

Abstract
Objective:
Genetic variants ε2/ε4 within the APOE gene are established risk factors for lobar intracerebral hemorrhage (ICH). Published preliminary data suggest a potential role for APOE ε4 in risk of nonlobar ICH. We therefore investigated the role of APOE in recurrent nonlobar ICH, and sought to clarify whether effects of APOE on circulating lipids mediate this association.
Methods:
Three hundred sixty-three survivors of nonlobar ICH were followed prospectively for ICH recurrence, with APOE genotype determined at enrollment. All participants had clinical, demographic, and laboratory data captured at time of index ICH and during follow-up. Using a multivariate model, we performed association and interaction analyses of the relationships among APOE genotype, lipid levels, and recurrent nonlobar ICH.
Results:
We observed 29 nonlobar ICH recurrences among 363 survivors. APOE ε4 was associated with recurrent nonlobar ICH (hazard ratio = 1.31; 95% confidence interval = 1.02–2.69; p = 0.038) after adjustment for age/sex/ethnicity and cardiovascular risk factors. Increasing low-density lipoprotein (LDL) levels were associated with decreased risk of recurrent nonlobar ICH (p = 0.027), as were decreasing HDL levels (p = 0.046). LDL levels modified the association of APOE ε4 with recurrent nonlobar ICH (mediation p < 0.05). No associations were identified between APOE ε2 and recurrent nonlobar ICH.
Conclusion:
APOE ε4 is associated with recurrent ICH in nonlobar brain regions, providing further evidence for its causal role in ICH unrelated to cerebral amyloid angiopathy. LDL levels modulated this effect, suggesting that circulating lipid levels may mediate a portion of the role of APOE ε4 in nonlobar ICH.
Primary intracerebral hemorrhage (ICH) is the most lethal acute cerebrovascular disorder, its mortality rate ranging from 40% to 50% at 90 days from the initial bleeding event.1 Survivors are often burdened with substantial disability and face a high risk of recurrent ICH.2 Previous studies demonstrated that survivors of ICH in the lobar brain regions are at higher risk of recurrence, and multiple predictors of recurrent lobar ICH have been identified.3,4 Among these, the ε2/ε4 variants of the APOE gene are presumed to modify lobar ICH recurrence risk via their role in small vessel disease associated with cerebral amyloid angiopathy (CAA).3,5
A recent, large meta-analysis of case-control studies conclusively demonstrated that APOE ε2 and ε4 increase risk of ICH in the lobar brain regions.6 An intriguing preliminary finding that emerged as well was an association between APOE ε4, but not ε2, and nonlobar ICH.6 This finding warrants further exploration of its potential underlying biology, as nonlobar ICH is typically associated with arteriolosclerosis rather than CAA.7 Total cholesterol (TC) levels and cholesterol fractions (low-density lipoprotein [LDL]/high-density lipoprotein [HDL]) have been found to be associated with ICH risk in prior studies, particularly nonlobar ICH.8,9 Given that APOE gene products participate extensively in lipid metabolism, lipid fractions may mediate the effect of APOE ε4 on nonlobar ICH.
Reasoning that demonstrating an effect of APOE ε4 on recurrent nonlobar ICH would provide substantial confirmation of the previously published association of ε4 with risk of nonlobar ICH, we investigated whether survivors of ICH with APOE ε4 were at elevated risk of recurrent nonlobar ICH in a prospective cohort. Furthermore, given the established role of APOE in lipid metabolism, we performed mediation analyses to clarify whether lipid levels alter any detected effect of ε4.
METHODS
Patient recruitment and baseline data capture.
All participants were enrolled in an ongoing longitudinal cohort study of primary ICH at Massachusetts General Hospital.3 Individuals selected for the present study were consecutive patients aged 18 years or older, presenting to Massachusetts General Hospital from January 1, 2001, to December 31, 2011, and diagnosed with acute nonlobar ICH (onset of symptoms <24 hours before presentation). Nonlobar ICH was characterized as intraparenchymal hemorrhage selectively involving the thalami, basal ganglia, or brainstem. Given known biological heterogeneity of cerebellar ICH, cerebellar location was excluded from analysis in the present study in order to focus on non-CAA-related effects of APOE variants.3,6 To qualify for longitudinal analyses, patients were required to (1) meet criteria for primary ICH (as previously described)3; (2) have survived at least 90 days after index nonlobar ICH; and (3) have ICH location confirmed on admission CT scan. Patients with a recurrent lobar ICH were excluded from analysis. We excluded a total of 11 patients who did not survive 90 days after nonlobar ICH to avoid capturing early rebleeding events (whose pathophysiology may differ from long-term ICH recurrence).3 Pre-ICH clinical and demographic data were collected by trained study staff via in-person interview at time of enrollment. All admission CT scans were reviewed by study investigators blinded to clinical and genetic data to confirm ICH location. APOE genotype was determined by analyzing DNA extracted from blood samples provided by participants, in accordance with previously published methods.3,6
Longitudinal follow-up.
Trained study staff contacted and interviewed by telephone patients and/or their caregivers at 6 months after index ICH and every 6 months thereafter. Investigators inquired about and collected imaging and medical records data pertaining to ICH recurrence (both lobar and nonlobar), death, and medication use/dosing. Telephone-based collection of data was supplemented by manual review of longitudinal electronic medical records (EMRs) at Massachusetts General Hospital (including primary care, specialty outpatient, and inpatient records). EMRs were used to capture lipid levels during follow-up. We prespecified data capture targets of ≥1 lipid panel per every 12-month period. Only lipid data obtained >90 days after acute ICH were considered valid for analysis during the first year of follow-up. We elected to use both fasting and nonfasting lipid measurements for all analyses, and both actual measured (when available) and calculated LDL levels were included. A total of 15 participants failed to meet these data availability requirements and were removed from all analyses without altering results (data not shown).
EMR review was also used to obtain/confirm detailed information on medication use and interval clinical history. Discrepancies between telephone-collected and EMR-collected medication exposure data resulted in removal of 5 participants from the present study, and their removal did not alter results (data not shown). If participant and/or caregiver reported new neurologic symptoms, ischemic stroke, ICH, or hospital admission, the relevant medical records and radiographic images were reviewed by a study investigator blinded to other clinical and genetic data to assess the presence/absence of recurrent ICH and its location. Participants' data were censored in case of (1) recurrent ICH confirmed by neuroimaging, (2) death, (3) loss to follow-up, or (4) follow-up period reaching the predetermined deadline of December 31, 2013 (thus guaranteeing at least 24 months of follow-up for all analyzed participants). For individuals who had multiple recurrent lobar ICH during follow-up, data were censored at time of first recurrence.
Statistical methods.
Age at index ICH was analyzed as a continuous variable. Participants' sex, ethnicity, education, and pre-ICH medication exposure were analyzed as static categorical variables. Medication exposures during follow-up were analyzed as time-dependent categorical variables (with exposure time determined from phone call/EMR data). In line with prior studies, APOE genotype was analyzed using 2 categorical variables indicating number of ε2 alleles (0, 1, or 2) and number of ε4 alleles (0, 1, or 2) with the ε3/ε3 genotype serving as reference.
We generated a single continuous variable representing TC levels, and analyzed LDL and HDL levels as separate continuous variables. Of note, given substantial collinearity between LDL and HDL (identified based on methods described below), we generated separate multivariable models incorporating LDL or HDL as nonlobar ICH recurrence predictors. For lipid measurements, participants were assigned the TC/LDL/HDL value for each 12-month period (i.e., months 1–12 after index ICH, months 13–24 after index ICH, etc.) at all failure times falling within that same time window. Participants with ≥2 measurements in a given 12-month time period would be assigned, at each failure time, the closest available value.
Categorical variables were compared using Fisher exact test and continuous variables using the Mann–Whitney rank sum or unpaired t test as appropriate. We determined univariate predictors of ICH recurrence via significance testing by the log-rank test. Multivariate analyses of association between APOE ε2/ε4 and nonlobar ICH recurrence used a Cox regression analysis model. Candidate covariates included all variables showing a trend in association with recurrent nonlobar ICH in univariate analysis (p < 0.20). Backward elimination of nonsignificant variables (p > 0.05) generated a final minimal model. Because of its close relationship with lipid measurements, statin exposure was reintroduced in all multivariate models regardless of significance, without altering reported results (data not shown). Multicollinearity was assessed by computing variance inflation factors for all predictors and removing all variables with variance inflation factor >5.
We chose to perform mediation analyses to identify whether lipid levels influence association between APOE genotype and nonlobar ICH recurrence. These analyses identify whether an external variable (mediator, i.e., lipid levels in our study) carries the influence of a given independent variable/predictor (i.e., APOE genotype) to a given dependent variable/outcome (i.e., ICH recurrence); the percentage of predictor's effect on outcome carried by the mediator can also be quantified. Mediation analyses were performed using the mediation package in R (http://cran.r-project.org/web/packages/mediation/index.html); we prespecified intent to perform analyses to look for mediation effect of lipid measurements (TC, LDL, and HDL) on APOE genotype association with ICH recurrence if the following conditions were met: (1) APOE ε2 or ε4 genotype associated with nonlobar ICH recurrence; and (2) TC, and/or LDL, and/or HDL measurements associated with nonlobar ICH recurrence. We also prespecified inclusion of all other predictors of nonlobar ICH recurrence (in multivariate analyses) as potential competing mediators.
All analyses were performed with R software version 2.8.1 (The R Foundation for Statistical Computing). All significance tests were 2-tailed.
Standard protocol approvals, registrations, and patient consents.
This study was performed with approval of the institutional review board of the Massachusetts General Hospital. All participants or surrogates provided written informed consent for participation in this study.
RESULTS
Baseline cohort characteristics.
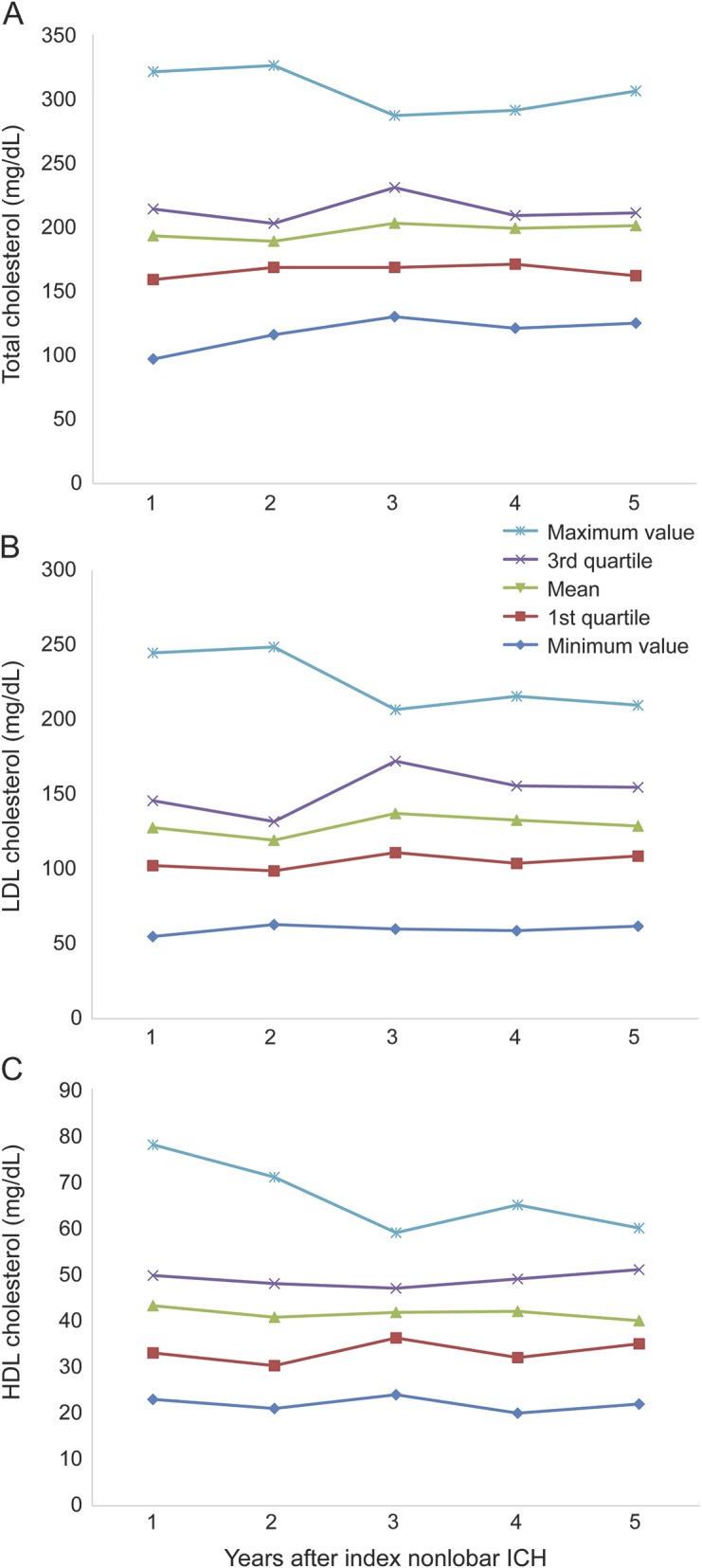
Baseline cohort characteristics for eligible participants included in the present study are summarized in table 1. Among 363 survivors of a nonlobar ICH, there were 29 nonlobar ICH recurrences and one lobar ICH recurrence during a median follow-up time of 34.6 months (interquartile range [IQR] 15.0–51.2), resulting in an incidence rate of 3.1% per year. APOE ε4 was associated with nonlobar ICH recurrence in univariate analysis, as were race/ethnicity, education level, medical history of ischemic heart disease, and a nonlobar ICH that had occurred before the index ICH (table 1). TC and lipid fraction measurements during follow-up time are presented in figure 1. We measured a mean TC measurement averaged over the entire study follow-up time of 202 mg/dL (IQR 166–221 mg/dL). Mean LDL cholesterol value for the entire study time was 122 mg/dL (IQR 108–163 mg/dL), and mean HDL value was 41 mg/dL (IQR 32–48 mg/dL).
Table 1.
Cohort characteristics and univariate analyses
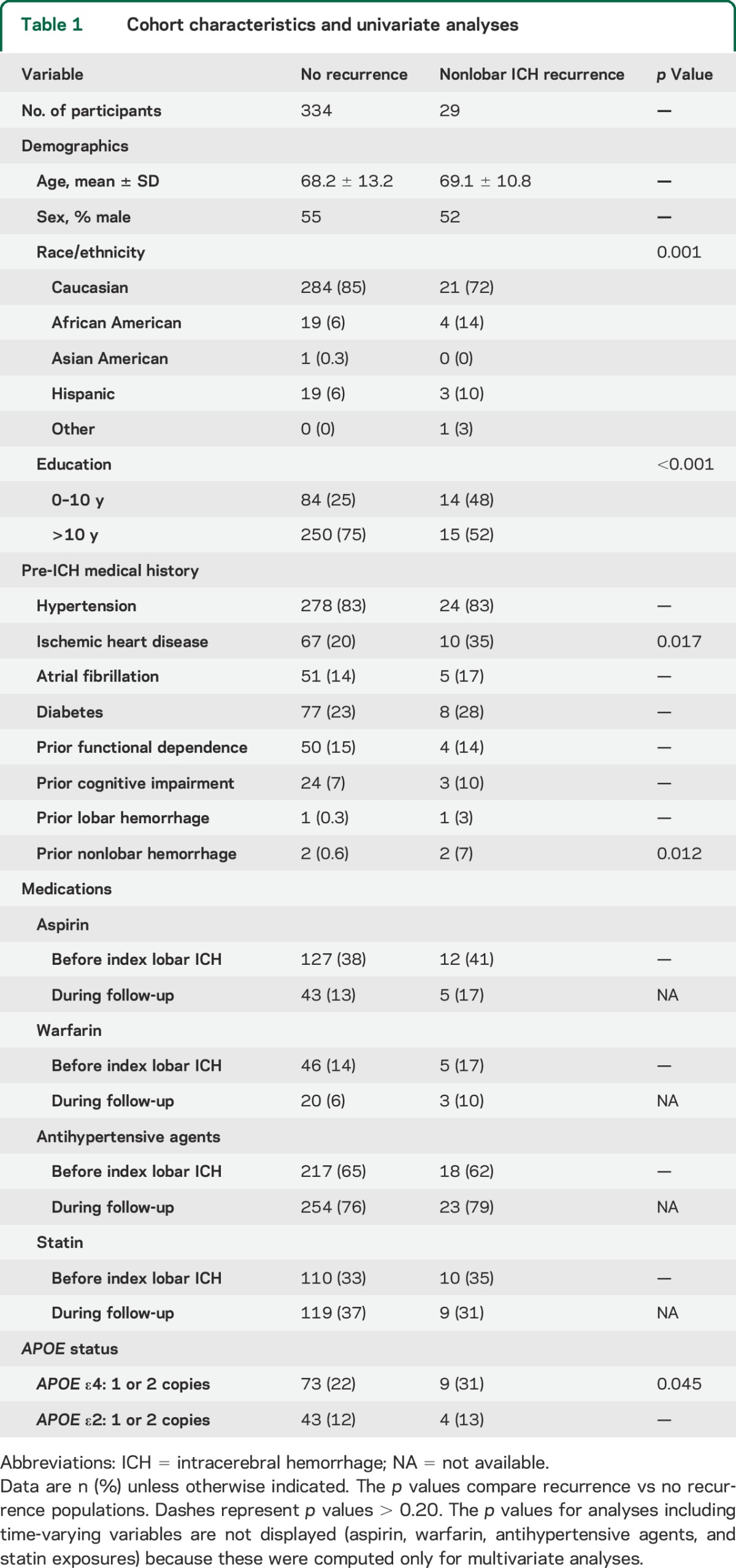
Figure 1. Total cholesterol and lipid fraction measurements during follow-up.
(A) Total cholesterol levels for entire study population during follow-up time. Individual lines connect minimum, first quartile, mean, third quartile, and maximum values per each year as detailed in the figure legend. (B) LDL cholesterol levels for all study participants during follow-up time (legend as in A). (C) LDL cholesterol levels for all study participants during follow-up time (legend as in A). HDL = high-density lipoprotein; ICH = intracerebral hemorrhage; LDL = low-density lipoprotein.
APOE genotype and lipid measurements association analyses.
Initial APOE multivariate analysis (table e-1 on the Neurology® Web site at Neurology.org) included univariate predictors of nonlobar ICH recurrence (as listed above), as well as time-varying medication exposures (antiplatelet agents, antihypertensive agents, anticoagulation, and statins were all initially entered in the model). Each allele copy of APOE ε4 was significantly associated with increased risk of nonlobar ICH recurrence (hazard ratio [HR] = 1.31, 95% confidence interval [CI] = 1.02–2.69, p = 0.038) (figure 2). There was no association between number of APOE ε2 alleles and risk of recurrent nonlobar hemorrhage.
Figure 2. APOE ε4 and risk of recurrent ICH in the nonlobar brain regions.

Kaplan–Meier plot displaying nonlobar ICH recurrence rates stratified by number of APOE ε4 allele copies during the first 5 years of follow-up. Univariate log-rank test p value = 0.031. Refer to main text for results of multivariate analyses. ICH = intracerebral hemorrhage.
We then performed multivariate analyses of nonlobar ICH recurrence including univariate predictors (except APOE genetic status) and TC/individual cholesterol fractions. There was no association between TC levels and nonlobar ICH recurrence (p > 0.20). Increasing LDL levels were significantly associated with decreased risk of nonlobar hemorrhage recurrence (HR = 0.88 per 10 mg/dL increase, 95% CI = 0.78–0.98, p = 0.027). In contrast, elevated HDL levels were associated with increased risk of nonlobar hemorrhage (HR = 1.12 per 10 mg/dL increase, 95% CI = 1.01–1.25, p = 0.042).
Lipid measurements modulate APOE ε4 association with nonlobar ICH recurrence.
Having demonstrated separate association in multivariate analyses for both APOE ε4 and lipid values (LDL and HDL) in accordance with our prespecified conditions, we proceeded to perform formal mediation analyses to explore potential modulating effects of lipid measurements on the association between APOE and nonlobar ICH recurrence. This analysis showed that 52% (95% CI = 31%–60%) of the observed effect of APOE on nonlobar ICH recurrence could be accounted for by combined effect of HDL and LDL levels (mediation p = 0.034). The results of mediation analyses including additional predictors of nonlobar ICH recurrence are detailed in figure 3. Separate mediation analyses identified significant effects for LDL cholesterol levels (42% of effect, 95% CI = 20%–72%, mediation p = 0.022), but not for HDL levels (13% of effect, 95% CI = 0%–37%, mediation p = 0.15).
Figure 3. Mediation analyses of the effect of APOE on risk of recurrent ICH in the nonlobar brain regions.

The percentages of effect size for APOE ε4 genotype association with nonlobar ICH recurrence mediated by other predictors of rebleeding risk are listed in the central rounded-edges rectangle column. Percentages refer only to the effect size attributed to APOE ε4 genotype (predicting ∼7% of overall nonlobar ICH recurrence risk). Only LDL + HDL levels mediated a proportion of the APOE effects on nonlobar ICH recurrence statistically different from 0%, as visually represented by the solid line connecting genotype and recurrence outcome through lipid data. Refer to main text for results of separate LDL and HDL mediation analyses. The remainder of APOE genotype predictive power not accounted for by percentages listed above is related to undetected mediated effects and/or direct (nonmediated) effects. CAD = medical history of coronary artery disease; CI = confidence interval; DM = medical history of diabetes mellitus; HDL = high-density lipoprotein; ICH = intracerebral hemorrhage; LDL = low-density lipoprotein.
We attempted to further characterize the presumed relationship between APOE ε4 and lipid measurements in determining nonlobar ICH risk by performing subset analyses based on lipid data. Our findings (figure 4) are consistent with progressively increasing LDL levels (averaged during follow-up time) being associated with decreased magnitude of association between APOE ε4 and nonlobar ICH (p value for trend test across categories = 0.019).
Figure 4. Nonlobar ICH recurrence risk by LDL levels and APOE genotype.

The p values are computed in each subset defined by LDL measurements using the log-rank test, and adjusting for race/ethnicity, education, CAD, and nonlobar ICH before index (enrollment event). LDL cutoffs represent mean values of all measurements during 5 years of follow-up, and were chosen to approximate LDL quartiles as identified in the overall dataset. CAD = coronary artery disease; ICH = intracerebral hemorrhage; LDL = low-density lipoprotein.
DISCUSSION
These results provide further robust evidence for an effect of APOE ε4 on risk of ICH in the nonlobar brain regions, raising the hypothesis that APOE influences the pathogenesis of the 2 most common forms of cerebral small vessel disease underlying ICH, CAA, and arteriolosclerosis.7 In addition, after finding that circulating LDL and HDL levels correlate with risk of recurrent nonlobar ICH, we identified preliminary evidence suggesting that LDL levels modulate the association between ε4 and ICH, with decreasing detrimental effect of ε4 as serum LDL levels increased.
By identifying an association between APOE ε4 and nonlobar ICH recurrence, we have confirmed and extended previous preliminary results suggesting a potential role for the APOE gene beyond lobar (presumably CAA-related) ICH.6 We have not identified an association between ε2 and nonlobar ICH recurrence; this finding mirrors identical null results from the aforementioned large case-control meta-analysis. Our statistical power to investigate a potential role for ε2 in nonlobar ICH is limited by low genetic variant frequency (especially compared with ε4), as well as lower effect sizes and ICH recurrence rates (comparing nonlobar ICH with lobar ICH). It is therefore plausible that we failed to identify an existing association between ε2 and nonlobar ICH due to chance alone. However, APOE ε2 alleles were in the past found to be associated with lobar ICH hematoma initial volume and expansion, functional outcome, and mortality, whereas no such associations were identified for nonlobar ICH.10 Negative findings for APOE ε2 in the present study may therefore be attributable to true biological heterogeneity of effects in different ICH pathophysiologic mechanisms (i.e., hypertensive vs CAA-related).
The ε4 allele of the APOE gene has been implicated in numerous different biological mechanisms beyond increasing amyloid burden.11–16 In light of the central role of APOE in lipid metabolism and accumulating evidence of lipid measurements being closely related to ICH risk,8,9 we hypothesized that lipid metabolism may explain the association between ε4 and nonlobar ICH. Our analyses are consistent with prior reports suggesting that increasing serum LDL levels and decreasing HDL levels are associated with decreased ICH risk (in the specific case of our study, decreased risk of nonlobar ICH recurrence). Adjustment for cardiovascular risk factors did not invalidate the association between LDL/HDL levels and nonlobar ICH recurrence. These findings suggest that lipid fractions have a biological role distinct from other clinical components of the metabolic syndrome in determining cerebral bleeding risk. Indeed, co-occurrence of high LDL and/or low LDL measurements with hypertension and elevated body mass index would have a priori suggested a detrimental effect on ICH recurrence.17
We therefore chose mediation analysis as an unbiased tool to look for other potential effects of lipid fractions on nonlobar ICH recurrence, i.e., modulating effects on APOE genetic variants. Of note, the direct effect of APOE ε4 on LDL/HDL levels (i.e., modest increase in LDL levels, and minimal decrease in HDL levels)18 cannot account for the observed associations with nonlobar ICH. Based on these findings alone, a small protective effect of ε4 against nonlobar ICH would in fact be expected. Our mediation analyses attributed approximately half of ε4 effect on nonlobar ICH recurrence to the combined action of LDL and HDL levels. Separate mediation analysis was statistically significant for LDL levels and not HDL, but our small sample size and decreased variance of HDL compared with LDL substantially limited our statistical power. Additional studies may well uncover a mediating role for HDL levels as well.
The exact mechanism by which LDL levels modulate APOE ε4 effect on risk of ICH recurrence remains unclear; our LDL quartile-stratified analyses suggest that high LDL levels cancel the association between ε4 and nonlobar ICH recurrence. These findings can be explained by hypothesizing that there exist multiple biologically detrimental effects of APOE ε4 relevant to nonlobar ICH pathophysiology (e.g., mitochondrial oxidative stress response, intracellular glucose metabolism, resistance to microvascular ischemia).11–13,16 These negative effects are then mitigated or nullified in the presence of elevated LDL levels. Lipid fractions may exert directly beneficial effects, or simply represent biomarkers of yet undiscovered protective biological pathways. Given the central role of lipids in cellular membrane trafficking and integrity, these represent areas of great interest for further investigation into the basic pathophysiology of nonlobar ICH.19,20 Ultimately, additional studies will be required to confirm and expand our findings.
Of note, our study identified an association between African American ancestry and increased risk of nonlobar ICH recurrence. Hispanic ancestry nonlobar ICH survivors were also noted to be at higher risk of recurrence, but with a non–statistically significant result. These findings are of particular interest because they persisted after adjustment for cardiovascular risk factors, which are often presumed to be the major driving factors behind health disparity in ICH recurrence/outcomes (especially for nonlobar ICH).21,22 Other, still unidentified biological mechanisms may also contribute to ICH outcome disparity in these minority populations, thus warranting further in-depth investigation.
Our study has several limitations. We were unable to precisely quantify alcohol consumption during follow-up. Since alcohol consumption is a known ICH risk factor and influences lipid levels, this represents a limitation of our study. We were unable to provide direct replication of our findings in a separate patient group. However, the concordance of findings between our recurrence study and published case-control analyses suggests that APOE ε4 does have a role in nonlobar ICH. Similarly, our association analyses for LDL/HDL levels are consistent with multiple prior reports, thus further supporting their validity. Our mediation analyses results, however, are in fact preliminary and will require further replication and validation. Despite our analyses identifying higher ICH recurrence risk for African American participants, the vast majority of our study participants are European American (i.e., Caucasian). Separate confirmatory analyses including larger numbers of non-European ancestry ICH survivors are therefore warranted. Perhaps most important, currently available data prevent us from further dissecting the relationships among APOE variants, nonlobar ICH, and lipid metabolism (particularly as they pertain to exploration of novel individual biological pathways).
In summary, we confirmed that APOE variant ε4 and lipid fraction levels have a significant role in determining nonlobar ICH recurrence. We also provided preliminary evidence suggesting that LDL levels modulate the association between ε4 and nonlobar ICH recurrence, by progressively decreasing its effect size with increasing LDL levels. Additional studies will be required to confirm these findings, and investigate in greater depth the biological mechanism linking APOE variants and lipid metabolism to nonlobar ICH pathophysiology.
Supplementary Material
GLOSSARY
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- EMR
electronic medical record
- HDL
high-density lipoprotein
- HR
hazard ratio
- ICH
intracerebral hemorrhage
- IQR
interquartile range
- LDL
low-density lipoprotein
- TC
total cholesterol
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept: M.R.R., A.B., C.D.A. Study design: M.R.R., A.B., C.D.A. Acquisition of data: M.R.R., A.B. Statistical analysis: A.B. Manuscript preparation: A.B., M.R.R., J.R., C.D.A. Manuscript review: M.R.R., A.B., A.M.A., T.W.K.B., S.M.G., A.W., J.R., C.D.A. Study supervision/coordination: M.R.R., A.B., A.M.A., T.W.K.B., S.M.G., A.W., J.R., C.D.A.
STUDY FUNDING
The authors' work on this study was supported by funding from the National Institute for Neurologic Disorders and Stroke (R01 NS063925, R01 NS059727, P50 NS051343, K23 NS086873, and AG26484). All funding entities had no involvement in study design, data collection, analysis, and interpretation, writing of the report, and in the decision to submit the paper for publication.
DISCLOSURE
M. Raffeld, A. Biffi, T. Battey, A. Ayres, and A. Viswanathan report no disclosures. S. Greenberg is supported by NIH-NINDS U10NS077360. J. Rosand is supported by NIH-NINDS U01NS069208, NIH-NINDS R01NS073344, and R01 NS059727. C. Anderson is supported by NIH-NINDS K23 086873, and the Anne B. Young Fellowship in Therapeutic Development, sponsored in part by Biogen Idec, Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Krishnamurthi RV, Moran AE, Forouzanfar MH, et al. The global burden of hemorrhagic stroke: a summary of findings from the GBD 2010 study. Glob Heart 2014;9:101–106. [DOI] [PubMed] [Google Scholar]
- 2.Poon MT, Fonville AF, Al-Shahi Salman R. Long-term prognosis after intracerebral haemorrhage: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2014;85:660–667. [DOI] [PubMed] [Google Scholar]
- 3.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010;75:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weimar C, Benemann J, Terborg C, et al. Recurrent stroke after lobar and deep intracerebral hemorrhage: a hospital-based cohort study. Cerebrovasc Dis 2011;32:283–288. [DOI] [PubMed] [Google Scholar]
- 5.Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol 2011;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biffi A, Sonni A, Anderson CD, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol 2010;68:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Badjatia N, Rosand J. Intracerebral hemorrhage. Neurologist 2005;11:311–324. [DOI] [PubMed] [Google Scholar]
- 8.Woo D, Deka R, Falcone GJ, et al. Apolipoprotein E, statins, and risk of intracerebral hemorrhage. Stroke 2013;44:3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Dong Y, Qi X, Huang C, Hou L. Cholesterol levels and risk of hemorrhagic stroke: a systematic review and meta-analysis. Stroke 2013;44:1833–1839. [DOI] [PubMed] [Google Scholar]
- 10.Biffi A, Anderson CD, Jagiella JM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol 2011;10:702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong JS, Kobayashi M, Hayashi H, et al. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J Biol Chem 2002;277:29919–29926. [DOI] [PubMed] [Google Scholar]
- 12.Fagan AM, Holtzman DM, Munson G, et al. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem 1999;274:30001–30007. [DOI] [PubMed] [Google Scholar]
- 13.Nathan BP, Bellosta S, Sanan DA, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 1994;264:850–852. [DOI] [PubMed] [Google Scholar]
- 14.Bellosta S, Nathan BP, Orth M, et al. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem 1995;270:27063–27071. [DOI] [PubMed] [Google Scholar]
- 15.Trommer BL, Shah C, Yun SH, et al. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport 2004;15:2655–2658. [DOI] [PubMed] [Google Scholar]
- 16.Gibson GE, Haroutunian V, Zhang H, et al. Mitochondrial damage in Alzheimer's disease varies with apolipoprotein E genotype. Ann Neurol 2000;48:297–303. [PubMed] [Google Scholar]
- 17.Biffi A, Cortellini L, Nearnberg CM, et al. Body mass index and etiology of intracerebral hemorrhage. Stroke 2011;42:2526–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan TA, Shah T, Prieto D, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta-analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. Int J Epidemiol 2013;42:475–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stancu CS, Toma L, Sima AV. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res 2012;349:433–446. [DOI] [PubMed] [Google Scholar]
- 20.Song Y, Kenworthy AK, Sanders CR. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Sci 2014;23:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty ML, Woo D, Haverbusch M, et al. Racial variations in location and risk of intracerebral hemorrhage. Stroke 2005;36:934–937. [DOI] [PubMed] [Google Scholar]
- 22.Labovitz DL, Halim A, Boden-Albala B, Hauser WA, Sacco RL. The incidence of deep and lobar intracerebral hemorrhage in whites, blacks, and Hispanics. Neurology 2005;65:518–522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.