

Introductory Paragraph
Pancreatic cancer is the fourth leading cause of cancer death in the developed world1. Both inherited high-penetrant mutations in BRCA22, ATM3, PALB24, BRCA15, STK116, CDKN2A7 and mismatch repair genes8 as well as low-penetrant loci are associated with increased risk9–12. To identify novel loci, we performed a genome-wide association study on 9,925 pancreatic cancer cases and 11,569 controls, including 4,164 newly genotyped cases and 3,792 controls in 9 studies from North America, Central Europe and Australia. Three newly associated regions were identified: 17q25.1 (LINC00673, rs11655237, OR=1.26, 95%CI:1.19–1.34, P=1.42×10−14), 7p13 (SUGCT, rs17688601, OR=0.88, 95%CI:0.84–0.92, P=1.41×10−8), and 3q29 (TP63, rs9854771, OR=0.89, 95%CI:0.85–0.93, P=2.35×10−8). Significant association was detected on 2p13.3 (ETAA1, rs1486134, OR=1.14, 95%CI:1.09–1.19, P=3.36×10−9), a region with prior suggestive evidence in the Han Chinese12. We replicate previously reported associations at 9q34.2(ABO)9, 13q22.1(KLF5)10, 5p15.33 (TERT, CLPTM1)10,11, 13q12.2 (PDX1)11, 1q32.1(NR5A2)10, 7q32.3(LINC-PINT)11, 16q23.1(BCAR1)11 and 22q12.1 (ZNRF3)11. Our study identifies novel loci associated with pancreatic cancer risk.
Main Text
We conducted a two-stage genome-wide association study (GWAS) of pancreatic cancer (Fig. 1). First, genome-wide genotyping of 8052 subjects from nine studies within the Pancreatic Cancer Case-Control Consortium (PanC4) (Supplementary Table 1) was conducted using the HumanOmniExpressExome-8v1 array. The overall study was approved by the Johns Hopkins Institutional Review Board (IRB). Each individual study obtained IRB approval from their parent institution. After quality control (Online Methods, Fig. 1, Supplementary Table 2), 7,956 individuals (4,164 cases and 3,792 controls) and 654,470 SNPs with call rates greater than 98% were analyzed. Unconditional logistic regression analysis adjusted for age and the first seven principal component eigenvectors was conducted under the log-additive genetic model (Fig. 2, Supplementary Fig. 1).
Figure 1.

Overview of Stage 1 and Stage 2 analyses
Figure 2.

Manhattan plot of PanC4 association analysis. Loci previously associated with pancreatic cancer in Caucasians are shown in black, 2p13.3 in blue and novel loci in red.
Analysis of 7,956 newly genotyped PanC4 individuals identified a novel locus at 17q25.1 (LINC00673, rs7214041, OR=1.38, 95%CI:1.26–1.51, P=1.95×10−10) significantly associated with pancreatic cancer risk (Table 1, column ‘PanC4’). In addition we replicate regions that had previously been reported to be associated with pancreatic cancer in the Caucasian population (Supplementary Table 3). These include: 9q34.29 (ABO, rs505922, OR=1.27, 95%CI:1.19–1.35, P=1.72×10−13), 13q22.110 (KLF5, rs9543325, OR=1.24, 95%CI:1.16–1.32, P=2.26×10−10), 5p15.3310 (CLPTM1, rs401681, OR=1.2, 95% CI:1.13–1.28, P=2.7×10−8), 13q12.211 (PDX1, rs9581943, OR=1.17, 95%CI:1.10–1.24, P=1.94×10−7), 1q32.110 (NR5A2, rs3790844, OR=0.83, 95%CI:0.77–0.90, P=3.05×10−6), 7q32.311 (LINC-PINT, rs6971499, OR=0.81, 95%CI:0.74–0.88, P=7.1×10−6), 5p15.3311 (TERT, rs2736098, OR=0.85, 95%CI: 0.78–0.93, P=2.31×10−5), 16q23.111 (BCAR1, rs7190458, OR=1.4, 95%CI=1.22–1.60, P=1.01×10−4), and 22q12.111 (ZNRF3, rs16986825, OR=1.14, 95% CI= 1.04–1.24, P= 2.72×10−3). In contrast, other than 2p13.3 (ETAA1, rs2035565, OR=1.15, 95%CI=1.07–1.25, P=2.69×10−4) (Supplementary Table 3) we observed no evidence of association (P>0.05) for SNPs previously reported to be associated (P<1×10−6) with pancreatic cancer in Asian populations12,13. While all ethnic groups were included in our analyses, over 92% of our study population reported Caucasian ancestry. We obtained similar results when analysis was limited to individuals reporting European ancestry. Because of limited sample sizes we did not conduct independent analysis of other ethnic groups(results not shown).
Table 1.
Significant (P<5×10−8) association results for pancreatic cancer
| Stage 1 | Stage 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Chra SNP Positionb Gene |
Effect Allele (Minor)/Reference Allele | Statistic | PanC4 4,164 cases 3,792 controls |
PanScan 1 1,856 cases, 1,890 controls |
PanScan 2 1,618 cases and 1,682 controls |
Combined Stage 1c 7,638 cases 7,364 controls |
PANDoRA 2,497 cases 4,611 controls |
Combined Stage 1&2d 9,925 cases 11,569 controls |
| 17q25.1h rs11655237 70,400,166 LINC00673 |
T/C | mafe cases;controls |
0.146; 0.110 | 0.139; 0.129 | 0.149; 0.116 | 0.135; 0.114 | ||
| infof | 0.963 | g | g | |||||
| OR (CI)g | 1.38 (1.26 – 1.52) | 1.09 (0.96 – 1.25) | 1.34 (1.16 – 1.55) | 1.27 (1.19 – 1.36) | 1.24 (1.10 – 1.40) | 1.26 (1.19 – 1.34) | ||
| p-value | 1.38×10−10 | 1.95×10−1 | 2.95×10−4 | 6.74×10−12 | 6.40×10−4 | 1.42×10−14 | ||
| 17q25.1h rs7214041 70,401,476 LINC00673 |
T/C | maf cases;controls |
0.148; 0.112 | 0.140; 0.133 | 0.150; 0.117 | 0.139; 0.117 | ||
| info | g | 0.966 | 0.96 | |||||
| OR (CI) | 1.38 (1.26 – 1.51) | 1.07 (0.93 – 1.22) | 1.33 (1.15 – 1.53) | 1.26 (1.18 – 1.35) | 1.25 (1.11 – 1.41) | 1.26 (1.19 – 1.34) | ||
| p-value | 1.95×10−10 | 3.36×10−1 | 3.69×10−4 | 2.67×10−11 | 3.37×10−4 | 2.88×10−14 | ||
| 2p13.3 rs1486134 67,639,769 ETAA1 (2236bp 3′) |
G/T | maf cases;controls |
0.302; 0.275 | 0.305; 0.292 | 0.305; 0.276 | 0.292; 0.273 | ||
| info | g | g | g | |||||
| OR (CI) | 1.14 (1.06 – 1.22) | 1.06 (0.96 – 1.18) | 1.15 (1.03 – 1.28) | 1.13 (1.08 – 1.19) | 1.16 (1.06 – 1.27) | 1.14 (1.09 – 1.19) | ||
| p-value | 5.96×10−5 | 1.57×10−1 | 5.18×10−3 | 8.35×10−7 | 9.42×10−4 | 3.36×10−9 | ||
| 7p13 rs17688601 40,866,663 SUGCT |
A/C | maf cases;controls |
0.241; 0.263 | 0.218; 0.254 | 0.237; 0.268 | 0.254; 0.277 | ||
| info | g | g | g | |||||
| OR (CI) | 0.89 (0.83 – 0.96) | 0.82 (0.73 – 0.91) | 0.85 (0.76 – 0.94) | 0.87 (0.82 – 0.91) | 0.91 (0.83 – 1.00) | 0.88 (0.84 – 0.92) | ||
| p-value | 1.98×10−3 | 1.66×10−4 | 8.72×10−3 | 9.77×10−8 | 3.93×10−2 | 1.41×10−8 | ||
| 3q29 rs9854771 189,508,471 TP63 |
A/G | maf cases;controls |
0.328; 0.362 | 0.336; 0.366 | 0.325; 0.356 | 0.341; 0.356 | ||
| info | g | 0.998 | 0.998 | |||||
| OR (CI) | 0.86 (0.81 – 0.92) | 0.88 (0.80 – 0.90) | 0.87 (0.79 – 0.97) | 0.87 (0.83 – 0.92) | 0.93 (0.86 – 1.01) | 0.89 (0.85 – 0.93) | ||
| p-value | 3.10×10−5 | 7.94×10−3 | 1.55×10−2 | 4.08×10−8 | 1.01×10−1 | 2.35×10−8 | ||
Cytogenetic regions according to NCBI Human Genome Build 37 and NCBI’s Map Viewer
SNP position according to NCBI Human Genome Build 37
Results from the Combined Stage 1 meta-analysis of PanC4, PanScan 1, and PanScan 2
Results from the Combined Stage 1 and 2 meta-analysis of PanC4, PanScan 1, PanScan 2, and PANDoRA
MAF- minor allele frequency
Quality of imputation metric. See online methods for more detail. If snp is genotyped and not imputed, a ‘g’ is reported
Allelic Odds Ratio and corresponding 95% Confidence Interval
R2>0.95
We then conducted a genome-wide meta-analysis of the PanC4 data with data from PanScan 19 and PanScan 210 (Combined Stage 1, Fig. 1). After quality control (Online Methods and Supplementary Table 4), we analyzed 528,179 SNPs and 3,746 individuals (1,856 cases and 1,890 controls) from PanScan 1 and 557,555 SNPs and 3,300 individuals (1,618 cases and 1,682 controls) from PanScan 2. Since the genotyping platforms differed across studies, missing genotypes were imputed using IMPUTE v214, with 1000 Genomes15 (release Dec 2013) and HapMap316 (release #2,2009) as reference panels. For PanScan 1 and PanScan 2, we conducted association analysis using unconditional logistic regression including age and the first four principal components eigenvectors as covariates. Data from PanC4, PanScan 1, and PanScan 2 were combined (7,638 cases and 7,364 controls and 866,891 SNPs) and analyzed using a fixed-effects model implemented in METAL17 (Fig. 3). A QQ plot (Supplementary Fig. 2) indicated appropriate control of type-1 errors, with λ values of 1.025 for PanC4, 0.998 for PanScan 1, and 1.017 for PanScan 2.
Figure 3.
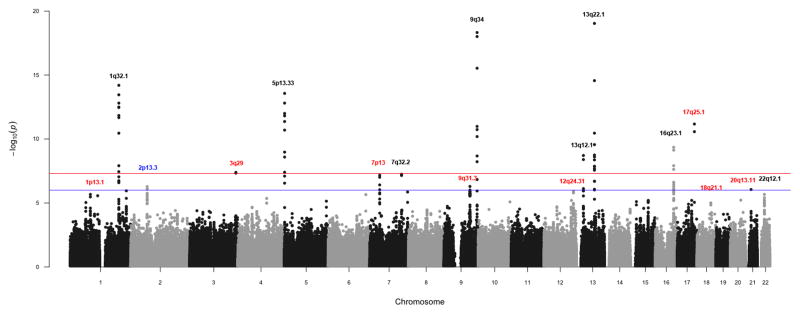
Manhattan plot of Combined Stage 1 association analysis. Loci previously associated with pancreatic cancer in Caucasians are shown in black, 2p13.3 in blue and novel loci in red.
The Combined Stage 1 Analysis (Table 1, column ‘Combined Stage 1’) yielded a second novel region of association at 3q29 (TP63, rs9854771, OR=0.87, 95%CI:0.83–0.92, P=4.08×10−8). A second SNP on 17q25.1 (rs11655237, OR=1.27, 95%CI:1.19–1.36, P=6.74×10−12), which is in high LD (r2=0.95) with rs7214041, also gave significant evidence of association in these combined data.
We next conducted a Stage 2 analysis in an independent set of 2,497 cases and 4,611 controls from the PANDoRA consortium18. We selected twenty-five SNPs from 23 independent regions (Supplementary Table 5) with p-values below 10−5 in either PanC4 or the Combined Stage 1 analyses. When multiple SNPs per region were associated, the most significant SNP was selected; SNPs on 17q25.1 and 2p13.3 were exceptions. After quality control (Online Methods, Supplementary Table 6), 2,287 cases and 4,205 controls from the PANDoRA study were analyzed. Age-adjusted association analyses by country were carried out, and results were combined using a fixed-effect model. We observed independent evidence of association at 17q25.1 in the PANDoRA study (Table 1, column ‘PANDoRA’: rs7214041, OR=1.25, 95%CI:1.11–1.41, P=3.37×10−4).
Combined analysis of the Stage 1 and 2 data for the 25 SNPs (Table 1 and Supplementary Table 5, column ‘Combined Stage 1&2’) revealed two additional significantly associated loci: 2p13.3(ETAA1, rs1486134, OR=1.14, 95%CI:1.09–1.19, P=3.36×10−9) and 7p13(SUGCT, rs17688601, OR=0.88, 95%CI:0.84–0.92, P=1.41×10−8). Promising signals (Supplementary Table 7) arose at 18q21.2(GRP, rs1517037, OR=0.87, 95%CI:0.83–0.92, P=3.17×10−7), 12q24.31(HNF1A, rs7310409, OR= 1.11, 95%CI:1.06–1.15, P=6.34×10−7), 1p13.1(WNT2B, rs351365, OR=0.89, 95%CI:0.85–0.93, P=7.39×10−7), and 20q13.11 (rs6073450, OR=1.11, 95%CI:1.06–1.15, P=9.21×10−7).
We identified and replicated a novel region for association on 17q25.1 (Fig. 4a). Two highly correlated variants (rs11655237 and rs7214041, r2=0.95) were associated with pancreatic cancer risk. Variant rs7214041 is to LINC00673 (long inter-genic non-protein coding RNA 673). rs11655237, a non-coding transcript variant, shows significant DNase hypersensitivity in multiple cancer cell lines and binds transcription factors including P300, FOXA1, FOXA2, and the DNA repair protein RAD21 according to HaploReg v219. HaploRegV2 also indicated rs7214041 alters regulatory motifs for HNF119. Interestingly, we also found suggestive evidence of an association with rs7310409 located at the HNF1A locus (12q24.31, Supplementary Fig. 3a and Supplementary Table 7). A recent study of the pancreatic cancer transcriptome suggests HNF1A may act as a tumor suppressor in pancreatic cancers20. Variation in HNF1A has been associated with risk of Type 2 diabetes21,22, a well-established risk factor for pancreatic cancer23–25, and maturity onset diabetes of the young (MODY)26. Furthermore, variants in HNF1A (in particular rs7310409) and HNF4A were identified as risk factors for pancreatic cancer in pathway-based and candidate-SNP-based analyses of the PanScan data27,28.
Figure 4.




Regional association and linkage disequilibrium (LD) plots for four novel genome-wide significant loci: (a) 17q25.1, (b) 3q29, (c) 2p13.3, and (d) 7p13. Association p-values are shown for three analyses: PanC4 only (black circles), Combined Stage 1 (PanC4, PanScan 1, and PanScan 2) (grey circles), and Combined Stage 1 and 2 (PanC4, PanScan 1, PanScan 2, and PANDoRA) (red circles). LD plots are based on 1000 Genomes European samples.
We also identified significant association for two variants in high LD (rs9854771 and rs1515496, r2=0.99) located in an intron of TP63 on 3q29 (Fig. 4b). p63 is a p53 homologue implicated in tumorigenesis and metastasis29 by playing a role in cell-cycle arrest and apoptosis. Overexpression of p63 can mimic p53 activation in certain experimental models30. Interestingly, different isoforms of p63 have opposing effects; TAp63 has tumor suppressive effects while DNp63 has oncogenic effects31. Danilov and colleagues suggested DNp63α was the predominant isoform in pancreatic cancer cell lines and promoted pancreatic cancer growth, motility and invasion32. Previous GWAS studies of lung cancer and bladder cancer have demonstrated significant evidence of association for SNPs in TP6333–37. HaploReg query of this region showed that both are predicted to be conserved elements via GERP, suggesting functional roles.
Our analysis revealed genome-wide significance in a region on 2p13.3 (rs1486134). A pancreatic cancer GWAS in Han Chinese subjects12 found suggestive evidence for another SNP on 2p13.3 (rs2035565) (Supplementary Table 3). High LD is present throughout this region (Fig. 4c), including strong LD between rs1486134 and rs2035565 in European and Asian populations based on 1000 Genomes15 samples (r2=0.91 and r2=0.90 respectively). This region includes the gene ETAA1 (Ewing tumor-associated antigen 1), alias ETAA16, that may function as a tumor-specific cell surface antigen in the Ewing’s family of tumors38.
We observed significant association on 7p13 for rs17688601, located in an intron of the SUGCT (succinyl-CoA:glutarate-CoA transferase) gene (alias c7orf10) (Fig. 4d). This variant is predicted in HaploREGV2 to alter binding of HNF1-4 and other DNA binding proteins19. The SUGCT protein is involved in glutarate metabolism and mutations in this gene are associated with glutaric aciduria39. While there is evidence of altered tricarboxylic acid cycle metabolism in pancreatic cancer40, the role of this gene in pancreatic cancer risk is unclear.
Combined Stage 1 and Stage 2 identified suggestive evidence of association (P<1×10−6) in four regions: 12q24.31(HNF1A) (Supplementary Fig. 3a), 18q21.2(GRP) (Supplementary Fig. 3b), 1p13.1(5′ of WNT2B) (Supplementary Fig. 3c), and 20q13.11 (Supplementary Fig. 3d). GRP (gastrin releasing peptide) production has been associated with pancreatic tumor growth in vitro41. WNT signaling plays an important role in pancreas development. WNT2B (Wingless-Type MMTV Integration Site Family, Member 2B) is overexpressed in pancreatic cancer and has been associated with decreased survival42. The 20q13.11 variant is located ~20kb of the HNF4A (MODY) gene, mutations of which are associated with early-onset diabetes43.
In the PanC4 study we observed 11 SNPs on chromosome 9q31.3 (Supplementary Fig. 3e) in moderate to high LD (r2 values between 0.6 and 1) with p-values from 7.00×10−8 to 2.73×10−6, including rs10991043 (OR=1.19, 95%CI:1.12–1.26, P=7.00×10−8) nearby the SMC2 (structural maintenance of chromosome 2) gene. This gene plays an important role in DNA repair in humans. While there was no evidence of association in the other study populations examined, the strong signal across multiple SNPs in PanC4 suggest that this region merits further investigation.
Further functional characterization of these associated regions is needed, including examining if these SNPs are functional through eQTL. Performing eQTL analysis of pancreatic tissues is challenging. Normal pancreatic tissue is primarily comprised of acinar cells (>90%), but pancreatic ductal adenocarcinoma has a ductal phenotype, and the appropriate normal tissue to analyze is debatable because the cell of origin of pancreatic ductal adenocarcinomas is debated. eQTL analysis of pancreatic tumor tissue is also problematic because the tumor tissue of a pancreatic ductal adenocarcinomas contains a variable mixture of cell types including fibroblasts, multiple types of immune cells, non-neoplastic pancreatic cells and cancer cells, with cancer cells representing only a minority of the total cell population. Furthermore, gene expression analysis of normal pancreatic tissue is often limited by the RNA degradation associated with high level RNAase expression in pancreatic acinar cells. An ideal study of pancreatic eQTLs for pancreatic cells would take into account these challenges.
Smoking is a well-established risk factor for pancreatic cancer44–47. For all nine SNPs identified in Table 1 and Supplementary Table 7, we conducted an analysis stratified by smoking status (ever smoker vs. never smoker) in PanC4 samples. No qualitative differences in effect size between current smokers and never smokers were observed (results not shown). Furthermore, when we included an interaction term in the model, this term was not significant at the 0.05 level.
We estimated the heritability of pancreatic cancer due to common GWAS SNPs using data from PanC4 samples of Caucasian ancestry using only directly genotyped SNPs(3,828 cases, 3,551 controls and 620,357 SNPs) as well as the combined dataset (7,032 cases 6,866 controls 268,681 SNPs). Using a disease prevalence of 0.0149, reflecting the lifetime risk of pancreatic cancer, we estimated that 16.4% (95%CI: 10.4%–22.4%) in PanC4 and 13.1% (95%CI 9.9%–16.3%) for the combined dataset of the total phenotypic variation was explained by genome-wide common SNPs. The established associated regions (loci in Table 1 and Supplemental Table 3), accounted for 3.0% (95%CI: 2.0%-3.9%) and 2.1%(95%CI 1.7%-3.1%) of the total phenotypic variation in the Panc4 population and the combined dataset, respectively.
We identified several novel regions involved in pancreatic cancer susceptibility, and provided additional evidence to support many of the established associations. While it is of interest that many of these highly associated variants are located in the introns of genes, these associations could be due to more distant genomic effects. Follow-up studies, including functional studies, are needed to fully understand how these variants (either directly or indirectly) impact risk of pancreatic cancer. Our work highlights the importance of common variation in pancreatic cancer risk.
Online Methods
Stage 1 Methods
PanC4 Quality Control
In total, 8052 individuals were selected for genotyping from studies participating in the Pancreatic Cancer Case-Control Consortium (PanC4). Participating sites included: The Central Europe study coordinated by the International Agency for Research on Cancer (IARC/Central Europe)48, Johns Hopkins Hospital49,50, Mayo Clinic51, MD Anderson Cancer Center52, Memorial Sloane-Kettering Cancer Center53, University of Toronto54, Queensland55, University of California San Francisco (UCSF)56, and Yale University57 (Supplementary Table 1). Cases were defined as individuals with adenocarcinoma of the pancreas. DNA samples from these individuals from PanC4, 180 study duplicates, 176 HapMap control samples, and 26 replicates from the previous pancreatic cancer GWAS PanScan 210, were genotyped on the IlluminaHumanOmniExpressExome-8v1 array at the Johns Hopkins Center for Inherited Disease Research (CIDR). Genotypes were called using GenomeStudio version 2011.1, Genotyping Module 1.9.4 and GenTrain version 1.0.
Genotyping results were inspected for quality by assessing the missing call rate, allelic imbalance, heterozygosity, discordance in reported versus genotyped gender, relatedness, ancestry and chromosomal anomalies. Unexpected relatedness between pairs of samples was assessed using the method of moments58 implemented in SNPRelate59. The median genotype call rate was 99.9%, with all individuals having a call rate greater than 98%. After removing individuals with excessive allele sharing, duplicates and subjects with incomplete information on age, 7,956 subjects (4,164 cases and 3,792 controls) were available for statistical analyses (Supplementary Table 2). SNPs with the following characteristics were excluded from statistical analyses: positional duplicates, more than two discordant calls in study duplicates, technical failures or missing call rate greater than 2%, more than one Mendelian error in HapMap control trios, Hardy-Weinberg equilibrium p-value<10−6, sex difference in allele frequency greater than 0.2 for autosomes/XY in samples of European ancestry, and minor allele frequencies (MAF) less than 0.005. Overall 654,470 SNPs passed the quality control filters applied; the median missing call rate was 0.024% and 98% of SNPs had a missing call rate less than 1% (Supplementary Table 2).
PanScan 1 and PanScan 2 Quality Control
PanScan 1 and PanScan 2 data were obtained from dbGAP60,61 (dbGaP study accession: phs000206.v4.p3). Data from all participating sites apart from Group Health (which required a separate data sharing agreement) were included in the analysis. Previously published PanScan 19 and PanScan 210 studies used the Illumina HumanHap550 and Illumina Human 610-Quad chips respectively. Quality control was performed as described above for PanC4. Forty-five unexpected duplicates between PanScan 1, PanScan 2, and PanC4 were identified and removed from analyses of the PanScan datasets. After data cleaning, 528,179 SNPs and 3,746 individuals (1,856 cases, 1,890 controls) remained in PanScan 1, and 557,555 SNPs and 3,300 individuals (1,618 cases and 1,682 controls) remained in PanScan 2 (See Supplementary Table 4).
Association Analysis
To investigate population structure, principal components analysis (PCA) was conducted separately for PanC4, PanScan 1 and PanScan 2 using SNPRelate59 (Supplementary Fig. 4). Genotype imputation was performed separately for PanScan 1, PanScan 2 and PanC4 using IMPUTE v214. Since PanScan 1 and PanScan 2 SNPs were originally mapped using an older genome assembly (NCBI build 36), we converted their genome position to genome assembly NCBI build 37 by using LIFTOVER. Markers not identified in the build 37 assembly were removed. To decrease computational time, we pre-phased genotypes to produce best-guess haplotypes using SHAPEIT v2 software62. Both 1000 Genomes15 Phase I-integrated haplotypes (release Dec 2013) and HapMap316 (release #2,2009) were used as reference panels during imputation.
After imputation, SNPs with quality scores < 0.3 were excluded from all subsequent analysis. Only SNPs directly genotyped in either PanC4, PanScan 1, or PanScan 2 and passing quality control filters were retained for analysis. This resulted in 866,891 SNPs in the Combined Stage 1 analysis. The expected genotype counts were then analyzed using the frequentist test option of SNPTEST63. Decade of age and eigenvectors from PCA were included as covariates. The number of eigenvectors to include was chosen based on inspection of the scree plot and p-values from association between eigenvectors and pancreatic cancer status. The results from each study were then combined using a fixed-effects inverse standard error approach implemented by METAL17 (see Supplementary Table 5, column ‘Combined Stage 1’). Test statistic inflation (λ), was estimated to be 1.025 for PanC4, 0.998 for PanScan 1, and 1.017 for PanScan 2. Test statistics for PanC4 and PanScan 2 were adjusted to account for small amounts of population stratification using METAL’s genomic control option. Our sample size gives us over 80% power to detect an odds ratio of 1.2 for SNPs with a minor allele frequency greater than 0.20. Manhattan and QQ plots of PanC4 GWAS are shown in Fig. 2 and Supplementary Fig. 1, respectively. Manhattan and QQ plots showing association results from the Combined Stage 1 analysis are shown in Fig. 3 and Supplementary Fig. 2. To examine whether our association results were confounded by population stratification, we conducted a secondary analysis, restricting our samples to those of European ancestry based on a PCA analysis performed with PanC4 and Hapmap3 samples. The loci identified through association testing did not change, and their odds ratios and p-values did not vary significantly (results not shown).
Stage 2 Methods
PANDoRA Replication Study
Twenty-five SNPs from 23 independent regions identified as showing evidence of association (P<1×10−5) in either the PanC4 analysis or the Combined Stage 1 analysis, were genotyped in samples from the PANcreatic Disease ReseArch (PANDoRA)18 consortium with TaqMan technology. These samples were drawn from case-control studies in six European countries: Czech Republic, Germany, Greece, Italy, Lithuania, and Poland. In total, 2497 cases with pancreatic cancer and 4611 controls were genotyped. 8% of samples were duplicated and overall concordance was >99%. Supplementary Table 6 shows the features of the PANDoRA dataset. Samples missing more than 2 SNPs (~15%) or missing covariate information were excluded from analyses. In total, 2,287 cases and 4,205 controls from the PANDoRA study remained after quality control.
Because PANDoRA is a collection of samples from various centers, we analyzed each country separately. Logistic regression models with additive effects of each allele were fit, as implemented in PLINK64 (Supplementary Table 5, column ‘PANDoRA’). Two SNPs, rs16867971 for Greece and rs10850078 for Lithuania, showed evidence of departure from HWE in controls (P<0.001). The SNP violating HWE was not analyzed for that country. A final fixed-effects meta-analysis of PanC4, PanScan 1, PanScan 2, and PANDoRA (Combined Stage 1 and 2 analysis) was then conducted using METAL17 on the 25 SNPs chosen for inclusion in Stage 2. Results are in Supplementary Table 5, column ‘Combined Stage 1&2’. To further interrogate these regions we examined all 1000G imputed SNPs (as described above) in regions with significant or suggestive (P<1×10−6) evidence of association (Fig. 4 and Supplemental Fig. 3).
In our Combined Stage 1 analysis (Supplementary Table 5, column ‘Combined Stage 1’), two SNPs selected for replication were observed to have heterogeneity p-values below 0.001. When restricting our analysis to individuals of European ancestry, heterogeneity p-values from the meta-analysis remained virtually unchanged, implying that the heterogeneity was not due to population stratification. One SNP, rs16867971, was directly genotyped in PanC4 and imputed in PanScan 1 and PanScan 2. We found evidence of association for rs16867971 in PanC4, but not in PanScan 1 or PanScan 2 (P>0.05). The second SNP, rs6706539, was also directly genotyped in PanC4 and imputed in PanScan 1 and PanScan 2. For rs6706539, allele A was associated with increased risk in PanScan 1 and PanScan 2 (P=0.008 and P=0.04, respectively) but protective in PanC4 (P=3.4×10−6). Upon examination of the imputed and non-imputed SNPs adjacent to rs6706539, we found that r2 values between this SNP and other SNPs within 10kb were low, ranging from 0.05 to 0.18 in 1000 Genomes CEU samples. It is possible that this low LD made imputation of this SNP rather difficult. Additionally, inspection of the alleles coded as reference and alternate alleles for this SNP in PanC4 and 1000 Genomes suggests that this oddity is not due to differences in strand alignment.
Forest plots of our top hits (Supplemental Fig. 5a–5i) showing the magnitude of odds ratios for each study population show that in the majority of the “top” SNPs, those with p-value <1×10−6, the direction of the effect was consistent across populations. Additionally, none of the top SNPs showed significant heterogeneity (at the 0.05 level) in the Combined Stage 1 and Stage 2 analysis. However, in many instances the effect size in the PanScan I population was smaller than the association observed in PanScan II, PanC4 and PANDoRA. We conducted a random effects meta-analysis as well and overall the results were consistent with the results observed from the fixed-effect meta-analysis (results not shown).
Heritability Analysis
Heritability analysis was performed using GCTA65 software. This analysis estimates the percentage of phenotypic variance explicated by common SNPs. We assumed a prevalence of 0.0149 (risk to age 90 in the US Caucasian population; SEER data collected in 2009–2011). We excluded individuals not clustering with HapMap16 CEU (CEPH- Utah residents with ancestry from northern and western Europe) samples in PCA analysis as well as individuals with estimated relationships > 0.05 or missing genotype rate >0.01. SNPs with missing rate>0.05, MAF <0.01 and HWE p-value<5×10−4 were also excluded. We estimated the overall heritability in the PanC4 study using SNP data, as well as the heritability attributed to the 12 regions with significant evidence of association in the Caucasian population plus the 6 suggestive regions identified.
HaploReg
HaploReg is a tool used for exploring functional annotations of non-coding variants. For each variant and region identified in this study, we used HaploReg to gain insight into functional annotations including chromatin state (promoters and enhancers), conserved regions, variant effect on regulatory motifs and protein binding sites. Regions were defined by SNPs with r2>0.8 to the associated SNP.
Supplementary Material
Acknowledgments
This work was supported by RO1 CA 154823. Genotyping Services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to the Johns Hopkins University, contract number HHSN268201100011I.
The IARC/Central Europe study was supported by a grant from the US National Cancer Institute at the National Institutes of Health (R03 CA123546-02) and grants from the Ministry of Health of the Czech Republic (NR 9029-4/2006, NR9422-3, NR9998-3, MH CZ-DRO-MMCI 00209805).
The work at Johns Hopkins University was supported by the NCI Grants P50CA62924 and R01CA97075. Additional support was provided by Susan Wojcicki and Dennis Troper.
The Mayo Clinic Molecular Epidemiology of Pancreatic Cancer study is supported by the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701).
The Memorial Sloan Kettering Cancer Center Pancreatic Tumor Registry is supported by P30CA008748, the Geoffrey Beene Foundation, the Arnold and Arlene Goldstein Family Foundation, and the Society of MSKCC.
The Queensland Pancreatic Cancer Study was supported by a grant from the National Health and Medical Research Council of Australia (NHMRC) (Grant number 442302). RE Neale is supported by a NHMRC Senior Research Fellowship (#1060183).
The UCSF pancreas study was supported by NIH-NCI grants (R01CA1009767, R01CA109767-S1) and the Joan Rombauer Pancreatic Cancer Fund. Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program; the NCI’s SEER Program under contract HHSN261201000140C awarded to CPIC; and the CDC’s National Program of Cancer Registries, under agreement #U58DP003862-01 awarded to the California Department of Public Health.
The Yale (CT) pancreas study is supported by National Cancer Institute at the U.S. National Institutes of Health, grant 5R01CA098870. The cooperation of 30 Connecticut hospitals, including Stamford Hospital, in allowing patient access, is gratefully acknowledged. The Connecticut Pancreas Cancer Study was approved by the State of Connecticut Department of Public Health Human Investigation Committee. Certain data used in that study were obtained from the Connecticut Tumor Registry in the Connecticut Department of Public Health. The authors assume full responsibility for analyses and interpretation of these data.
Studies included in PANDoRA were partly funded by: the Czech Science Foundation (No. P301/12/1734), the Internal Grant Agency of the Czech Ministry of Health (IGA NT 13 263); the Baden-Württemberg State Ministry of Research, Science and Arts (Prof. H. Brenner), the Heidelberger EPZ-Pancobank (Prof. M.W. Büchler and team: Prof. T. Hackert, Dr. N. A. Giese, Dr. Ch. Tjaden, E. Soyka, M. Meinhardt; Heidelberger Stiftung Chirurgie and BMBF grant 01GS08114), the BMBH (Prof. P. Schirmacher; BMBF grant 01EY1101), the “5×1000” voluntary contribution of the Italian Government, the Italian Ministry of Health (RC1203GA57, RC1303GA53, RC1303GA54, RC1303GA50), the Italian Association for Research on Cancer (Prof. A. Scarpa; AIRC n. 12182), the Italian Ministry of Research (Prof. A. Scarpa; FIRB - RBAP10AHJB), the Italian FIMP-Ministry of Health (Prof. A. Scarpa; CUP_J33G13000210001), and by the National Institute for Health Research Liverpool Pancreas Biomedical Research Unit, UK. We would like to acknowledge the contribution of Dr Frederike Dijk and Prof. Oliver Busch (Academic Medical Center, Amsterdam, the Netherlands).
Assistance with genotype data quality control was provided by Cecelia Laurie and Cathy Laurie at University of Washington Genetic Analysis Center.
Footnotes
URLs
PanC4: www.panc4.org
GLOBOCAN: globocan.iarc.fr/Default.aspx
PLINK: pngu.mgh.harvard.edu/~purcell/plink/
LIFTOVER: genome.ucsc.edu/cgi-bin/hgLiftOver
SHAPEIT: mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html
IMPUTE2: mathgen.stats.ox.ac.uk/impute/impute_v2.html
SNPTEST: mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html
METAL: csg.sph.umich.edu/abecasis/metal/
HAPLOREG: www.broadinstitute.org/mammals/haploreg/haploreg.php
Author Contributions
D.C, G.M.P, H.A.R, and A.P.K organized and designed the study. F.C and A.P.K organized and supervised the genotyping of samples. E.J.C, E.M, D.C, and A.P.K designed and conducted the statistical analysis. E.J.C, E.M, M.G.G, and A.P.K drafted the first version of the manuscript. P.M.B, S.G, M.G.G, D.L, R.N., S.H.O, G.S, L.T.A, W.R.B, M.F.B, A.B, M.B, P.B, H.B, H.B.B-d-M, F.C, G.C, G.M.C, K.G.C, S.J.C, S.P.C, M.C, L.F, C.F, N.F, M.G, M.H, J.M.H, I.H, E.A.H, R.N.H, R.J.H, V.J, T.J.K, J.K, R.C.K, S.L, L.L, E.M-P, A.M, B.M-D, J.P.N, A.L.O, I.O, C.P, R.P, C.R, A.S, A.S, R.Z.S-S, O.S, F.T, Y.K.V, P.V, B.M.W, H.Y, G.M.P, H.A.R, and A.P.K contributed samples for the GWAS and/or the replication analysis. All authors contributed to the final version of the manuscript.
Competing Financial Interests
Under a licensing agreement between Myriad Genetics, Inc., and the Johns Hopkins University, M. Goggins and A.P. Klein are entitled to a share of royalty received by the University on sales of products related to PALB2. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
References
- 1.Ferlay J, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. International Agency for Research on Cancer; 2013. (online). http://globocan.iarc.fr. [Google Scholar]
- 2.Goggins M, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–4. [PubMed] [Google Scholar]
- 3.Roberts NJ, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–6. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones S, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson D, Easton DF, Consortium BCL. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–65. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 6.van Lier MG, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:1258–64. doi: 10.1038/ajg.2009.725. author reply 1265. [DOI] [PubMed] [Google Scholar]
- 7.Vasen HF, et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87:809–11. [PubMed] [Google Scholar]
- 8.Lynch HT, Voorhees GJ, Lanspa SJ, McGreevy PS, Lynch JF. Pancreatic carcinoma and hereditary nonpolyposis colorectal cancer: a family study. Br J Cancer. 1985;52:271–3. doi: 10.1038/bjc.1985.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amundadottir L, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986–90. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen GM, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–8. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolpin BM, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46:994–1000. doi: 10.1038/ng.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu C, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2012;44:62–6. doi: 10.1038/ng.1020. [DOI] [PubMed] [Google Scholar]
- 13.Low SK, et al. Genome-wide association study of pancreatic cancer in Japanese population. PLoS One. 2010;5:e11824. doi: 10.1371/journal.pone.0011824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campa D, et al. Genetic susceptibility to pancreatic cancer and its functional characterisation: the PANcreatic Disease ReseArch (PANDoRA) consortium. Dig Liver Dis. 2013;45:95–9. doi: 10.1016/j.dld.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoskins JW, et al. Transcriptome analysis of pancreatic cancer reveals a tumor suppressor function for HNF1A. Carcinogenesis. 2014 doi: 10.1093/carcin/bgu193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B. The hepatic nuclear factor-1alpha G319S variant is associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab. 1999;84:1077–82. doi: 10.1210/jcem.84.3.5528. [DOI] [PubMed] [Google Scholar]
- 23.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. Jama. 1995;273:1605–9. [PubMed] [Google Scholar]
- 24.Li D, et al. Diabetes and risk of pancreatic cancer: a pooled analysis of three large case-control studies. Cancer Causes Control. 2011;22:189–97. doi: 10.1007/s10552-010-9686-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chari ST, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology. 2005;129:504–11. doi: 10.1053/j.gastro.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamagata K, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–8. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 27.Pierce BL, Ahsan H. Genome-wide “pleiotropy scan” identifies HNF1A region as a novel pancreatic cancer susceptibility locus. Cancer Res. 2011;71:4352–8. doi: 10.1158/0008-5472.CAN-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33:1384–90. doi: 10.1093/carcin/bgs151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergholz J, Xiao ZX. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron. 2012;5:311–22. doi: 10.1007/s12307-012-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–73. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 31.Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487–99. doi: 10.1038/cdd.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danilov AV, et al. DeltaNp63alpha-mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS One. 2011;6:e26815. doi: 10.1371/journal.pone.0026815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Figueroa JD, et al. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum Mol Genet. 2014;23:1387–98. doi: 10.1093/hmg/ddt519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan Q, et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet. 2012;44:1330–5. doi: 10.1038/ng.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiraishi K, et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat Genet. 2012;44:900–3. doi: 10.1038/ng.2353. [DOI] [PubMed] [Google Scholar]
- 36.Miki D, et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat Genet. 2010;42:893–6. doi: 10.1038/ng.667. [DOI] [PubMed] [Google Scholar]
- 37.Rothman N, et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat Genet. 2010;42:978–84. doi: 10.1038/ng.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borowski A, et al. Structure and function of ETAA16: a novel cell surface antigen in Ewing’s tumours. Cancer Immunol Immunother. 2006;55:363–74. doi: 10.1007/s00262-005-0017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marlaire S, Van Schaftingen E, Veiga-da-Cunha M. C7orf10 encodes succinate-hydroxymethylglutarate CoA-transferase, the enzyme that converts glutarate to glutaryl-CoA. J Inherit Metab Dis. 2014;37:13–9. doi: 10.1007/s10545-013-9632-0. [DOI] [PubMed] [Google Scholar]
- 40.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avis I, et al. Effect of gastrin-releasing peptide on the pancreatic tumor cell line (Capan) Mol Carcinog. 1993;8:214–20. doi: 10.1002/mc.2940080403. [DOI] [PubMed] [Google Scholar]
- 42.Jiang H, et al. Expression of Gli1 and Wnt2B correlates with progression and clinical outcome of pancreatic cancer. Int J Clin Exp Pathol. 2014;7:4531–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Yamagata K, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384:458–60. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs CS, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med. 1996;156:2255–60. [PubMed] [Google Scholar]
- 45.Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393:535–45. doi: 10.1007/s00423-007-0266-2. [DOI] [PubMed] [Google Scholar]
- 46.Jang JH, Cotterchio M, Borgida A, Gallinger S, Cleary SP. Genetic variants in carcinogen-metabolizing enzymes, cigarette smoking and pancreatic cancer risk. Carcinogenesis. 2012;33:818–27. doi: 10.1093/carcin/bgs028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talamini R, et al. Tobacco smoking, alcohol consumption and pancreatic cancer risk: a case-control study in Italy. Eur J Cancer. 2010;46:370–6. doi: 10.1016/j.ejca.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Urayama KY, et al. Body mass index and body size in early adulthood and risk of pancreatic cancer in a central European multicenter case-control study. Int J Cancer. 2011;129:2875–84. doi: 10.1002/ijc.25959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein AP, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Research. 2004;64:2634–2638. doi: 10.1158/0008-5472.can-03-3823. [DOI] [PubMed] [Google Scholar]
- 50.Brune KA, et al. Importance of Age of Onset in Pancreatic Cancer Kindreds. Journal of the National Cancer Institute. 2010;102:119–126. doi: 10.1093/jnci/djp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McWilliams RR, et al. Polymorphisms in DNA repair genes, smoking, and pancreatic adenocarcinoma risk. Cancer Res. 2008;68:4928–35. doi: 10.1158/0008-5472.CAN-07-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hassan MM, et al. Risk factors for pancreatic cancer: case-control study. Am J Gastroenterol. 2007;102:2696–707. doi: 10.1111/j.1572-0241.2007.01510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olson SH, et al. Allergies, variants in IL-4 and IL-4R alpha genes, and risk of pancreatic cancer. Cancer Detect Prev. 2007;31:345–51. doi: 10.1016/j.cdp.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Eppel A, Cotterchio M, Gallinger S. Allergies are associated with reduced pancreas cancer risk: A population-based case-control study in Ontario, Canada. Int J Cancer. 2007;121:2241–5. doi: 10.1002/ijc.22884. [DOI] [PubMed] [Google Scholar]
- 55.Tran B, et al. Association between ultraviolet radiation, skin sun sensitivity and risk of pancreatic cancer. Cancer Epidemiol. 2013;37:886–92. doi: 10.1016/j.canep.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 56.Duell EJ, et al. Detecting pathway-based gene-gene and gene-environment interactions in pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:1470–9. doi: 10.1158/1055-9965.EPI-07-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Risch HA. Etiology of pancreatic cancer, with a hypothesis concerning the role of N-nitroso compounds and excess gastric acidity. J Natl Cancer Inst. 2003;95:948–60. doi: 10.1093/jnci/95.13.948. [DOI] [PubMed] [Google Scholar]
- 58.Purcell S, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81 doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng X, et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–8. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mailman MD, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tryka KA, et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–9. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 63.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 64.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.