas is achieved, as genetic abrogation of other therapeutic pathways did not impair tumor growth in the absence of p53 (Navas et al, 2012). Furthermore, here we show that downregulation of Trf1 can also block the growth and metastatic potential of both mouse and human lung cancer cell lines derived from already established K-Ras-induced lung carcinomas by using xenograft models.
We find that the mechanisms through which Trf1 deletion impairs cancer progression are related to its previously described roles in telomere capping, telomere replication, and mitosis (Martinez et al, 2009; Sfeir et al, 2009). In this regard, we show that Trf1 deficiency results in a high burden of telomeric DNA damage, genetic instability, proliferation defects, apoptosis, and mitotic catastrophe.
Importantly, we demonstrate here that a long-term systemic depletion of TRF1 in healthy adult tissues does not compromise organism viability, although we observed decreased cellularity in some highly proliferative compartments, such as the hematopoietic compartment and blood, which were recovered upon tamoxifen removal. Together, these findings suggest a therapeutic window for TRF1 inhibition in cancer.
Inspired by the above notion, we have identified compounds that disrupt TRF1 binding to telomeres illustrating the feasibility of chemically targeting shelterin proteins. Furthermore, we have shown that “in vivo” treatment of already established lung adenocarcinomas with one of the identified compounds, ETP-47037, results in decreased TRF1 signal in vivo and the impairment of tumor progression in the absence of decreased mouse viability.
In summary, the results described here are proof of concept that TRF1 abrogation is an effective therapeutic strategy to block the growth of aggressive lung carcinomas independently of telomere length and p53 status and that it is possible to achieve this by small molecules that are able to target TRF1 in vivo. Finally, as this strategy relies on a universal mechanism, namely induction of telomere uncapping, we speculate that it could be applied in many other cancer types.
Materials and Methods
Mice
K-Ras+/LSLG12Vgeo (Guerra et al, 2003), and Trf1lox/lox (Martinez et al, 2009), p53−/− (Jackson Labs, http://jaxmice.jax.org/strain/002101.html) strains were crossed to obtain K-Ras+/LSLG12Vgeo Trf1lox/lox p53−/− mice. To generate Trf1lox/lox hUBC-CreERT2 mice, we crossed our Trf1lox/lox (Martinez et al, 2009) with a mouse strain that carries a ubiquitously expressed, tamoxifen-activated recombinase, hUBC-CreERT2 mice (Ruzankina et al, 2007). Trf1lox/lox hUBC-CreERT2 mice were fed ad libitum for 7 weeks with tamoxifen-containing diet (Tekland CRD Tam400/CreER). For allograft experiments, 7-week-old athymic nude females were obtained from Harlan. All mice were maintained at the Spanish National Cancer Center under specific pathogen-free conditions in accordance with the recommendations of the Federation of European Laboratory Animal Science Associations (FELASA). All animal experiments were approved by the Ethical Committee and performed in accordance with the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, developed by the Council for International Organizations of Medical Sciences (CIOMS).
Adenovirus intratracheal infection
Eight- to ten-week-old mice were treated once with intratracheal adeno-Cre (Gene Vector Core, University of Iowa, 1 × 1010 pfu/ml) instillation with 1 × 108 PFU/mouse of virus after anesthesia by intraperitoneal injection of ketamine–medetomidine (Domitor, 1 mg/ml; Orion Corporation). To wake up the mice after the instillation, they were injected with 0.05 mg of atipamezole (Antisedan, 5 mg/ml; Orion Corporation).
In vivo imaging by computed tomography (CT) and positron emission tomography (PET)
Nine weeks after inoculation, an in vivo follow-up of tumor growth was achieved by six computed tomographies (CT) every 15 days. PET was performed 22nd week post-inoculation, and the mice were sacrificed (24th week post-inoculation). CT and PET analyses were performed as previously described (Ambrogio et al, 2014). For PET quantification, tumor regions of interest (ROIs) were selected in the PET-CT overlapped image. In these ROIs, the standardized 18FDG-glucose uptake value (SUV) was calculated using the following formula: SUV = tumor FDG concentration (MBq)/(injected dose/body weight).
Telomere length analyses on tissue sections
Quantitative telomere fluorescence in situ hybridization (Q-FISH) directly on tumor sections was performed as previously described (Flores et al, 2008) and analyzed by Definiens software.
Chemical library
The Experimental Therapeutics Program at CNIO, ETP-CNIO, owns a chemical library of about 50,000 single compounds built as a result of the consolidation of several sub-libraries selected attending to different criteria such as chemical diversity, kinase-targeted focus, potential to disrupt protein–protein interactions, and the presence of low molecular weight compounds to facilitate fragment-based drug discovery. The drug-likeness of the whole library was also ensured by the application of filters such as “rule of five” (Lipinski, 2004). The compounds were selected from commercial origin as well as from internally newly designed and synthesized chemical matter. Representative libraries of the whole 50K library with smaller sizes were defined after clustering, based on similarity analysis, and selection of representative compounds from each cluster. A 640-compound library, subject of the currently reported screening campaign, is the minimum size set of compounds representing the chemical ETP-CNIO collection.
Screening for identification of TRF1 inhibitors
We tested the CNIO-640 library previously described. iPS cells expressing eGFP-TRF1 were seeded in 0.1% gelatin-pretreated cell-carrier black 384-well microplates (Perkin Elmer) at a density of 1.25 × 104 cells per well 24 h before adding the compounds. Compounds were weighed out and diluted in dimethyl sulfoxide (DMSO) to a final concentration of 10 mM (mother plate). From here, an intermediate dilution plate was prepared. The appropriate volume (μl) of each compound solution was added automatically (Beckman FX 96 tip) from the intermediated plate to the media of plated cells to get a 12.5-μM final concentration for each compound assayed in duplicate. Cell viability was previously tested in a dose curve with increasing concentrations of DMSO. After 8-h incubation, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature and washed three times with PBS. Those compounds that killed cells at 12.5 μM at 8 h were not considered as positive hits.
For quantitative measurement of eGFP-TRF1 foci levels, pictures of fixed cells were automatically acquired from each well by the Opera High Content Screening (HCS) system (Perkin Elmer). Sixty images of random fields per well, with a 40× magnification lens, were taken under non-saturating conditions. At least 1 × 103 cells were analyzed for each well. Briefly, images were segmented using the DAPI staining to generate masks matching cell nuclei from which eGFP-TRF1 foci were analyzed. SPSS software was used for statistical analysis as follows: Within each plate, the eGFP-TRF1 intensities of control eGFP-Trf1KI/KI cells were distributed by quartiles (Q). First-quartile distribution (Q1) was taken as threshold to distinguish low- or high-intensity eGFP-TRF1 foci. Percentage of low vs. high GFP-TRF1 levels was normalized using the average of negative and positive controls as minimum and maximum reference levels. The number obtained was taken as relative TRF1 inhibition for each compound.
The paper explained
Problem
Unlimited cell division in cancer requires activation of mechanisms that ensure maintenance of telomere length. Targeting of telomeres in human cancer has been approached via targeting telomerase activity. A caveat of therapeutic strategies based on telomerase inhibition to treat cancer is that they will be effective only when telomeres shorten below a minimum length. We have addressed whether induction of telomere dysfunction independently of telomere length by targeting a shelterin component could be applied as a more universal way to rapidly impair the growth of dividing cells.
Results
We demonstrate that acute telomere uncapping owing to inhibition of the TRF1 shelterin component has therapeutic activity in blocking the growth of p53-deficient K-Ras-induced lung tumors by inducing DNA damage at telomeres. This anti-tumorigenic activity of TRF1 inhibition is independent of telomere length. In parallel, we show that whole-body partial TRF1 depletion, although resulting in moderate loss of cellularity in the bone marrow in few Trf1-deleted mice, did not impair organismal viability and survival. Importantly, we identify small molecules that disrupt TRF1 binding in vivo, and that effectively block the growth of already established p53-deficient K-Ras-induced lung carcinomas through induction of DNA damage and cell arrest, again in the absence of deleterious effects in mouse survival or viability.
Impact
This represents the first demonstration that targeting the TRF1 shelterin component may represent a novel therapeutic approach for cancer treatment.
In vivo treatment with compound ETP-47037
K-Ras+/LSLG12Vgeo Trf1lox/lox p53−/− tumors were induced by intratracheal adeno-Cre instillation as described above. Once the lung tumors developed, mice were daily dosed orally with 75 mg/kg of ETP-47037 formulated in 10% N-methyl-pyrrolidone and 90% polyethylene-glycol 300 for 10 days and 2 days of resting. The reduction in number and size of the tumors was analyzed by computed tomography (CT).
Acknowledgments
We are indebted to R. Serrano for animal care. We thank C. Guerra, R. Blasco, and D. Santamaria for scientific and technical advice. M.A.B.'s laboratory is funded with the Spanish Ministry of Science and Innovation, projects SAF2008-05384 and 2007-A-200950 (TELOMARKER), European Research Council Advanced grant GA#232854, the Körber Foundation, Fundación Botín, and Fundación Lilly.
Author contributions
MAB, PM, MMP, and MGB performed the experiments and wrote the manuscript. SM, CBA, and JP aided in the chemical screening. CA and MB generated the tumor-derived mouse cell line. FM did the in vivo imaging. DM assisted with microscopy techniques. MC and JMF performed the histopathological analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Ambrogio C, Carmona FJ, Vidal A, Falcone M, Nieto P, Romero OA, Puertas S, Vizoso M, Nadal E, Poggio T, et al. Modeling lung cancer evolution and preclinical response by orthotopic mouse allografts. Cancer Res. 2014;74:5978–5988. doi: 10.1158/0008-5472.CAN-14-1606. [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Armstrong GN, Gramatges MM, Bertuch AA, Jhangiani SN, Doddapaneni H, Lewis L, Tombrello J, Tsavachidis S, Liu Y, et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2015;107:384. doi: 10.1093/jnci/dju384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012;120:2990–3000. doi: 10.1182/blood-2012-03-418038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellon M, Datta A, Brown M, Pouliquen JF, Couppie P, Kazanji M, Nicot C. Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int J Cancer. 2006;119:2090–2097. doi: 10.1002/ijc.22026. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007;3:640–649. doi: 10.1038/nchembio.2007.38. [DOI] [PubMed] [Google Scholar]
- Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, Baccarini M, Barbacid M. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–663. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA, Shen HC, Smart CE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boue S, Paramonov I, Barrero MJ, Izpisua Belmonte JC. Analysis of human and mouse reprogramming of somatic cells to induced pluripotent stem cells. What is in the plate? PLoS ONE. 2010;5:e12664. doi: 10.1371/journal.pone.0012664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan SK, Wang Q, Tressler R, Harley C, Go N, Bassett E, Huff CA, Jones RJ, Matsui W. Telomerase inhibition targets clonogenic multiple myeloma cells through telomere length-dependent and independent mechanisms. PLoS ONE. 2010;5:e12487. doi: 10.1371/journal.pone.0012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Moller M, Eriksen JA, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55:1553–1564. doi: 10.1007/s00262-006-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3:1271–1274. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- Buseman CM, Wright WE, Shay JW. Is telomerase a viable target in cancer? Mutat Res. 2012;730:90–97. doi: 10.1016/j.mrfmmm.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba I, Takahashi T, Nau MM, D'Amico D, Curiel DT, Mitsudomi T, Buchhagen DL, Carbone D, Piantadosi S, Koga H, et al. Mutations in the p53 gene are frequent in primary, resected non-small cell lung cancer. Lung Cancer Study Group. Oncogene. 1990;5:1603–1610. [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141:81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, Varmus HE. Structure and variability of human chromosome ends. Mol Cell Biol. 1990;10:518–527. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Chang S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab Invest. 2007;87:1071–1076. doi: 10.1038/labinvest.3700673. [DOI] [PubMed] [Google Scholar]
- Elkin M, Vlodavsky I. Tail vein assay of cancer metastasis. Curr Protoc Cell Biol. 2001 doi: 10.1002/0471143030.cb1902s12. 12:19.2:19.2.1-19.2.7. [DOI] [PubMed] [Google Scholar]
- Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22:654–667. doi: 10.1101/gad.451008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto R, Kamata N, Taki M, Yokoyama K, Tomonari M, Nagayama M, Yasumoto S. Gene expression of telomerase related proteins in human normal oral and ectocervical epithelial cells. Oral Oncol. 2003;39:445–452. doi: 10.1016/s1368-8375(03)00003-4. [DOI] [PubMed] [Google Scholar]
- Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, Orr N, Rhie SK, Riboli E, Feigelson HS, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45:392–398. doi: 10.1038/ng.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet. 2000;26:114–117. doi: 10.1038/79089. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- Harvey DM, Levine AJ. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 1991;5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Delcuve GP, Davie JR. Histone deacetylase is a component of the internal nuclear matrix. J Biol Chem. 1991;266:21936–21942. [PubMed] [Google Scholar]
- Herbert B, Pitts AE, Baker SI, Hamilton SE, Wright WE, Shay JW, Corey DR. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA. 1999;96:14276–14281. doi: 10.1073/pnas.96.25.14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- Huang FC, Chang CC, Lou PJ, Kuo IC, Chien CW, Chen CT, Shieh FY, Chang TC, Lin JJ. G-quadruplex stabilizer 3,6-bis(1-methyl-4-vinylpyridinium)carbazole diiodide induces accelerated senescence and inhibits tumorigenic properties in cancer cells. Mol Cancer Res. 2008;6:955–964. doi: 10.1158/1541-7786.MCR-07-0260. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, Wright WE, Shay JW, Go NF. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–9504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Kachatrian L, Takai H, Mercer K, Hingorani S, Jacks T, de Lange T. Targeted deletion reveals an essential function for the telomere length regulator Trf1. Mol Cell Biol. 2003;23:6533–6541. doi: 10.1128/MCB.23.18.6533-6541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Maraver A, Fernandez-Marcos PJ, Herranz D, Canamero M, Munoz-Martin M, Gomez-Lopez G, Mulero F, Megias D, Sanchez-Carbayo M, Shen J, et al. Therapeutic effect of gamma-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell. 2012;22:222–234. doi: 10.1016/j.ccr.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsutani N, Yokozaki H, Tahara E, Tahara H, Kuniyasu H, Haruma K, Chayama K, Yasui W. Expression of telomeric repeat binding factor 1 and 2 and TRF1-interacting nuclear protein 2 in human gastric carcinomas. Int J Oncol. 2001;19:507–512. [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40:1404–1406. doi: 10.1038/ng.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin BS, Nordfjall K, Andersson U, Roos G. hTERT cancer risk genotypes are associated with telomere length. Genet Epidemiol. 2012;36:368–372. doi: 10.1002/gepi.21630. [DOI] [PubMed] [Google Scholar]
- Mocellin S, Verdi D, Pooley KA, Landi MT, Egan KM, Baird DM, Prescott J, De Vivo I, Nitti D. Telomerase reverse transcriptase locus polymorphisms and cancer risk: a field synopsis and meta-analysis. J Natl Cancer Inst. 2012;104:840–854. doi: 10.1093/jnci/djs222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh BK, Kim YJ, Park C, Park YN. Up-regulation of telomere-binding proteins, TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am J Pathol. 2005;166:73–80. doi: 10.1016/S0002-9440(10)62233-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyashiki JH, Hayashi S, Yahata N, Iwama H, Ando K, Tauchi T, Ohyashiki K. Impaired telomere regulation mechanism by TRF1 (telomere-binding protein), but not TRF2 expression, in acute leukemia cells. Int J Oncol. 2001;18:593–598. doi: 10.3892/ijo.18.3.593. [DOI] [PubMed] [Google Scholar]
- Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973;41:181–190. doi: 10.1016/0022-5193(73)90198-7. [DOI] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera SA, Maser RS, Xia H, McNamara K, Protopopov A, Chen L, Hezel AF, Kim CF, Bronson RT, Castrillon DH, et al. Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis. 2008;29:747–753. doi: 10.1093/carcin/bgn050. [DOI] [PubMed] [Google Scholar]
- Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, Jakobsdottir M, Helgadottir H, Thorlacius S, Aben KK, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay AJ, Quesada V, Foronda M, Conde L, Martinez-Trillos A, Villamor N, Rodriguez D, Kwarciak A, Garabaya C, Gallardo M, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013;45:526–530. doi: 10.1038/ng.2584. [DOI] [PubMed] [Google Scholar]
- Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, Pooley KA, Pritchard AL, Tiffen JC, Petljak M, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet. 2014;46:478–481. doi: 10.1038/ng.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, van Bodegom PC, Bos JL. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988;48:5738–5741. [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RP, Garrobo I, Foronda M, Palacios JA, Marion RM, Flores I, Ortega S, Blasco MA. TRF1 is a stem cell marker and is essential for the generation of induced pluripotent stem cells. Nat Commun. 2013;4:1946. doi: 10.1038/ncomms2946. [DOI] [PubMed] [Google Scholar]
- Seger YR, Garcia-Cao M, Piccinin S, Cunsolo CL, Doglioni C, Blasco MA, Hannon GJ, Maestro R. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell. 2002;2:401–413. doi: 10.1016/s1535-6108(02)00183-6. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010;584:3819–3825. doi: 10.1016/j.febslet.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac-de Paillerets B, Nagore E, Avril MF, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482–486. doi: 10.1038/ng.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin-ya K, Wierzba K, Matsuo K, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123:1262–1263. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 2002;21:4338–4348. doi: 10.1093/emboj/cdf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, Gaudernack G. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol Immunother. 2011;60:809–818. doi: 10.1007/s00262-011-0991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanasoula M, Escandell JM, Martinez P, Badie S, Munoz P, Blasco MA, Tarsounas M. p53 prevents entry into mitosis with uncapped telomeres. Curr Biol. 2010;20:521–526. doi: 10.1016/j.cub.2010.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- Wang ES, Wu K, Chin AC, Chen-Kiang S, Pongracz K, Gryaznov S, Moore MA. Telomerase inhibition with an oligonucleotide telomerase template antagonist: in vitro and in vivo studies in multiple myeloma and lymphoma. Blood. 2004;103:258–266. doi: 10.1182/blood-2003-02-0546. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kowalski J, Tsai HL, Marik R, Prasad N, Somervell H, Lo PK, Sangenario LE, Dyrskjot L, Orntoft TF, et al. Differentiating alternative splice variant patterns of human telomerase reverse transcriptase in thyroid neoplasms. Thyroid. 2008;18:1055–1063. doi: 10.1089/thy.2008.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- Wu X, Smavadati S, Nordfjall K, Karlsson K, Qvarnstrom F, Simonsson M, Bergqvist M, Gryaznov S, Ekman S, Paulsson-Karlsson Y. Telomerase antagonist imetelstat inhibits esophageal cancer cell growth and increases radiation-induced DNA breaks. Biochim Biophys Acta. 2012;1823:2130–2135. doi: 10.1016/j.bbamcr.2012.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File