

Abstract
Most variants implicated in common human disease by Genome-Wide Association Studies (GWAS) lie in non-coding sequence intervals. Despite the suggestion that regulatory element disruption represents a common theme, identifying causal risk variants within indicted genomic regions remains a significant challenge. Here we present a novel sequence-based computational method to predict the effect of regulatory variation, using a classifier (gkm-SVM) which encodes cell-specific regulatory sequence vocabularies. The induced change in the gkm-SVM score, deltaSVM, quantifies the effect of variants. We show that deltaSVM accurately predicts the impact of SNPs on DNase I sensitivity in their native genomic context, and accurately predicts the results of dense mutagenesis of several enhancers in reporter assays. Previously validated GWAS SNPs yield large deltaSVM scores, and we predict novel risk SNPs for several autoimmune diseases. Thus, deltaSVM provides a powerful computational approach for systematically identifying functional regulatory variants.
Sequence variation in DNA regulatory elements is hypothesized to contribute substantially to risk for common diseases. Variants associated with human disease by GWAS predominantly lie in non-coding genomic regions1 and occur within putative regulatory elements far more often than expected by chance2,3, suggesting that disruption of regulatory function is a common mechanism by which non-coding sequence variants contribute to human disease. Linkage disequilibrium (LD), and the absence of regulatory vocabularies, complicates the discrimination of regulatory risk variants from other variation within disease-associated intervals. Therefore there is a pressing need for methods to predict the impact of regulatory sequence variation, expediting targeted functional validation and the exploration of disease-implicated pathways. However, few formal computational methods have been developed to predict the impact of Single Nucleotide Polymorphisms (SNPs) on regulatory element activity4,5.
Regulatory elements modulate the expression of their target genes through direct binding of sequence-specific transcription factors (TFs)6. While consensus on the mechanisms of regulatory element activity is emerging, we lack a predictive model capable of (1) specifying the cell types and environmental conditions under which an element would modulate the expression of its target gene(s), and (2) describing how specific mutations to that sequence would influence its activity. Here, we develop a computational model that addresses the latter: given a regulatory element active in a specific cell type, compute the effect of a given DNA sequence variation within the element. When trained on a set of putative regulatory sequences, our established gapped k-mer Support Vector Machine (gkm-SVM)7 identifies sequence features within these regulatory regions which determine their cell-type dependent activity. We then use this gkm-SVM to quantify the effect of sequence changes within regulatory elements via a metric we term deltaSVM (See overview in Fig. 1). In this systematic, quantitative approach we leverage high quality catalogs of human regulatory elements, generated using DNase I Hypersensitivity, distinctive histone modifications, and TF binding8,9. For example, if the gkm-SVM is trained on DNaseI Hypersensitive Sites (DHSs), it identifies the sequence features that determine chromatin accessibility in the corresponding cellular context. Our method is, however, blind to extant databases or binding motif data, and consequently can uncover novel motifs, combinatorial constraints, and key accessory factors, and quantify the significance of their individual contributions to regulatory element activity.
Figure 1. Overview of our deltaSVM method.

[left] The first step in calculating deltaSVM is to train a gkm-SVM classifier using a positive training set of putative regulatory sequences (identified by DNase I hypersensitivity, for example) and a negative training set of matched negative control sequences. The gkm-SVM generates a regulatory sequence vocabulary – a weighted list of all possible 10-mers, where each 10-mer receives an SVM weight that quantifies its contribution to the prediction of regulatory function. [right] After training, this regulatory sequence vocabulary can be used to score the predicted impact of any sequence variant on regulatory activity, as shown here for a single nucleotide substitution in a melanocyte enhancer of the Tyrp1 enhancer.
Results
Model training and validation
We previously demonstrated that a properly trained SVM can predict cell-type specific regulatory elements from primary genome sequence alone7,10–12. To test whether this SVM-based approach could be adapted to predict the functional consequence of sequence variation within regulatory elements, we first took advantage of a large set of dsQTLs (DNase I Sensitivity Quantitative Trait Loci) identified in a collection of human Lymphoblastoid Cell Lines (LCLs)13–15. These are SNPs within putative regulatory regions (marked by DNase I Hypersensitivity) and are associated with altered DNase I sensitivity therein. We first trained a gkm-SVM on the top DHSs in the LCL GM128788. The gkm-SVM produces a scoring function characterized by a set of weights quantifying the contribution of each possible 10-mer to a region’s DNase I sensitivity in GM12878 cells. We then can calculate deltaSVM, the predicted impact of any Single Nucleotide Variant (SNV) on chromatin accessibility in LCLs, by summing the change in weight between alleles for each of the ten 10-mers encompassing the SNV, as shown in Fig. 2a for the dsQTL rs495322313. Here, the indicted SNP allele disrupts a NF-κB binding site, which in our model reduces the strong positive contribution of several 10-mers. Two neighboring SNPs do not make significant changes to the weights, as shown graphically in Fig. 2b, and the score of each allele is the sum of the weights across this region. Similarly, we can extend this method to INDELs and multiple substitutions by summing weights across all affected bases.
Figure 2. deltaSVM can accurately predict SNPs associated with DNaseI Hypersensitivity.

(a) An example of a deltaSVM calculation using a known dsQTL SNP (rs4953223). (b) 10-mer gkm-SVM scores across the dsQTL locus containing rs4953223 are shown. Only the functional SNP produces dramatic changes in gkm-SVM scores. (c) Effect sizes of dsQTL SNPs from Ref. 13 are well correlated with their deltaSVM scores. (d–e) deltaSVM predicts dsQTLs with far greater accuracy than existing methods. Discriminative powers are compared between various methods using 50x larger control SNP set. (d) ROC curve. (e) Precision-Recall curve.
To systematically assess the ability of deltaSVM to predict the impact of SNPs on DNase I sensitivity, we compared deltaSVM to the set of dsQTLs13, quantified by the effect-size, beta. The correlation between deltaSVM and effect-size for the 579 SNPs within 100 bp of a DHS is highly significant, with a Pearson correlation coefficient C = 0.721 (t-distribution P = 7.68e-94) (Fig. 2c). This correlation falls off rapidly with distance (Supplementary Figure 1), thus our analysis is consistent with local action of dsQTLs. However, if our predictions are accurate, deltaSVM analyses on non-dsQTL SNPs should also yield low scores in order to limit false positive predictions. We chose a 50x larger negative set of non-dsQTL SNPs with comparable levels of DNase I sensitivity as a negative set, since there are typically 50–100 SNPs within a single LD block15. In Fig. 2d we show the Receiver Operating Characteristic (ROC) curve, plotting True Positive rate (TP/P) vs. False Positive rate (FP/P), and in Fig. 2e we show the Precision-Recall (PR) curve, plotting precision (true positives over predicted positives, TP/PP = TP/(TP+FP)) vs. recall (TP/P), for our method (gkm-SVM deltaSVM) compared to four other methods4,5,10,16.
Here, as is typically the case for genomic predictions where the search space is large, the lower left corner of the ROC curve, where the FP rate is low, has the most dramatic effect on the accuracy (precision) of the predictions17. At a recall of 10%, the gkm-SVM predictions are 55.9% accurate, ~5x more accurate than deltaSVM based on smaller 6-mers (kmer-SVM)10 as shown in Fig. 2e, because while the kmer-SVM can predict full regions very accurately by averaging many weights, the kmer weights needed to evaluate SNPs are determined from a small set of support vectors and are noisy. By contrast the gkm-SVM reduces the false positive rate significantly by using much more statistically robust gapped-kmer weights18. Additionally, in comparison to conservation (GERP score16), and to two recently published methods integrating functional genomic datasets to predict the deleteriousness of noncoding variants (CADD4 and GWAVA5) the gkm-SVM is ≥ 10x more accurate than any of these existing methods at 10% recall (Fig. 2e).
Three key features contribute to our dramatically improved accuracy. First, we train gkm-SVM on set of regulatory elements whose activity is specific to the relevant cell type. Second, this large training set (thousands) includes both positive and negative elements to statistically determine the DNA sequence elements required for activity, rather than relying on the precise state of any specific regulatory element in a specific assay. Thirdly, we identify a complete catalog of both positive and negative sequence features, as many SNPs result in a significant deltaSVM based on what the variant changes to, rather than what it was in the reference/assayed genome. In our discriminative approach, gkm-SVM identifies these negative sequence elements by their presence in the negative set and their absence in the positive set. This is critical for accurately assessing the effect of variants, as many of our predicted functional SNPs modulate intermediate strength binding sites.
Ultimately we are interested in how a variant modulates the expression of its target genes. 125 of 579 dsQTLs are also eQTLs19 (variants associated with differential gene expression), but some dsQTLs are anti-correlated with eQTLs13. Both classes of dsQTLs are strongly positively correlated with deltaSVM (Fig. 3a–c). Thus surprisingly, but consistent with earlier analysis13, we find that as 22% of the dsQTLs become more accessible, they repress target gene expression. We also analyzed the relationship between deltaSVM and evolutionary sequence conservation. Interestingly, although bases predicted to either reduce or increase DNase I sensitivity when mutated are more conserved than bases predicted to be neutral, we found that negative deltaSVM bases are much more conserved than positive deltaSVM bases (Fig. 3d and Supplementary Figure 2).
Figure 3. deltaSVM is strongly positively correlated with dsQTL effect size, and positively or negatively correlated with eQTL effect size depending on the sign of the correlation of dsQTL and eQTL.
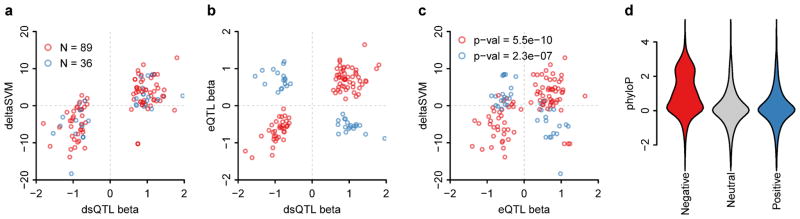
Degner et al reported that 16% of the dsQTLs were also eQTLs, but that 30% of the eQTL dsQTLs were anti-correlated with the expression change. Our predictions are consistent with this observation: (a) deltaSVM is always positively correlated with dsQTL effect size (beta), (b) but because eQTL beta and dsQTL beta are anti-correlated 30% of the time, (c) deltaSVM and eQTL beta are only correlated (positively and negatively) if we treat the activating dsQTLs (red) and repressive dsQTLs (blue) separately. (d) Bases predicted to reduce the activity of functional regions are evolutionarily constrained.
gkm-SVM predicts functional impact of enhancer variants
To directly test the ability of our SVM-based approach to predict the functional consequence of sequence variation on enhancer activity, we first turned to well-characterized enhancers of the pigmentation genes Tyr and Tyrp120,21 (Fig. 4a,b). We trained a melanocyte-specific gkm-SVM on a large set of putative melanocyte enhancers marked by EP300 and H3K4me112, and scored all possible SNVs in the Tyr and Tyrp1 enhancers, selecting and synthesizing more than 40 SNVs, across a range of deltaSVM scores, and tested each variant independently in luciferase reporter assays. For both enhancers, deltaSVM is strongly correlated with the observed difference in luciferase reporter activity between mutant and wild-type enhancer constructs (Pearson C = 0.778, P < 2e-5 for Tyr, and C = 0.529, P < .0095 for Tyrp1; Fig. 4c,d).
Figure 4. deltaSVM accurately predicts change in luciferase expression in targeted mutagenesis of Tyr and Tyrp1 melanocyte enhancers.

(a,b) Base by base evaluation of all possible substitutions as scored by deltaSVM. Black circles mark substitutions that were tested in luciferase assays. Orange bars show positions of the previously characterized binding sites. (c,d) Correlation of deltaSVM prediction and observed normalized luciferase expression. Blue circles indicate previously tested binding site20,21. Error bar is one standard deviation of the changes in luciferase expression (4 biological replicates per variant).
Despite their depth, our analyses of the Tyr and Tyrp1 enhancers tested only a subset of all possible variants therein, and relied on in vitro reporter assays. Therefore, we turned to a dataset in which all possible variants within a 259 bp liver-specific enhancer of the ALDOB gene were tested using a massively parallel reporter assay in vivo in mouse liver22. We trained a gkm-SVM on large set of putative liver enhancers marked by DNase I hypersensitivity and H3K4me1 signal in adult mouse liver23. We then compared deltaSVM for each of the tested mutant regions to the observed functional output. Again we see a very high correlation (C = 0.630, P < 3.24e-81) between the predicted impact of the mutation using our sequence-based model and the observed change in enhancer activity relative to wild-type sequence (Fig. 5a). If we further use the “aggregate score” model22 averaging deltaSVM for each of the 3 possible base substitutions, this correlation reaches C = 0.691 (Supplementary Figure 3).
Figure 5. deltaSVM accurately predicts change of expression in massively parallel reporter assays.
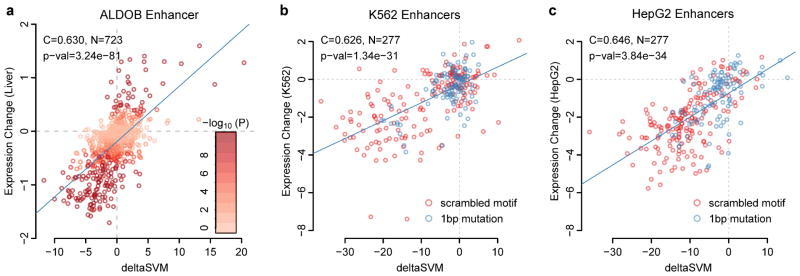
(a) Correlations of deltaSVM predictions and observed in vivo mutation effect size in the ALDOB enhancer in mice22. (b) Correlation of deltaSVM and mutated enhancers in K562 cells24. (c) Correlation of deltaSVM and mutated enhancers in HepG2 cells24.
We next asked how deltaSVM performed predicting functional variants in diverse sets of enhancers. We analyzed data from another massively parallel reporter assay using targeted mutation of enhancers predicted to be active in K562 and HepG2 cells24. For each wild-type construct that was expressed significantly in either cell line, we separately scored all 1 bp and motif scrambling mutations using a gkm-SVM trained on K562 and HepG2 DHS regions8, and compared the measured expression change to the predicted deltaSVM score in each cell line (Fig. 5b,c). For both datasets again we find high correlation (C = 0.626, P < 1.34e-31 for K562 and C = 0.646, P < 3.84e-34 for HepG2). Since all elements were tested and scored in both cell types, this high correlation underscores the accuracy of deltaSVM’s cell-type specific predictions and is further supported by the low correlation of deltaSVMs scores from gkm-SVMs trained on non-relevant cell-types (Table 1).
Table 1. Cell-type specificity of deltaSVM predictions.
We compare weights trained on all five cell types to the measured expression changes in all five cell lines. P-values are in parenthesis. Although some TFs are active in more than one cell type, we generally observe that deltaSVM predictions are cell-type specific.
| Gkm-SVM | LCL-dsQTL | Tyr | Tyrp1 | Aldob | K562 enhancers | HepG2 enhancers |
|---|---|---|---|---|---|---|
| GM12878 DHS | 0.721 (7.68e-94) | 0.302 (0.172) | 0.117 (0.595) | 0.112 (0.00256) | 0.204 (0.00062) | 0.201 (0.00076) |
| Mouse melan-a EP300 | 0.245 (2.19e-9) | 0.78 (2.0e-5) | 0.53 (0.0095) | 0.147 (7.42e-5) | 0.204 (0.00062) | 0.194 (0.00116) |
| Mouse liver DHS | 0.131 (0.00157) | 0.282 (0.203) | 0.056 (0.798) | 0.630 (3.24-e81) | −0.329 (2.04e-8) | 0.551 (2.07e-23) |
| K562 DHS | 0.581 (1.45e-53) | 0.390 (0.0726) | 0.104 (0.638) | 0.092 (0.0137) | 0.626 (1.34e-31) | −0.042 (0.483) |
| HepG2 DHS | 0.518 (3.84e-41) | 0.551 (0.00791) | 0.166 (0.450) | 0.547 (1.01e-57) | −0.184 (.0021) | 0.646 (3.84e-34) |
Causal SNP predictions for human disease
In a final test of its potential, we asked whether deltaSVM could predict the functional consequences of studied disease-associated sequence variants. We compared deltaSVM values for three experimentally validated SNPs, each of which has been shown to alter expression leading to increased disease risk or pertinent traits: Rfx6 (rs339331, prostate cancer)25, Bcl11a (rs1427407, fetal hemoglobin levels)26, and Sort1 (rs12740374, LDL cholesterol levels)27. We trained three separate gkm-SVMs with DHSs from cell lines appropriate to each phenotype (LNCaP, mouse MEL, and HepG2 hepatocytes). We then scored all loci with deltaSVM trained on all three cell-types. In each case, we found that the validated SNPs are only scored higher than flanking SNPs when deltaSVM was trained on the appropriate cell type (Fig. 6a). Specifically, the SORT1 variant only has high deltaSVM when trained on HepG2 cells, relevant for liver, as shown in the 3rd column of Figure 6a.
Figure 6. deltaSVM only identifies validated causal SNPs when trained on the appropriate cell type.

(a) Three validated GWAS SNPs from Rfx6 (1st column), Bcl11a (2nd column), and Sort1 (3rd column) and flanking negative SNPs were each scored with deltaSVM trained on all three relevant cell-types. The validated SNPs are properly identified from among flanking SNPs when trained on the appropriate cell type (red) but not other cell types (blue). (b,c) Scoring autoimmune GWAS loci with deltaSVM trained on Th1 yields high confidence causal SNPs listed in Table 2. BACH2 locus is shown in (b) as an example. Error bar in (c) is one standard deviation of the expected binomial distribution.
Since the set of these validated regulatory SNPs is limited, we next examined 413 SNPs associated with 11 autoimmune diseases enriched in T helper cell type 1 (Th1) H3K27Ac regions28. We similarly trained a gkm-SVM on Th1 DHSs8, and for each disease associated locus, scored the lead SNPs and an additional 2700 SNPs in tight LD (as defined in methods), and random SNPs including equivalent size flanking sets as a control. An example locus in BACH2, associated with several autoimmune diseases, is shown in Fig. 6b. We identified high scoring deltaSVM SNPs for 17 independent disease associations (Table 2), which we predict to be expression perturbing SNPs with high confidence (P < .02), while at this threshold random sampling produced 8 SNPs (Fig. 6c, Supplementary Figure 4). Most of these high scoring SNPs are not the lead SNP, and thus represent novel predictions for the causal SNP.
Table 2. deltaSVM predicted causal autoimmune SNPs.
Scoring autoimmune GWAS loci with deltaSVM trained on Th1 yields predictions for high confidence causal SNPs.
| Disease | Lead SNP | Predicted SNP | CHR | Lead SNP Position | Predicted SNP Position | Distance | Nearset Gene | |deltaSVM| | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| VIT | rs853308 | rs860475 | chr8 | 133929917 | 133929799 | −118 | TG | 16.797 | 29 |
| VIT | rs16872571 | rs4697651 | chr4 | 10726853 | 10721433 | −5420 | . | 13.978 | 29 |
| T1D | rs7020673 | rs4380994 | chr9 | 4291747 | 4282536 | −9211 | GLIS3 | 13.591 | 30 |
| CRO | rs6908425 | rs7748720 | chr6 | 20728731 | 20689945 | −38786 | CDKAL1 | 13.177 | 31,32 |
| MS | rs1782645 | rs1250568 | chr10 | 81048611 | 81045280 | −3331 | ZMIZ1 | 11.039 | 33 |
| CEL | rs1250552 | rs1250568 | chr10 | 81058027 | 81045280 | −12747 | ZMIZ1 | 11.039 | 34 |
| CRO | rs10801047 | rs6665749 | chr1 | 191559356 | 191577594 | 18238 | . | 10.646 | 35 |
| CRO | rs2797685 | rs2797685 | chr1 | 7879063 | 7879063 | 0 | PER3 | 10.411 | 31 |
| Allergy | rs962993-T | rs962993 | chr10 | 9053132 | 9053132 | 0 | . | 10.008 | 36 |
| PBC | rs10931468 | rs3771317 | chr2 | 191538562 | 191543962 | 5400 | NAB1 | 9.999 | 37 |
| CEL | rs7753008 | rs72928038 | chr6 | 90809639 | 90976768 | 167129 | BACH2 | 9.787 | 38 |
| RA | rs72928038 | rs72928038 | chr6 | 90976768 | 90976768 | 0 | BACH2 | 9.787 | 39, 40, 33 |
| ATD | |||||||||
| MS | |||||||||
| MS | rs564976 | rs485789 | chr3 | 159729059 | 159730148 | 1089 | AK097161 | 8.782 | |
| MS | rs733724 | rs733724 | chr6 | 105223864 | 105223864 | 0 | HACE1 | 7.360 | 41 |
| PBC | rs3024921 | rs3024921 | chr2 | 191943272 | 191943272 | 0 | STAT4 | 6.931 | 42 |
Discussion
One of the most significant challenges facing contemporary genetics and precision medicine is the interpretation of noncoding sequence variation. This challenge remains in the face of significant advances in genome sequence technologies that now make possible the generation of huge quantities of noncoding sequence data. Despite the wealth of evidence linking noncoding sequence variation to disease risk, we have been left wanting in our ability to directly interpret primary noncoding sequence data and hence to infer the biological consequences of disease-associated variation. Our efforts in this study take significant strides in creating a framework within which we can computationally develop and utilize regulatory lexicons, making robust predictions of the consequences of variation in functional noncoding sequence modules.
To ensure very high confidence predictions, we have limited our initial analyses to the highest deltaSVM scores in Table 2. However, comparison with validated SNPs (Fig. 6a) shows that many more moderate deltaSVM scoring SNPs will also perturb regulatory activity, but likely with relatively diminished effect. In this sense our random control sampling is highly conservative, as the positive loci are all known to be associated with disease. The high accuracy and low false positive rate of deltaSVM (Fig. 2e) is crucial to identify these causal SNPs with high accuracy. Together with our observations at lymphoblastoid dsQTLs, the Tyr and Tyrp1 enhancers, the ALDOB enhancer analyses and those performed in K562, and HepG2, these results clearly demonstrate that deltaSVM can broadly predict the empirically measured, cell-type specific functional consequences of enhancer sequence variants, given an appropriate training substrate.
Our results strongly support the hypothesis that non-coding disease associated SNPs that disrupt DNase HS/enhancer function do so directly through modulation of local TF-DNA interactions, leading to concomitant changes in chromatin state and gene expression. Many of these sequence determinants are recognizable as TF binding sites. Additionally gkm-SVM is also in principle capable of capturing sequence determinants of structural properties of TF-DNA interactions e.g. constrained sequence flanking TFBS that may contribute to the activity or stability of the enhancer/promoter regulatory complex. Precise variant evaluation requires an accurate assessment of the relative contribution of moderate and weak binding sites or other variants which affect chromatin accessibility, which we estimate requires at least ~1000 training elements and a robust classifier. We have shown that these sequence features are robust predictors of chromatin accessibility, and are also predictive of gene expression changes when enhancer/promoter connections are established. But chromatin accessibility is sometimes negatively correlated with gene expression changes, and our results suggest that enhancer/enhancer and enhancer promoter interactions at a larger scale will ultimately determine a sequence variant’s impact on gene expression.
In its application, both gkm-SVM and deltaSVM reinforce the recognized importance of cell type, developmental time, and biological state when connecting disease mechanisms to molecular events. Here, the biological state defines a set of active nuclear TFs in the cell, which in turn map sequence features to their target regulatory elements through combinatorial binding. It’s understandable therefore that the predictive power of deltaSVM for a given variant indicted in any given disease process is heavily dependent on the availability a biologically appropriate substrate on which to train the gkm-SVM. Consistent with this expectation, we have demonstrated that deltaSVM predictions are highly cell type dependent, i.e. deltaSVM from weights trained on one cell type are weak predictors of expression changes in other cell types (Table 1). Further, deltaSVM only identifies the validated disease associated SNPs shown in Fig. 6a if trained on an appropriate cell type. While the ENCODE8,23 and Roadmap9 projects have provided a wealth of such training data, our results indicate that future progress in common disease etiology will be greatly facilitated by coupling sequence based computational analysis with the generation of functional genomics data targeting disease relevant developmental stages and cell types. What we provide here is evidence that such integration of computational and disease-informed biological and strategies can be used to illuminate the roles played by noncoding regulatory variation in disease.
Materials and Methods
gkm-SVM and deltaSVM
We trained a gkm-SVM by following previously reported methods with minor modifications7,10–12. Briefly, we first defined a positive training set by using publically available DnaseI-seq and ChIP-seq datasets, as discussed in greater detail below. We then generated a negative training set by randomly sampling from the genome equal number of regions that match length, GC and repeat fractions of the positive set. To remove false negative regions as much as possible, we excluded any regions with P < 1e-5 (MACS43) from sampling. We then trained a gkm-SVM with default parameters (l = 10, k = 6, and d = 3 with truncated filter), and measured the classification performance using ROC curves with five-fold cross validation. Scaling of performance with gkm-SVM feature length is shown in Supplementary Figure 5. To calculate deltaSVM, 10-mer SVM scores were used as a proxy for weights. We generated the final weights by averaging gkm-SVMs trained on five independently generated 1x negative sets. When we compare deltaSVM between different training sets we normalize weights by the standard deviation of the weight distribution, but we have reported raw weights here for simplicity. This correction is typically a small effect (< 50%).
Training set for DNaseI Hypersensitive regions in lymphoblastoid cell lines
GM12878 DNaseI-seq peaks were first defined by MACS43 (P < 1e-9) for each replicate independently. We then chose peaks that were consistently found in both replicates. These peaks were further trimmed and 300 bp central DHSs that maximize the DNase I hypersensitive signals were determined. We also excluded any regions with repeats > 70% and regions overlapping with dsQTLs, to avoid possible overfitting when scoring dsQTLs. We ultimately obtained 22,384 300 bp DHSs as the positive training set.
Training set for mouse melanocyte enhancers
To train gkm-SVM appropriate for Tyr and Tyrp1 enhancers in mouse melanocytes, we determined 4,337 EP300 bound regions in the mouse melanocyte cell line melan-Ink4a-Arf 12 as the positive training set by following the above protocol with some adjustments (MACS P < 0.002). Promoter proximal regions and repeats were excluded from the training set. Since this positive set is much smaller than the others, we generated 10x larger negative sets in order to obtain more robust weights for deltaSVM analysis.
Training set for mouse liver enhancers
Similar to the training set for DHSs in LCLs, we defined a positive training set (n = 19,590) relevant to the ALDOB enhancer by integrating DNaseI-seq and H3K4me1 ChIP-seq on adult mouse liver tissue23. To specify liver enhancers, we additionally excluded all promoter proximal DHSs (defined as regions with distances to the nearest known transcription start sites (TSS) < 2 kbp) from the training set, after determining the 300 bp core DHSs as described above. We further selected DHSs that overlap with H3K4me1 ChIP-seq peaks, which are well-known markers for enhancer activity44,45, and defined these as the positive training set.
DeltaSVM analysis of dsQTL SNPs
We used the dsQTL tables and the raw data files downloaded from the GEO database (accession number GSE31388) to define the positive and control sets of dsQTL SNPs. Because association alone does not necessarily imply the causation due in part to LD problem, we further applied more stringent rules to determine the most likely causal dsQTL SNPs. We first restricted to 1,296 SNPs within their associated 100 bp DHSs to ensure that the changes in DNase I sensitivities are physically linked to the changes in their DNA sequences. We also applied a more strict association P-value threshold (P < 1e-5) to reduce false positive associations, finally resulting in 579 SNPs. As a control SNP set, we generated a 50x larger set of common random SNPs (N = 28,950; minor allele frequency > 5%) sampled only from the top 5% DHS regions that had been used to identify dsQTLs in the previous study13. To reduce false negative SNPs, we excluded from sampling any DHSs that had been found to be significantly associated with any of the dsQTL SNPs. Weights from gkm-SVM and kmer-SVM trained on the GM12878 DHSs were then used to calculate deltaSVM scores. We confirmed that training a gkm-SVM on negative sequences constrained to match the positive sequences distance to TSS distribution did not affect overall performance (Supplementary Figure 6). We further confirmed that using negative dsQTL control SNPs constrained to match the positive dsQTL distance to TSS and LD distribution did not affect overall performance (Supplementary Figure 7). As a comparison, we considered three different scoring metrics; Combined-Annotation-Dependent Depletion (CADD4), Genome-Wide Annotation of Variants (GWAVA5), and conservation scores (Genomic Evolutionary Rate Profiling: GERP16). We downloaded pre-computed CADD scores for all 1000 Genome variants, from which the scores for the dsQTLs and control SNPs were extracted. We also extracted the corresponding GWAVA scores from the pre-calculated table downloaded from the GWAVA website. We analyzed all three different GWAVA models (region, tss, and unmatched) and chose the best one (region), as determined by AUC, for the main analysis. The GERP scores were also extracted from the same GWAVA result files. To do a fair comparison, we only considered SNPs for which all the five scores are available, resulting in 574 positive SNPs and 27,735 control SNPs. The entire prediction results are available in Supplementary Table 1. eQTL beta was calculated using quantile-normalized gene expression from the eQTL website.
Melanocyte Luciferase Assay and deltaSVM analysis
We selected 22 and 23 SNVs for functional testing in the Tyr (mm10 coordinates chr7: 87508164–87508388; 226 bp) and Tyrp1 (mm10 coordinates chr4:80819561–80819851; 291 bp), respectively. These SNVs were randomly selected as follows: 10 SNVs in each enhancer predicted to reduce the enhancer’s activity (negative deltaSVM), 4 SNVs in each enhancer predicted to increase the enhancer’s activity (positive deltaSVM), 4 in each enhancer SNVs predicted to have a neutral impact on the enhancer’s activity (deltaSVM near 0), and 4 (Tyr) or 5 (Tyr) additional SNVs that overlap with key motifs identified in previous reports20,21. Reference and SNV enhancer sequences were synthesized (Genewiz; South Plainfield, NJ), verified by sanger sequencing, and cloned into a luciferase reporter plasmid containing a minimal promoter and a luciferase reporter gene. For each SNV, we performed 4 biological replicates (each with an independent plasmid DNA clone) in order to control for differences that might arise from random mutations in the plasmid backbone or from variation in the quality of plasmid preps. We transfected each reporter plasmid into the mouse melanocyte cell line melan-Ink4a-Arf, and measured luciferase activity 24 hours later using the Dual-Luciferase Reporter Assay System (Promega; Madison, WI). We compared the activity of each variant enhancer sequence to the activity of the reference sequence (normalized to 1), and were thus able to quantitate the impact of each SNV on the enhancer’s activity. The entire deltaSVM predictions and the luciferase assay results are provided as Supplementary Table 2 and 3, respectively.
deltaSVM Analysis of Massively Parallel Reporter Assays
To compare with exhaustive single nucleotide mutagenesis of the ALDOB enhancer22, we trained a gkm-SVM on adult mouse liver DHS as described above and scored each single nucleotide variant with deltaSVM and compared them with its measured in vivo expression changes (Supplementary Table 4)22. To compare with the directed mutagenesis of putative K562 and HepG2 enhancers24, we trained K562 an HepG2 specific gkm-SVMs on the top 10000 500 bp DHS regions in K562 and HepG2 cells8, after excluding regions that were DHS in more than 30% of human ENCODE cell lines, or near promoters (< 2 kb from TSS), against an equal size GC and repeat matched training set. We compared deltaSVM and the expression change for pair of mutant wild-type constructs for each wild-type construct significantly expressed in either cell line (mean normalized expression>3.5) which yielded 175 wild-type constructs and 277 mutant constructs: 102 of these are single base pair mutations and 175 are motif scrambling (8–17 bp changed) (Supplementary Table 5). For the motif scrambling mutations we summed all 10-mer scores spanning the mutated motif.
Training set for validated enhancers
For each appropriate cell line, we trained on the top 10,000 500 bp DHS regions, after excluding regions that were DHS in more than 30% of human/mouse ENCODE cell lines/tissues, or near promoters (< 2 kb from TSS), against an equal size GC and repeat matched training set. The cell lines chosen were human LNCaP8 for Rfx6, mouse erythroleukemia (MEL)23 cells for Bcl11a, and HepG28 cells for Sort1.
Scoring of Autoimmune variants
We selected 11 autoimmune traits enriched in Th1 H3K27Ac as shown in Fig 3 of Ref. 28. We made predictions for 413 lead SNPs associated with 11 autoimmune diseases enriched in Th1 H3K27Ac regions (T1D: Type 1 Diabetes, CRO: Crohn’s Disease, MS: Multiple Sclerosis, CEL: Celiac Disease, PBC: Primary Biliary Cirrhosis, RA: Rheumatoid Arthritis, Allergy, ATD: Autoimmune Thyroid Disease, UC: Ulcerative Colitis, VIT: Vitiligo, SLE: Systemic Lupus Erythematosus)28. We trained a gkm-SVM on the top 10,000 500 bp Th1 DHS regions, after excluding regions that were DHS in more than 30% of human ENCODE cell lines, or near promoters (< 2 kb from TSS), against an equal size GC and repeat matched training set. We scored the lead SNP and all flanking off-lead candidates in LD as defined by (R2 > .5 and PICS28 probability > .0275), yielding 3113 total SNPs. Since the significance of the maximum deltaSVM score in a locus will depend on the number of SNPs in that locus, as a random control we scored random SNPs and equal size flanking sets. To determine the cutoff, we first determined 2nd percentile deltaSVM score from 10,000 random permutations for each number of flanking SNPs (1~30), and then calculated mean and standard deviation of the 100 repeated experiments as the final cutoff. We identified 17 high scoring deltaSVM SNPs which we predict to be expression perturbing SNPs with high confidence (P < .02), while at this threshold random sampling produced 8 SNPs (binomial test P < 0.004, Supplementary Figure 4). deltaSVM scores for all 3113 SNPs are provided as Supplementary Table 6.
Supplementary Material
Supplementary Table 1: All predictions of the 574 dsQTL SNPs and the 27735 control SNPs.
Supplementary Table 2: deltaSVM predictions of all possible point mutations in Tyr and Tyrp1 enhancers.
Supplementary Table 3: Experimental validation results of randomly selected deltaSVM predictions from Tyr and Typr1 enhancers.
Supplementary Table 4: deltaSVM predictions of all possible single point mutations in ALDOB enhancer and the corresponding in vivo effect size.
Supplementary Table 5: deltaSVM predictions of mutations in K562 and HepG2 enhancers.
Supplementary Table 6: deltaSVM predictions of all 3113 autoimmune disease associated SNPs.
Acknowledgments
This research was supported in part by US National Institutes of Health grant R01 NS62972 to ASM, and by grant R01 HG007348 to MAB.
Footnotes
URLs
All gkm-SVM weights and source code used in this study are available at http://www.beerlab.org/deltasvm. CADD, GWAVA, and eQTL data was downloaded from http://cadd.gs.washington.edu, ftp://ftp.sanger.ac.uk/pub/resources/software/gwava/, and http://eqtl.uchicago.edu/RNA_Seq_data/results.
Author Contributions
MAB, ASM, DL, and DUG designed the study and wrote the paper. DUG and MB performed the experiments and analyzed the data. DL and MAB developed the computational algorithms and analyzed the data. BJS and AL contributed computational analysis. DL and DUG contributed equally to this work.
Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maurano MT, et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gusev A, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ritchie GRS, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghandi M, Lee D, Mohammad-Noori M, Beer MA. Enhanced Regulatory Sequence Prediction Using Gapped k-mer Features. PLoS Comput Biol. 2014;10:e1003711. doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ENCODE Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011;21:2167–2180. doi: 10.1101/gr.121905.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletez-Brant C, Lee D, McCallion AS, Beer M. A kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res. 2013;41:W544–W556. doi: 10.1093/nar/gkt519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorkin DU, et al. Integration of ChIP-seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res. 2012;22:2290–2301. doi: 10.1101/gr.139360.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degner JF, et al. DNase_I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davydov EV, et al. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Dongwon, Beer Michael A. In: Genome Analysis: Current Procedures and Applications. Poptsova MS, editor. Horizon Scientific Press; 2014. [Google Scholar]
- 18.Ghandi M, Mohammad-Noori M, Beer MA. Robust k-mer frequency estimation using gapped k-mers. J Math Biol. :1–32. doi: 10.1007/s00285-013-0705-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickrell JK, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murisier F, Guichard S, Beermann F. A conserved transcriptional enhancer that specifies Tyrp1 expression to melanocytes. Dev Biol. 2006;298:644–655. doi: 10.1016/j.ydbio.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Murisier F, Guichard S, Beermann F. The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment Cell Res Spons Eur Soc Pigment Cell Res Int Pigment Cell Soc. 2007;20:173–184. doi: 10.1111/j.1600-0749.2007.00368.x. [DOI] [PubMed] [Google Scholar]
- 22.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue F, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kheradpour P, et al. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23:800–811. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Q, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet. 2014;46:126–135. doi: 10.1038/ng.2862. [DOI] [PubMed] [Google Scholar]
- 26.Bauer DE, et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farh KKH, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Y, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.International Multiple Sclerosis Genetics Consortium (imsgc) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubois PCA, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parkes M, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinds DA, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. 2013;45:907–911. doi: 10.1038/ng.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mells GF, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trynka G, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eyre S, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper JD, et al. Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet. 2012;21:5202–5208. doi: 10.1093/hmg/dds357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gourraud PA, et al. A genome-wide association study of brain lesion distribution in multiple sclerosis. Brain J Neurol. 2013;136:1012–1024. doi: 10.1093/brain/aws363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu JZ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2012;44:1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 45.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: All predictions of the 574 dsQTL SNPs and the 27735 control SNPs.
Supplementary Table 2: deltaSVM predictions of all possible point mutations in Tyr and Tyrp1 enhancers.
Supplementary Table 3: Experimental validation results of randomly selected deltaSVM predictions from Tyr and Typr1 enhancers.
Supplementary Table 4: deltaSVM predictions of all possible single point mutations in ALDOB enhancer and the corresponding in vivo effect size.
Supplementary Table 5: deltaSVM predictions of mutations in K562 and HepG2 enhancers.
Supplementary Table 6: deltaSVM predictions of all 3113 autoimmune disease associated SNPs.