

Abstract
Objective:
To assess the influence of common mitochondrial DNA (mtDNA) sequence variation on multiple sclerosis (MS) risk in cases and controls part of an international consortium.
Methods:
We analyzed 115 high-quality mtDNA variants and common haplogroups from a previously published genome-wide association study among 7,391 cases from the International Multiple Sclerosis Genetics Consortium and 14,568 controls from the Wellcome Trust Case Control Consortium 2 project from 7 countries. Significant single nucleotide polymorphism and haplogroup associations were replicated in 3,720 cases and 879 controls from the University of California, San Francisco.
Results:
An elevated risk of MS was detected among haplogroup JT carriers from 7 pooled clinic sites (odds ratio [OR] = 1.15, 95% confidence interval [CI] = 1.07–1.24, p = 0.0002) included in the discovery study. The increased risk of MS was observed for both haplogroup T (OR = 1.17, 95% CI = 1.06–1.29, p = 0.002) and haplogroup J carriers (OR = 1.11, 95% CI = 1.01–1.22, p = 0.03). These haplogroup associations with MS were not replicated in the independent sample set. An elevated risk of primary progressive (PP) MS was detected for haplogroup J participants from 3 European discovery populations (OR = 1.49, 95% CI = 1.10–2.01, p = 0.009). This elevated risk was borderline significant in the US replication population (OR = 1.43, 95% CI = 0.99–2.08, p = 0.058) and remained significant in pooled analysis of discovery and replication studies (OR = 1.43, 95% CI = 1.14–1.81, p = 0.002). No common individual mtDNA variants were associated with MS risk.
Conclusions:
Identification and validation of mitochondrial genetic variants associated with MS and PPMS may lead to new targets for treatment and diagnostic tests for identifying potential responders to interventions that target mitochondria.
Mitochondrial dysfunction occurs early in multiple sclerosis (MS)1–7 and plays a substantial role in the axonal degeneration and demyelination observed in the disease.1 Mitochondrial structural changes, altered mitochondrial gene expression and enzyme activities, increased free radical production, and oxidative damage have all been reported in patients with MS.2 In addition, mitochondrial DNA (mtDNA) alterations and defective mtDNA repair have been observed in MS2; multiple deletions of mtDNA gray matter are putative important contributors to neurodegeneration in MS.5 Oxidative damage to mtDNA in active MS lesions also leads to mitochondrial dysfunction and a related decrease in complex I activity3 as well as axonal degeneration.3,4
To date, several small studies have reported individual mtDNA mutation associations with MS; however, these studies focused on selected mutations (see review8). The largest studies focused specifically on examining mtDNA associations with MS did not examine variation across the mitochondrial genome but rather emphasized specific haplogroup-defining variants, yielding inconclusive results.9–11 For this study, we investigated the role of common sequence variation across the mitochondrial genome on MS, with emphasis on the risk of developing a primary progressive (PP) MS disease course, in cases from several datasets within the International Multiple Sclerosis Genetics Consortium (IMSGC) and performed replication of significant findings in an independent sample.
METHODS
We analyzed the unreported mtDNA variants from a previously published genome-wide association study (GWAS)12 conducted as part of the Wellcome Trust Case Control Consortium 2 (WTCCC2) project and replicated significant findings in an independent US study of MS. Cases for the discovery study were recruited through the International Multiple Sclerosis Genetics Consortium (IMSGC) and compared with the WTCCC2 common control set complemented by data from the control arms of the GWAS. Details of case ascertainment, processing and genotyping, and sample and genotyping quality control were previously described.12 We excluded samples with ≥10% missing mtDNA single nucleotide polymorphism (SNP) data as well as SNPs with >10% missing calls. Of 136 mtDNA SNPs genotyped across GWAS arrays, 115 passed quality measures (missing rate <10%). All patients with MS met established diagnostic criteria13 and were non-Hispanic whites. Controls were also white and of European descent and reported no history of chronic diseases personally or in their nuclear family.
A previous study of MS10 demonstrated that mtDNA associations are especially sensitive to poor case-control matching due to population substructure, which led us to examine only WTCCC2-IMSGC datasets with cases and controls identified from the same country (Finland, Italy, Norway, Sweden, United Kingdom, United States, and Germany). In total, 7,391 cases and 14,568 controls from 7 countries were analyzed for haplogroup and SNP associations. Significant haplogroup and SNP associations were tested for replication in 3,720 cases and 879 controls from a single site (University of California, San Francisco) not included in the discovery GWAS. Replication SNPs were genotyped using custom TaqMan SNP genotyping assays conducted in 384-well plates on the ABI 7900HT Sequence Detection System using SDS 2.3 software. mtDNA variants were also examined for associations following stratification of cases according to disease course (relapsing-remitting MS and PPMS), which was adjudicated from 5 of the 7 discovery countries (Finland, Italy, United Kingdom, United States, and Germany) and the US replication study.
The major European haplogroups were defined using PhyloTree.14 MS and PPMS risk were examined among 2 European super haplogroups (JT and UK), 9 haplogroups (H, V, J, T, U, K, I, W, and X), and 26 mtDNA variants with minor allele frequencies (MAFs) ≥5%. Unconditional logistic regression was used to obtain odds ratios (ORs) and 95% confidence intervals (CIs) for MS and PPMS. Disease risk was examined for each haplogroup using the remaining haplogroups as a reference. Individual variant associations were examined using logistic regression to test risk associated with the minor allele compared with the common allele. All analyses were adjusted for sex and study country (SAS v 9.4, SAS Institute, Inc., Cary, NC), and analyses of common variants were additionally adjusted for 6 eigenvectors of mitochondrial genetic ancestry derived from principal component analysis (PCA).15
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review boards at each study site for all experiments using human participants. Written informed consent was obtained from all patients participating in these studies.
RESULTS
MS cases (n = 7,391) and controls (n = 14,568) from 7 countries were analyzed for haplogroup and SNP associations. Significant haplogroup and SNP associations were tested for replication in 3,720 MS cases and 879 controls, and control frequencies for each study population are detailed in table 1. Haplogroup frequencies among 7 discovery study populations (table 2) are consistent with previously observed haplogroup frequencies.16 An elevated risk of MS (table 3) was detected among haplogroup JT carriers from the pooled discovery datasets (OR = 1.15, 95% CI = 1.07–1.24, p = 0.0002) after adjustment for multiple comparisons (9 haplogroups, critical α = 0.006). The increased risk of MS (table 3) was observed for both haplogroup J (OR = 1.11, 95% CI = 1.01–1.22, p = 0.03) and haplogroup T carriers (OR = 1.17, 95% CI = 1.06–1.29, p = 0.002). None of the haplogroup associations with MS replicated in 3,720 cases and 879 controls from the independent sample set (table 3), although power to detect a small effect was limited.
Table 1.
Multiple sclerosis case and control distributions in discovery and replication populations
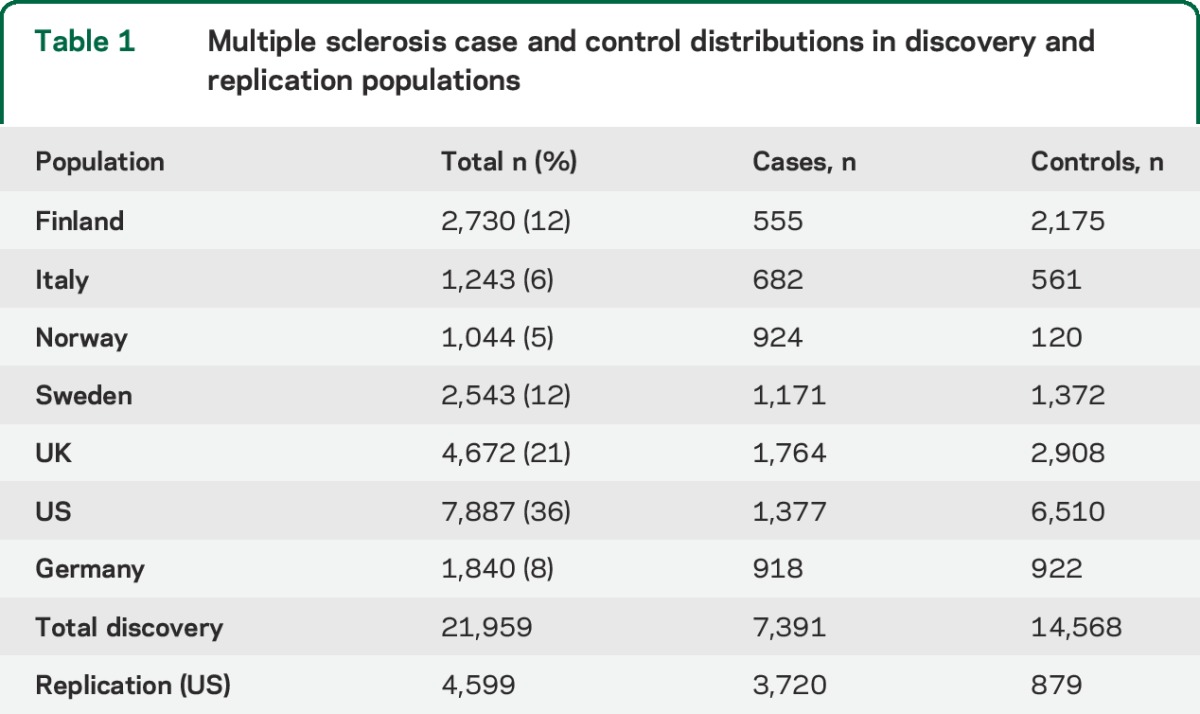
Table 2.
Mitochondrial haplogroup frequencies among multiple sclerosis cases and controls in 7 discovery pooled populations
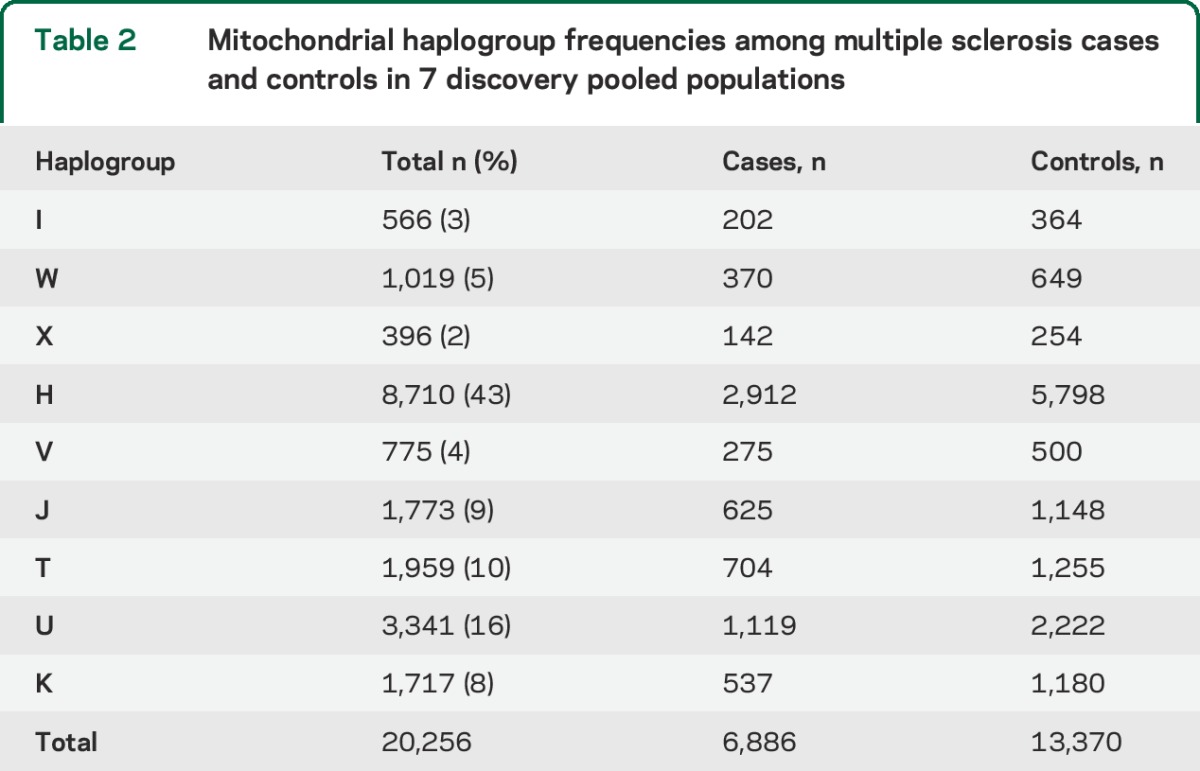
Table 3.
Statistically significant mitochondrial haplogroup associations with MS
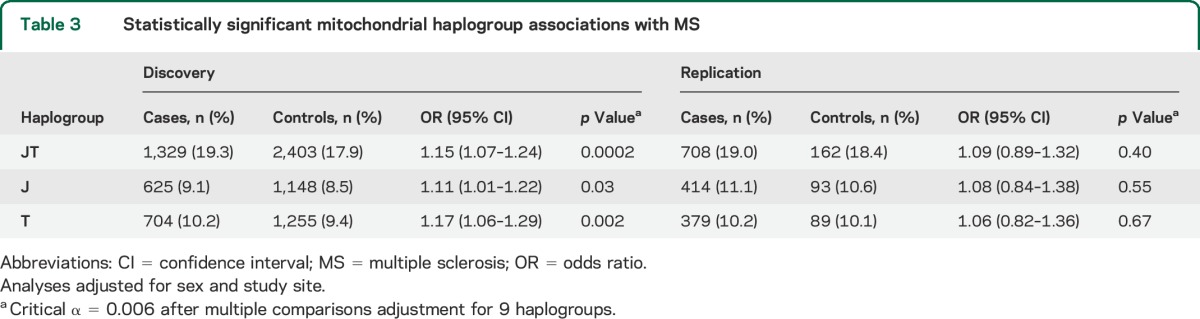
Nominal associations with MS were detected for haplogroups UK (1,656 cases and 3,402 controls; OR = 0.93, 95% CI = 0.87–1.00, p = 0.04) and W (OR = 1.17, 95% CI = 1.03–1.34, p = 0.019), although these associations did not maintain significance after adjustment for multiple comparisons. No common individual mtDNA variants (MAF 5%) were associated with MS risk in the analysis of any single or the 7 pooled discovery populations after adjustment for multiple comparisons (26 SNPs, critical α = 0.002).
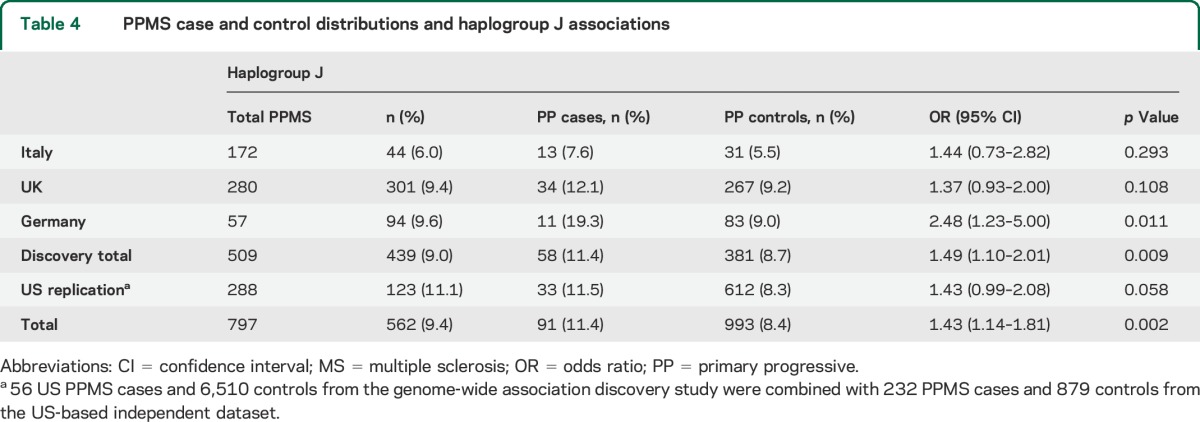
When the cases were stratified according to disease course, an elevated risk for PPMS among 509 cases from 3 non-US discovery populations with adjudicated PPMS (Italy, United Kingdom, and Germany) was detected for haplogroup J (OR = 1.49, 95% CI = 1.10–2.01, p = 0.009, table 4). PPMS represented 15.5% of the total MS group in the dataset. No haplogroup J carriers were detected among 24 PPMS cases from the Finnish population, so these samples did not contribute to the discovery analysis. To increase the power of PPMS replication, 56 US PPMS cases and 6,510 controls from the GWAS discovery study were combined with 232 PPMS cases and 879 controls from the US-based replication study. Risk for PPMS for haplogroup J among 288 US replication cases was borderline significant (OR = 1.43, 95% CI = 0.99–2.08, p = 0.058, table 4). Altogether, an elevated risk of PPMS was identified among 797 cases from the pooled discovery and replication studies (OR = 1.43, 95% CI = 1.14–1.81, p = 0.002, table 4).
Table 4.
PPMS case and control distributions and haplogroup J associations
DISCUSSION
We examined common SNPs across the mitochondrial genome in >7,000 MS cases and >14,000 controls from 7 countries and identified significant haplogroup associations with MS and PPMS. The elevated risk of MS among haplogoup JT carriers in the discovery samples was observed in both haplogroup J and T carriers independently, although the association with haplogroup T was stronger. An elevated risk of PPMS among haplogroup J carriers was identified in 3 discovery populations, with a very strong trend in the replication study. Two large studies previously identified MS associations among haplogroup T (m.4917A>G) carriers from 523 MS cases11 and haplogroup J (m.13708G>A) carriers from 1,099 MS cases.10
Determining which mtDNA SNPs contribute to haplogroup associations is confounded by both the number of variants that are diagnostic for each haplogroup and the repeated occurrence of the same mutations on different mtDNA backgrounds. Haplogroup J is defined by SNPs in ND3 (m.10398A>G), ND5 (m.12612A>G and m.13708G>A), HV regions 1 (m.16069C>T) and 2 (m.295C>T), and a noncoding region (m.489T>C).14 Haplogroup T is defined by 10 mtDNA SNPs located in ND2, ND5, CytB, ATP6, the 12S and 16S ribosomal RNAs, 2 transfer RNAs, and HV region 1.14 This makes determining function related to genotypes especially challenging, as the clusters of haplogroup-defining SNPs are in complete (or nearly complete) linkage disequilibrium. Thus, cell-based assays will not rule out the effects of each diagnostic SNP. A previous study identified increased survival in haplogroup JT septic patients and showed that survival may be due to an SNP defining haplogroup JT and not haplogroup J– or T–defining variants.17 Another study18 compared European haplogroup J and H mtDNA using cytoplasmic hybrids or “cybrids” (experimental hybrid cells containing mtDNA from different sources placed in a uniform nuclear DNA background), and haplogroup J cybrids exhibited increased mitochondrial transcription levels and a greater than 2-fold increase in mtDNA copy number compared with haplogroup H cybrids. This is one of the few examples demonstrating functional consequences for variants underlying specific haplogroups. Of the several haplogroup J–defining SNPs, the investigators hypothesized that the HV2 m.295C>T variant was the causal SNP, as this is the regulatory region that is responsible for mtDNA replication and stability.18 It may appear counterintuitive that haplogroup J would be associated with increased MS risk based on the association with increased mtDNA copy number. Enhanced DNA replication may amplify the expansion of heteroplasmic deleterious mutations (a mixture of normal and mutated mtDNA molecules in a cell) with potentially detrimental long-term consequences.19,20 In a previous cohort study of US elders,21 carriers of haplogroup T had a significantly higher risk of developing dementia, and haplogroup J carriers had a significantly steeper longitudinal decline in cognitive function, suggesting that cell-based phenotypes of mitochondrial function may not reflect the complexities of human disease phenotypes.
The m.295C>T SNP may not be the only potentially functional haplogroup J–defining variant: m.13708G>A, which is also diagnostic for haplogroup J, encodes the p.A458T, ND5 substitution and is considered a secondary Leber hereditary optic neuropathy (LHON) mutation.22 As previously mentioned, an earlier study of MS10 identified a highly significant association for the haplogroup J–defining m.13708G>A SNP. By sequencing a subset of study participants, the authors concluded that the association is due to m.13708G>A and not the other haplogroup J SNPs (including m.295C>T) linked to this variant. The m.13708G>A variant was also significantly associated with pediatric acquired demyelination syndrome (PD-ADS),23 providing further evidence to suggest that m.13708G>A is the SNP underlying the haplogroup J associations with MS and PPMS identified in the current study.
The lack of independent replication for the haplogroup J association with MS may have several interpretations. Although it is possible that carriers of haplogroup J are not at increased risk of MS, previous m.13708G>A SNP associations with MS10 and PD-ADS23 in combination with the replicated PPMS association reported herein all suggest that haplogroup J is a risk factor for MS. The contribution of specific mtDNA variants to disease risk could vary in different ancestral groups due to geographic differences in subhaplogroup structure, leading to inconsistencies between studies.24 In addition, mtDNA variants that fail to replicate may also show regional variation in interactions with other mtDNA SNPs, nuclear DNA, or environmental factors.24–26 For example, it has been suggested that haplogroup J status may increase the penetrance of primary LHON mutations and the risk of disease,27 although this hypothesis remains controversial.26,28,29 Finally, the observed enrichment (but not exclusivity) of haplogroup J in PPMS suggests an association with rapid disease progression and, accordingly, likely heterogeneity within the typical relapsing-remitting course group, in which the J haplogroup could be associated primarily with rapid transition to the secondary progressive phase. The available dataset was not suitable to test this hypothesis, as large cohorts with extended follow-up will be required.
The current study offered distinct advantages over previous studies examining specific mtDNA SNPs. We examined common variation across the entire mitochondrial genome and adjusted for mitochondrial genetic ancestry derived from PCA.15 As previously described,10 appropriate control matching and adjustment for population substructure (e.g., PCA used herein) are critical components for avoiding population stratification and founder effects that can bias mtDNA association studies.15 For example, the common individual mtDNA SNPs identified in previous smaller studies9–11 were not associated with MS risk in the current analysis. By examining MS cases and well-matched controls from 7 countries, we identified associations that were consistent across multiple independent populations.
Many of the SNPs that define the European haplogroups J and T occur in genes encoding subunits of oxidative phosphorylation complex I, including the previously identified MS risk SNPs m.4917A>G, ND211 (haplogroup T) and m.13708G>A, ND510 (haplogroup J). Complex I deficiency is the most frequent cause of bioenergetic dysfunction30 and accounts for many rare clinical presentations31 and multiple degenerative diseases beyond MS,1–7,32 including Alzheimer disease,33 amyotrophic lateral sclerosis,34 Parkinson disease,35 and aging.36 Complex I is a major contributor to cellular reactive oxygen species (ROS) production,37 and this oxidative damage in active MS lesions specifically leads to mitochondrial dysfunction and a related decrease in complex I activity3 as well as axonal degeneration.3,4 If validated in additional studies, our findings may suggest novel therapeutic targets for MS and PPMS that affect ROS production,38 apoptosis,39 and ATP generation.40 Identifying genetic markers of MS risk and progression may also be useful for classifying individuals who would most benefit from pharmacologic interventions that target the mitochondria.
GLOSSARY
- CI
confidence interval
- GWAS
genome-wide association study
- IMSGC
International Multiple Sclerosis Genetics Consortium
- LHON
Leber hereditary optic neuropathy
- MAF
minor allele frequency
- MS
multiple sclerosis
- mtDNA
mitochondrial DNA
- OR
odds ratio
- PCA
primary component analysis
- PD-ADS
pediatric acquired demyelination syndrome
- PPMS
primary progressive MS
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphism
- WTCCC2
Wellcome Trust Case Control Consortium 2
AUTHOR CONTRIBUTIONS
Gregory J. Tranah: contributed to the design of the study, analysis and interpretation of the data, and drafting/revising the manuscript for intellectual content. Adam Santaniello: contributed to data management. Stacy J. Caillier: contributed to sample management and genotyping. Sandra D'Alfonso: contributed data and drafted/revised the manuscript for intellectual content. Filippo Martinelli Boneschi: contributed data and drafted/revised the manuscript for intellectual content. Stephen L. Hauser: contributed to the design and conceptualization of the study. Jorge R. Oksenberg: contributed to the design of the study, analysis and interpretation of the data, and drafting/revising the manuscript for intellectual content.
STUDY FUNDING
Genetic data were provided by the International Multiple Sclerosis Genetics Consortium and the Wellcome Trust Case Control Consortium. This study was supported by the NIH (R01NS26799, R01NS4947703, and R03AG032498) and the Italian Foundation for Multiple Sclerosis [FISM grants “Progetto Speciale Immunochip,” 2011/R/14, Fondazione cariplo (n° 2010-0728)].
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Mahad D, Lassmann H, Turnbull D. Review: mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol 2008;34:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta 2010;1802:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu F, Selak M, O'Connor J, et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci 2000;177:95–103. [DOI] [PubMed] [Google Scholar]
- 4.Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 2006;59:478–489. [DOI] [PubMed] [Google Scholar]
- 5.Campbell GR, Ziabreva I, Reeve AK, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 2011;69:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahad DJ, Ziabreva I, Campbell G, et al. Mitochondrial changes within axons in multiple sclerosis. Brain 2009;132:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Kleist-Retzow JC, Hornig-Do HT, Schauen M, et al. Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficient cells but efficient compensation of energetic disadvantage by enhanced anaerobic glycolysis due to low ATP steady state levels. Exp Cell Res 2007;313:3076–3089. [DOI] [PubMed] [Google Scholar]
- 8.Andalib S, Talebi M, Sakhinia E, et al. Multiple sclerosis and mitochondrial gene variations: a review. J Neurol Sci 2013;330:10–15. [DOI] [PubMed] [Google Scholar]
- 9.Ban M, Elson J, Walton A, et al. Investigation of the role of mitochondrial DNA in multiple sclerosis susceptibility. PLoS One 2008;3:e2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Koczan D, Sulonen AM, et al. mtDNA nt13708A variant increases the risk of multiple sclerosis. PLoS One 2008;3:e1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vyshkina T, Sylvester A, Sadiq S, et al. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin Immunol 2008;129:31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 2009;30:E386–E394. [DOI] [PubMed] [Google Scholar]
- 15.Biffi A, Anderson CD, Nalls MA, et al. Principal-component analysis for assessment of population stratification in mitochondrial medical genetics. Am J Hum Genet 2010;86:904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saxena R, de Bakker PI, Singer K, et al. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet 2006;79:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorente L, Iceta R, Martin MM, et al. Severe septic patients with mitochondrial DNA haplogroup JT show higher survival rates: a prospective, multicenter, observational study. PLoS One 2013;8:e73320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suissa S, Wang Z, Poole J, et al. Ancient mtDNA genetic variants modulate mtDNA transcription and replication. PLoS Genet 2009;5:e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payne BA, Wilson IJ, Hateley CA, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet 2011;43:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durham SE, Samuels DC, Chinnery PF. Is selection required for the accumulation of somatic mitochondrial DNA mutations in post-mitotic cells? Neuromuscul Disord 2006;16:381–386. [DOI] [PubMed] [Google Scholar]
- 21.Tranah GJ, Nalls MA, Katzman SM, et al. Mitochondrial DNA sequence variation associated with dementia and cognitive function in the elderly. J Alzheimers Dis 2012;32:357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon DK, Johns DR. Mitochondrial disorders: clinical and genetic features. Annu Rev Med 1999;50:111–127. [DOI] [PubMed] [Google Scholar]
- 23.Venkateswaran S, Zheng K, Sacchetti M, et al. Mitochondrial DNA haplogroups and mutations in children with acquired central demyelination. Neurology 2011;76:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet 2007;81:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 2004;303:223–226. [DOI] [PubMed] [Google Scholar]
- 26.Carelli V, Achilli A, Valentino ML, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet 2006;78:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torroni A, Petrozzi M, D'Urbano L, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 1997;60:1107–1121. [PMC free article] [PubMed] [Google Scholar]
- 28.Oostra RJ, Bolhuis PA, Wijburg FA, Zorn-Ende G, Bleeker-Wagemakers EM. Leber's hereditary optic neuropathy: correlations between mitochondrial genotype and visual outcome. J Med Genet 1994;31:280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikoskelainen EK, Huoponen K, Juvonen V, Lamminen T, Nummelin K, Savontaus ML. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations. Ophthalmology 1996;103:504–514. [DOI] [PubMed] [Google Scholar]
- 30.Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2001;2:342–352. [DOI] [PubMed] [Google Scholar]
- 31.Loeffen JL, Smeitink JA, Trijbels JM, et al. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat 2000;15:123–134. [DOI] [PubMed] [Google Scholar]
- 32.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 2006;86:369–408. [DOI] [PubMed] [Google Scholar]
- 33.Grazina M, Pratas J, Silva F, Oliveira S, Santana I, Oliveira C. Genetic basis of Alzheimer's dementia: role of mtDNA mutations. Genes Brain Behav 2006;5(suppl 2):92–107. [DOI] [PubMed] [Google Scholar]
- 34.Coussee E, De Smet P, Bogaert E, et al. G37R SOD1 mutant alters mitochondrial complex I activity, Ca(2+) uptake and ATP production. Cell Calcium 2011;49:217–225. [DOI] [PubMed] [Google Scholar]
- 35.Haas RH, Nasirian F, Nakano K, et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol 1995;37:714–722. [DOI] [PubMed] [Google Scholar]
- 36.Stefanatos R, Sanz A. Mitochondrial complex I: a central regulator of the aging process. Cell Cycle 2011;10:1528–1532. [DOI] [PubMed] [Google Scholar]
- 37.Hirst J. Towards the molecular mechanism of respiratory complex I. Biochem J 2009;425:327–339. [DOI] [PubMed] [Google Scholar]
- 38.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 2010;45:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li N, Ragheb K, Lawler G, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 2003;278:8516–8525. [DOI] [PubMed] [Google Scholar]
- 40.Tarnopolsky MA, Simon DK, Roy BD, et al. Attenuation of free radical production and paracrystalline inclusions by creatine supplementation in a patient with a novel cytochrome b mutation. Muscle Nerve 2004;29:537–547. [DOI] [PubMed] [Google Scholar]