

Key Points
Adoptive transfer of T cells genetically modified to express anti-TSLPR chimeric antigen receptors can cure B-ALL in xenograft models.
Anti-TSLPR CAR constructs containing a CH2CH3 spacer domain were inactive against TSLPR-overexpressing B-ALL.
Abstract
Adoptive transfer of T cells genetically modified to express chimeric antigen receptors (CARs) targeting the CD19 B cell–associated protein have demonstrated potent activity against relapsed/refractory B-lineage acute lymphoblastic leukemia (B-ALL). Not all patients respond, and CD19-negative relapses have been observed. Overexpression of the thymic stromal lymphopoietin receptor (TSLPR; encoded by CRLF2) occurs in a subset of adults and children with B-ALL and confers a high risk of relapse. Recent data suggest the TSLPR signaling axis is functionally important, suggesting that TSLPR would be an ideal immunotherapeutic target. We constructed short and long CARs targeting TSLPR and tested efficacy against CRLF2-overexpressing B-ALL. Both CARs demonstrated activity in vitro, but only short TSLPR CAR T cells mediated leukemia regression. In vivo activity of the short CAR was also associated with long-term persistence of CAR-expressing T cells. Short TSLPR CAR treatment of mice engrafted with a TSLPR-expressing ALL cell line induced leukemia cytotoxicity with efficacy comparable with that of CD19 CAR T cells. Short TSLPR CAR T cells also eradicated leukemia in 4 xenograft models of human CRLF2-overexpressing ALL. Finally, TSLPR has limited surface expression on normal tissues. TSLPR-targeted CAR T cells thus represent a potent oncoprotein-targeted immunotherapy for high-risk ALL.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common oncologic diagnosis in children. Substantial progress has been made in upfront therapy for pediatric ALL such that >80% of patients will be cured.1-3 Nonetheless, ALL remains a significant cause of death from cancer in children because of initial refractoriness or relapse of leukemia that no longer responds to cytotoxic chemotherapy.4,5 Furthermore, long-term therapy–induced morbidity is a major issue, particularly in patients deemed to be at high risk for relapse who are thus treated with more intensive regimens under current risk-adapted protocols.6 ALL occurs less frequently in adults than in children, but the prognosis for adult ALL is much worse despite treatment with multiagent cytotoxic chemotherapy.7 Treatment of young adults on pediatric-type regimens has improved outcome, but not to the level achieved in children.8 Thus, for multiple reasons, there remains the need for new treatments for ALL for all age groups that is not based on intensification of cytotoxic chemotherapy.
After more than a decade of study with modest clinical progress, immune-based treatment of cancer has shown much promise in recent years. In particular, the adoptive cell transfer of T cells genetically modified to express chimeric antigen receptors (CARs) targeting antigens expressed on lymphoid cells have demonstrated potent activity in B-cell malignancies, including ALL, with reported remission rates of 70% to 90% in multiple-relapse or chemotherapy-refractory patients.9-14 The surface protein targeted in the majority of these trials is the CD19 antigen that is expressed on both malignant and nonmalignant B cells. However, not all patients respond to CD19-redirected CAR T cells, and relapses have been observed, in some cases because of loss of CD19 expression.14 Loss of CD19 has also been observed after treatment with a bispecific antibody-based reagent targeting CD19 and CD3.15 Alternative cell surface targets are thus needed. In principle, the optimal target for immunotherapy is a cell-surface protein that is functionally important for the leukemic cell such that relapse as a result of antigen loss is less likely to occur.
Substantial progress in leukemia genomics has resulted in the identification of genes and pathways that are dysregulated in ALL.3 One such category of pathways are those associated with cytokine signaling, including IL-7 receptor pathways, in particular CD127 (encoded by IL7RA).16 Thymic stromal lymphopoietin (TSLP) is a cytokine that binds to the TSLPR, a heterodimeric receptor complex comprised of a TSLPR subunit (encoded by the CRLF2 gene) and a CD127 subunit, and induces JAK/STAT pathway signaling. Overexpression of the TSLPR has been identified in 5% to 15% of children and adults with B-ALL, largely because of CRLF2 translocations or deletions resulting in alternative promoters.17-22 Importantly, in a number of studies, CRLF2 rearrangements have been associated with poor prognosis in both pediatric and adult ALL,23-29 and emerging studies have identified activation of the TSLPR signaling pathway as biologically important for ALL blasts.30,31 Recent comprehensive genomic analyses of high-risk B-ALL cases demonstrated that CRLF2 rearrangements in ALL are frequently associated with a gene-expression profile highly similar to that of Philadelphia chromosome–positive ALL, but without the BCR-ABL1 fusion. Patients with these “Philadelphia chromosome–like” (Ph-like) leukemias respond poorly to conventional chemotherapy and have high rates of relapse.17 Multiple groups have now demonstrated that CRLF2 rearrangements account for half of Ph-like ALL genomic alterations and are also highly associated with concomitant JAK1 and JAK2 point mutations.17,18,20,21,32 We maintain that the TSLPR functions as an ALL oncoprotein given its cell surface expression and association with poor clinical outcomes and thus may be an ideal immunotherapeutic target. Furthermore, TSLPR expression in normal tissues appears to be restricted. We demonstrate that a TSLPR CAR can completely eradicate human CRLF2-rearranged (TSLPR-overexpressing) ALL in multiple model systems, including patient-derived xenografts (PDX).
Materials and methods
Cells and culture conditions
The following B-cell acute lymphoblastic leukemia (B-ALL) cell lines were used: MUTZ5 (DSMZ ACC 490), REH-TSLPR (native REH transduced with human TSLPR), and nontransduced REH as a TSLPR-negative control. Cell lines were cultured in media supplemented with 10% heat-inactivated fetal bovine serum (Gemini Bioproducts), 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Invitrogen). The 293T retroviral vector packaging cell line (Clontech) was cultured in Dulbecco’s modified Eagle medium (Invitrogen). In addition, PDX models of CRLF2-rearranged Ph-like ALL (de novo specimens JH331, JH352, and NH362 and relapse specimen ALL4364; Table 1) were used for in vivo testing of TSLPR CAR T cells. PDX models were created as previously described33,34 using viably cryopreserved Ph-like ALL specimens banked in the Children’s Oncology Group or Children’s Hospital of Philadelphia leukemia biorepositories under institutional review board–approved research protocols. For these studies, secondary and tertiary PDX models were used for relapse and de novo ALL specimens, respectively. Briefly, primary leukemia cells from peripheral blood or bone marrow were IV injected into nonobese diabetic/severe combined immunodeficient (NOD.Cg-Prkdcscid) Il2rgtm1wjl/SzJ (NSG) mice (106 cells/mouse). Splenocytes harvested from successfully engrafted xenografts were reinjected (106 cells/mouse) to create secondary and tertiary xenografts for treatment trials. Human peripheral blood mononuclear cells from healthy donors were obtained from the Department of Transfusion Medicine at the National Institutes of Health (NIH) Clinical Center under an institutional review board–approved protocol. All research specimens from human subjects were obtained with informed consent in accordance with the Declaration of Helsinki.
Table 1.
Genomic alterations in CRLF2-rearranged Ph-like ALL specimens used for patient-derived xenograft models
| Specimen | USI | Source | CRLF2 rearrangement | JAK mutation | Other mutations |
|---|---|---|---|---|---|
| JH331 | PAMDKS | de novo | IGH@-CRLF2 | JAK2 R683G | IKZF1, PAX5, CDKN2A |
| JH352 | PALJCF | de novo | P2RY8-CRLF2 | JAK1 L624_R629 > W | CDKN2A/B |
| NH362 | PALTWS | de novo | IGH@-CRLF2 | Negative | IKZF1, PAX5, CDKN2A |
| ALL4364 | n/a | relapse | P2RY8-CRLF2 | JAK2 R683G |
n/a, not available; USI, unique specimen identifier (Children’s Oncology Group).
Construction of TSLPR chimeric antigen receptors
TSLPR binding single-chain fragment variable (scFv) sequences were determined from the anti-TSLPR–producing hybridoma 3G11 obtained from the MD Anderson Cancer Center as described in supplemental Methods, available on the Blood Web site. For construction of the long CAR constructs, the CH2CH3 domains from IGHG1 (gb|AAC82527.1 aa 98-329) were included. The leader sequence for the scFv coding for T-cell surface glycoprotein CD8 α chain was included to facilitate membrane trafficking. The CAR-encoding amino acid sequences were reverse-translated, codon-optimized, and synthesized as single constructs (DNA 2.0). These constructs were then subcloned into a third-generation lentiviral plasmid (pELNS-BBzeta) containing a CD8 transmembrane domain, a 4-1BB (CD137)-signaling domain, and a CD3zeta domain (kindly provided by Dr Carl June at the University of Pennsylvania35 and previously described36).
Lentiviral vector production and T-cell transduction
TSLPR CAR-encoding lentiviral vectors were produced by transient transfection of the 293T cell line as previously described.35 Briefly, 293T cells were plated into poly-D lysine–coated 15-cm plates (BD Biosciences). The following day, 293T cells were transfected using lipofectamine 2000 (Life Technologies) with plasmids encoding the TSLPR CAR along with packaging and envelope vectors (pMDLg/pRRE, pMD.2G, and pRSV-Rev, kindly provided by Dr Richard Morgan of the Surgery Branch, Center for Cancer Research, NCI, NIH). Lentiviral supernatants were collected 48 to 72 hours posttransfection, centrifuged at 3000 rpm for 10 minutes to remove cell debris, then stored at −80°C. Human peripheral blood mononuclear cells from normal donors were activated with a 1:1 ratio of CD3/CD28 microbeads (Life Technologies) in AIM-V media containing 40 IU/mL recombinant IL-2 (teceleukin, rhIL-2; Roche) for 24 hours. Activated T cells were resuspended at 2 million cells per 3 mL of lentiviral supernatant plus 1 mL of fresh AIM-V media with 10 μg/mL protamine sulfate and 40 IU/mL IL-2 and cultured in 6-well plates. Plates were centrifuged at 1000g for 2 hours at 32°C and then incubated at 37°C overnight. A second transduction was performed the following day. On the third day after transduction, the CD3/CD28 beads were removed and the cells were cultured at 300 000 cells/mL in AIM-V containing 100 IU/mL IL-2 with fresh IL2-containing media added every 2 to 3 days until harvest at day 8 or 9.
Flow cytometry analysis
Surface expression of CAR-transduced T cells was determined by flow cytometry using a TSLPR-Fc (R&D Systems) followed by incubation with PE-F(ab)2 or APC-F(ab)2 specific for human IgG-Fc (Jackson ImmunoResearch Laboratories). Alternatively, biotin-conjugated protein L (Thermo Scientific) was used to detect CAR expression after incubation with streptavidin-conjugated PE (BD Biosciences). Expression of CD19, CD22, and TSLPR on B-ALL lines were detected using the following anti-human antibodies: CD45-PerCP-Cy5.5 (eBioscience), CD19-Pacific Blue, CD19-APC-Cy7, CD10-PE-Cy7, CD22-PE, and TSLPR-APC (Biolegend). T cells were characterized with the following antibodies: CD3-APC-Cy7, CCR7-FITC (CD197), CD45RA-APC, CD4-Pacific Blue (BioLegend), CD45-PerCP-Cy5.5 (eBioscience), and CD8-V500 (BD Biosciences). The binding of the 3G11 hybridoma supernatant to the TSLPR-overexpressing ALL lines was detected with goat anti-mouse IgG-PE (BD Biosciences). Dead cells were excluded by staining with Fixable Viability Dye eFluor 506 (eBioscience).
Cellular cytotoxicity and cytokine assays
Target cells were labeled with 100 uCi 51Cr (PerkinElmer) for 1 hour. After washing, 5000 targets per well were coincubated for 4 to 6 hours with bead-purified (Pan T Cell II isolation kit; Miltenyi Biotec), transduced T cells at various effector to target (E:T) ratios. Assay supernatants were counted for 51Cr release using LumaPlates (PerkinElmer) and a Top Count Reader (Packard). Specific lysis was calculated as follows: % lysis = (experimental lysis − spontaneous lysis)/(maximum lysis − spontaneous lysis) × 100. 1 × 105 CAR T cell or control T cells were washed 3 times and then cocultured with target cell at 1:1 ratio in a 96-well plate. All samples were run in triplicate. The overnight culture supernatants were measured for cytokine levels with either IFNgγ enzyme-linked immunosorbent assay kits (R&D) or multiplex assay (Meso Scale Discovery) as per the manufacturer’s instructions.
In vivo studies
Animal studies were carried out under protocols approved by the NCI Bethesda Animal Care and Use Committee. B-ALL cell lines and previously xenografted human B-ALL specimens were IV-injected into NSG mice (NOD scid γ, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Jackson ImmunoResearch Laboratories) as previously described.33 For nonluciferase–expressing PDX models, animals were treated with TSLPR T cells once ≥5% human ALL was detectable by flow cytometry in peripheral blood. For luciferase-expressing lines, leukemia was detected using the Xenogen IVIS Lumina (Caliper Life Sciences). NSG mice were injected intraperitoneally with 3 mg d-luciferin (Caliper Life Sciences) and were imaged 6 minutes later with an exposure time of 3 minutes. Living Image Version 4.1 software (Caliper Life Sciences) was used to analyze the bioluminescent signal flux for each mouse as photons/s per cm2 per steradian. Leukemia burden in nonluciferase–expressing xenografts was measured by flow cytometry of peripheral blood, bone marrow, and spleen.
Tissue microarray
Tissue microarray (TMA) is a screening tool that allows hundreds of tissue samples to be analyzed quickly and conveniently. Formalin-fixed, paraffin-embedded tissue blocks from 48 healthy patients were selected for use as normal controls. Hematoxylin and eosin–stained slides from these tissue blocks were annotated for desired areas, and the slides were then used as guides for core selection. TMAs were constructed on a Beecher Manual Tissue Arrayer I (MTA I). For this TMA, 1 mm cores of normal formalin-fixed, paraffin-embedded tissue were punched from a donor block and arrayed into a recipient paraffin block. Cores were placed into the recipient block following a coordinate grid map. Each tissue type was represented by 3 cores when possible. A total of 138 cores were used in each TMA block. Tissue types represented were aorta, adipose, bladder, breast, cartilage, cervix, colon, connective tissue, diaphragm, epididymis, heart, kidney, liver, lung, mesentery, ovary, pancreas, small intestine, skeletal muscle, smooth muscle, spleen, testes, thymus, thyroid, tonsil, trachea, and skin. Immunohistochemistry with the anti-CRLF2 clone 109626 (Abcam) antibody was performed on the Ventana Discovery XT platform. The protocol included a CC1 standard retrieval process. The antibody was diluted using SignalStain enhancer (Cell Signaling) to a final dilution of 1:1500. CRLF2 was further detected using Ultramap anti Rabbit HRP and the Chromomap DAB detection kit. The TMA was scanned with a SCN400 Leica scanner at 40× original magnification.
Results
Cloning and construction of TSLPR-targeted chimeric antigen receptors and generation of TSLPR CAR–expressing T cells
We first confirmed binding of an anti-TSLPR antibody produced by the 3G11 hybridoma to CRLF2-rearranged (TSLPR-overexpressing) B-ALL cell lines and xenografted primary specimens (TSLPRhi ALL, Figure 1A). 3G11 does not bind murine TSLPR, thus precluding the ability to assess toxicity in murine models (not shown). The sequences for the heavy- and light-chain variable regions (Fv) were then determined (supplemental Methods). Single-chain Fv (scFv) sequences were constructed using a glycine linker and inserted into a chimeric antigen-receptor lentiviral vector backbone encoding CD8α hinge and CD8 transmembrane regions with CD3ζ and 41BB (CD137) intracellular domains (short CAR; Figure 1B). Because distance of the scFv from the T-cell surface may affect CAR function, a construct containing an immunoglobulin CH2CH3 spacer domain between the scFv and the transmembrane sequence was also generated (long CAR). Lentiviral vectors encoding the TSLPR CARs were then used to transduce CD3/CD28 bead–activated human T cells, resulting in a high efficiency of gene transfer as detected by both protein L and a TSLPR Fc fusion protein (Figure 1C). There were no differences in cell expansion between TSLPR short CAR or cells CD19 CAR or CD22 CAR–transduced cells (supplemental Figure 1). Importantly, TSLPR reportedly has limited expression in normal tissues outside of the immune system, where it has been found on dendritic cells (with the highest expression on a subset of myeloid DCs) and some T cells (a small subset of CD4+ T cells).37,38 To confirm this, we performed immunohistochemistry on a normal TMA (Figure 1D). As expected, there were scattered rare cells in lymphoid tissues and thymus, demonstrating TSLPR expression, possibly representing dendritic cells and developing T cells. There was also some staining in renal tubular cells and colonic mucosa, although the staining pattern in these tissues was not consistent with cell surface expression. In the liver, there was cytoplasmic staining in cells likely representing Kupffer cells. Finally, there were scattered cells in the basal layer of the skin with cytoplasmic staining. There was no staining in the myocardium. There was also no evidence for in vivo reactivity of TSLPR CAR T cells against a panel of normal tissue cell lines including cardiomyocytes or neurocytes induced from pluripotent stem cells (supplemental Figure 2). Importantly, as has been shown in primary CRLF2-rearranged B-ALL specimens,31 the TSLPRhi ALL cell line MUTZ5 and a human TSLPRhi PDX (JH331) express TSLPR at comparably high levels with those of CD19 and CD22 (Figure 1E). REH expresses very low levels of TSLPR relative to the TSLPRhi ALL and an REH line transduced with human TSLPR.
Figure 1.
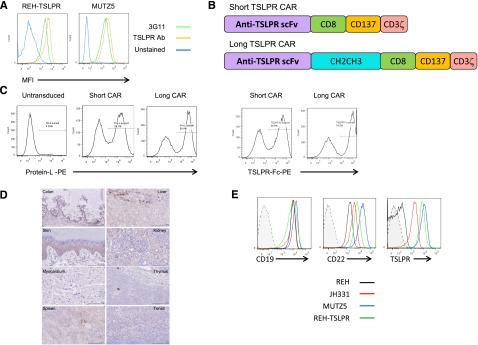
Construction of short and long anti-TSLPR CAR constructs and lentiviral transduction of T cells. (A) Anti-TSLPR hybridoma (3G11) supernatant binds to the surface of TSLPR-overexpressing ALL. Binding was detected using phycoerytherin-conjugated goat-anti-mouse antibody. Binding of a commercially available, directly conjugated anti-TSLPR antibody (Ab) is shown for comparison. (B) Schematic representation of anti-TSLPR chimeric antigen receptor constructs. Both constructs contain an anti-TSPLR single-chain fragment variable sequence from the 3G11 hybridoma, a CD8 transmembrane domain, a CD137 (41BB) costimulatory domain, and a CD3 ζ-signaling domain. The long version of the CAR also contains a spacer region derived from an immunoglobulin CH2CH3 domain. (C) Transduction efficiency of activated CD3/CD28 bead-expanded human T cells with lentiviral-based vectors expressing short and long anti-TSLPR constructs. The left panels show detection of CAR using Protein L. The right panels show detection using a TSLPR protein Fc construct. (D) TSLPR expression on normal pediatric tissues, representative of 48 healthy donors. Colon: Weak cytoplasmic granular staining in crypt base columnar cells and weak stromal tissue positivity can be seen. Liver: Mild-moderate diffuse cytoplasmic granular staining in liver sinusoid. Kupffer cells around the lining of liver sinusoid shows stronger granular staining compared with the surrounding parenchyma. Heart: Absent or negative staining on myocardial tissue. Thymus: Weak and diffuse granular staining over thymus tissue. Tonsil: Some, but not all, lymphoid tissue in the tonsil shows weak granular cytoplasmic staining. Spleen: Weak cytoplasmic staining over splenic cords. There is also hemosiderin-laden macrophages and lipofusin (yellow droplets). Kidney: Proximal tubules, distal tubules, and collecting ducts show mild-to-moderate cytoplasmic reaction, but the glomeruli are negative. Skin: Lower level of stratum spinosum and stratum basale (basal layer) show granular cytoplasmic staining. There is no staining of keratinized squamous cell layer and collagen tissue. (E) Expression level of TSLPR on B-ALL relative to expression of CD19 and CD22. JH331 is a patient-derived xenograft model of CRLF2-rearranged ALL with endogenous TSLPR overexpression. Shaded histograms represent isotype control.
Both short and long TSLPR CAR T cells demonstrate in vitro activity against TSLPRhi ALL
We next tested whether T cells transduced with the TSLPR CAR constructs demonstrated activity in vitro when incubated with the B-ALL cell line REH-transduced to express TSLPR (REH-TSLPR), as well as a naturally TSLPR-overexpressing CRLF2-rearranged ALL line (MUTZ5). As is shown in Figure 2A, both short and long CAR T cells produce high levels of interferon γ (IFNγ) and tumor necrosis factor-α (TNFα) when incubated with REH-TSLPR. We also found that, in addition to IFNγ and TNFα, substantial amounts of IL-2 and IL-8 were produced (supplemental Figure 3). When the lytic capacity of TSLPR CAR T cells was measured, the short and long constructs demonstrated equivalent activity against REH-TSLPR (Figure 2B). However, the short TSLPR CAR T cells appeared to have higher lytic capacity against MUTZ5 than the long CAR T cells despite comparable levels of TSLPR surface expression on both REH-TSLPR and MUTZ5. As an additional negative control, we also assessed TSLPR CAR T-cell reactivity against the parent (TSLPRlo) cell line. Minimal cytokine production (Figure 2A) or cell lysis (data not shown) was observed.
Figure 2.

Both short and long demonstrate activity in vitro but only the short CAR is active in vivo. (A) Production of IFNγ and TNFα by short and long anti-TSLPR CAR T cells as measured by enzyme-linked immunosorbent assay of supernatant after coincubation with TSLPRhi ALL (REH TSLPR or MUTZ5). (B) Lysis of REH-TSLPR and MUTZ5 TSLPRhi ALL by both short and long CAR T cells measured by 51Cr release after 4-hour coculture. Results show specific lysis calculated as described in Methods. (C) Short TSLPR CAR T cells (15 × 106) administered IV on day 4 after injection of luciferase-expressing REH-TSLPR demonstrate potent activity as measured by bioluminescent imaging, whereas long TSLPR CAR given at the same dose fail to alter leukemia progression. Control mice received the same dose of expanded GFP-transduced T cells. A single animal in the short TSLPR CAR group was sacrificed as the result of a wasting syndrome consistent with xenogeneic graft-versus-host disease. ACT, adoptive cell transfer. (D) In a separate experiment, mice were treated as in Figure 2C, and peripheral blood was analyzed on days 16 and 27 after injection of 15 × 106 short or long TSLPR CAR T cells or GFP-transduced control T cells. Representative dot plots showing increased persistence of short CD8+ TSLPR CAR T cells compared with long CAR T cells on days 16 and 27 after injection. (E) Significantly increased absolute number of short TSLPR CAR T cells at day 27 after injection compared with long CAR T cells as measured using a bead calibration as described in “Methods.”
Only short TSLPR CAR T cells are active against B-ALL in vivo, correlating with greater in vivo persistence than long TSLPR CAR T cells
We next tested the ability for TSLPR CAR T cells to eradicate ALL in vivo when injected into mice engrafted with TSLPR-expressing ALL. Four days after IV injection of luciferase-expressing REH-TSLPR, leukemia was detectable at low levels. Injection of 15 × 106 short CAR T cells completely eradicated ALL in this cell line model (Figure 2C). Interestingly, despite equivalent in vitro activity against REH-TSLPR, long CAR T cells had minimal impact on leukemia progression in mice. To determine the reason for the disparate activity of the long and short CAR constructs, we next looked at CAR T cell persistence by flow cytometry and found that short CAR T cells were present in greater numbers in the peripheral blood compared with long CAR T cells (Figure 2D-E). Interestingly, this was most notable at later time points despite progression of TSLPR-expressing ALL in recipients of long CAR T cells. Thus the marked increase in activity seen with the short TSLPR CAR construct compared with the long construct was associated with greater persistence of short CAR-expressing T cells.
Short CAR T cells eradicate TSLPR-overexpressing ALL when infused at high leukemic burden
To test the short TSLPR CAR T cells in higher leukemia burden models, CAR T-cell infusion was delayed until day 16 after REH-TSLPR injection in NSG mice (Figure 3A). Remarkably, 10 × 106 short TSLPR CAR T cells were still able to induce rapid clearance of highly-engrafted TSLPRhi ALL, with maintenance of leukemia clearance in the majority of mice through day 40 (Figure 3A). Importantly, there was no evidence for any alteration in the rapid progression of TSLPRlo ALL by TSLPR CAR T cells, demonstrating that activity is dependent on expression levels of the CAR target. CD8+ T cells generally exhibit greater in vitro lytic function and are thought to be the most important mediators of direct antitumor activity in vivo when compared with CD4+ T cells. Interestingly, although the in vitro expansion protocol used in these experiments resulted in a predominance of CD4+ T cells before infusion, CD8+ T cells had expanded markedly by day 50 and represented the largest T-cell subset in vivo (Figure 3B). As expected, this expansion of CD8+ CAR-expressing T cells was associated with expression of surface markers associated with effector phenotypes by day 50 (Figure 3 C-D). There was also a substantial percentage of CAR T cells with a CCR7+/CD45RA– phenotype, consistent with a central memory subset, thought to be important for persistence and sustained antitumor activity.
Figure 3.

Potent activity of short TSLPR CAR T cells is associated with a relative expansion of CD8+ CAR T cells in vivo. (A) Mice were injected with high-dose REH-TSLPR (5 × 106) followed by late injection of short TSLPR CAR T cells (10 × 106) on day 16. ACT, adoptive cell transfer. (B) Slightly increased relative number of CD4+ CAR T cells after CD3/CD28 bead-mediated expansion. This converts to a predominance CD8+/TSLPR CAR+ (measured by TSLPR Fc) at day 50 after injection. (C) Representative dot plots showing phenotype of short TSLPR CAR T cells before injection at day 20 after injection. (D) Relative percentages of naïve, central memory, and effector memory CAR T cells at day 20 after injection.
Quantitative assessment of short CAR T cells defines curative dose in established leukemia
We next performed an in vivo short CAR T-cell dose titration to define the range over which activity is observed and to determine whether the short TSLPR could cure animals with high levels of engrafted leukemia. As is shown in Figure 4, short TSLPR CAR cells were curative at 15 × 106 cells per mouse and with clear activity at 5 to 10 × 106 per mouse and some activity as low as 1 × 106 per mouse in well-engrafted animals, particularly noted as improved survival (Figure 4B). Again, there was minimal activity seen after infusion of the long CAR T cells, with only slight decrease in leukemic burden in the peripheral blood (Figure 4C) and luciferase activity at day 6 (Figure 4D) compared with green fluorescent protein (GFP) control T cells, but there was no difference between the 2 groups at any of the later time points. These data are consistent with the failure of the long CAR T cells to persist in vivo.
Figure 4.
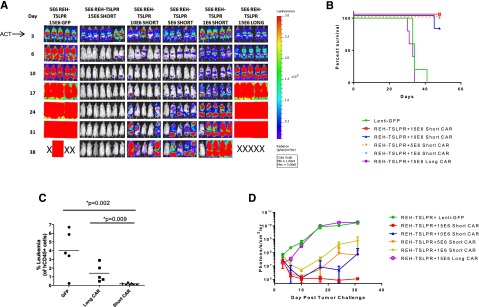
Short TSLPR CAR T cells demonstrate activity over a broad dose range and mediate durable eradication of TSLPRhi ALL at high leukemic burden. (A) Dose titration of short TSLPR CAR T cells given on day 3 after high-dose REH TSLPR ALL injection (5 × 106 cells/mouse). ACT, adoptive cell transfer. (B) Survival plot of mice treated in Figure 4A. (C) Flow cytometry of peripheral blood for the presence of CD45+ leukemia cells at day 12 after injection. (D) Quantitation of bioluminescence in mice treated in Figure 4A.
Short TSLPR CAR T cells eradiate pre–B-ALL that naturally overexpresses TSLPR
All of the results shown thus far used an engineered REH ALL cell line that was transduced to overexpress TSLPR, but also allowed assessment of the target specificity of the CAR by direct comparison with the non–TSLPR-overexpressing parent REH cell line (TSLPRlo). We thus next tested the short TSLPR construct in 3 PDX models of primary de novo Ph-like ALL with CRLF2 rearrangements (Figure 5A). As is shown in Figure 5B, the short TSLPR CAR eradicated leukemia in the luciferase-expressing human TSLPRhi ALL PDX model JH331, as measured by bioluminescent imaging. These responses were durable in a repeat experiment in which mice were imaged for 70 days (supplemental Figure 4). The short TSLPR CAR also eradicated leukemia in the blood, bone marrow, and spleens of 2 additional de novo TSLPRhi ALL PDX models (Figure 5C-D).
Figure 5.

Leukemia eradication in human CRLF2-rearranged Ph-like ALL patient-derived xenograft models by short TSLPR CAR T cells. (A) Flow cytometric surface expression of CD19 and TSLPR in human ALL cells from TSLPRhi PDX models JH352, NH362, and JH331. Shaded histograms denoted by dotted lines represent unstained controls. (B) Complete eradication of leukemia in a TSLPRhi luciferase–expressing PDX model (JH331) by short TSLPR CAR T cells. ACT, adoptive cell transfer. (C-D) Analysis of human CD45+ CD19+ ALL cells in peripheral blood (C) and bone marrow (D) of TSLPRhi PDX models JH352 and NH362 injected with 1 × 106 ALL cells per mouse, then treated 22 days later with 15 × 106 short TSLPR CAR T cells. (E-G) Tissue analyses of an aggressive relapsed TSLPRhi ALL PDX model (ALL4364) treated with TSLPR CAR T cells. One million ALL cells per mouse were injected IV on day 1, then animals were treated with 1.2 million of TSLPR CAR+ T cells IV on day 14. (E) Spleens from short TSLPR CAR-treated and untreated mice on days 35 and 42 after leukemia injection (2 and 3 weeks after CAR injection). (F) Representative dot plot of bone marrow, spleen, and peripheral blood on day 35 after leukemia injection. (G) Scatter plots of organ and blood leukemia infiltration at day 35 after TSLPRhi ALL injection comparing short TSLPR CAR treated and untreated mice. (H) Separate experiment showing survival analysis demonstrating prolonged survival of TSLPR CAR T cell–treated ALL4364 PDX animals (n = 5/group). ACT, adoptive cell therapy. (I) Expression of TSLPR on human CD45+/CD19+ leukemia cells in the bone marrow of a mouse with relapse after short TSLPR CAR treatment.
TSLPR overexpression has been associated with clinically high-risk ALL and, in many cases, an aggressive and chemotherapy-refractory phenotype. We thus tested the short TSLPR CAR against an aggressive relapsed CRLF2-rearranged ALL specimen (ALL4364) that is lethal by 60 days after IV injection into NSG mice. As is shown in Figure 5E-H, short TSLPR CAR T cells demonstrated potent activity against this aggressive TSLPRhi ALL, resulting in decreased splenomegaly and reduction in blast counts in the spleens and blood of xenografted animals as early as day 14 after CAR T-cell injection. Interestingly, although activity in the bone marrow was also observed, clearance of leukemia was less rapid and not statistically significant at this early evaluation time. However, CAR T-cell treatment was associated with eventual clearance of aggressive TSLPRhi ALL and prolonged survival (Figure 5H). Analysis of late relapses demonstrated retained expression of TSLPR, indicating that failure was not caused by loss of antigen (Figure 5I).
Comparable activity of TSLPR CAR with CD19 CAR and CD22 CAR
We next compared the activity of the short TSLPR CAR with a CD19 CAR containing the same scFv (FMC68) as those currently demonstrating potent clinical activity10 and a CD22 CAR construct with potent activity in human xenograft models.39 Importantly, all of the CAR constructs contained the same 41BB costimulatory domain (Figure 6A) and demonstrated comparable transduction efficiencies (Figure 6B). As is shown in Figure 6C-D, T cells transduced with the short TSLPR CAR, CD19 CAR, or CD22 CAR T cells were curative in the JH331 PDX model at a dose of 3 × 106 CAR T cells, with durable responses with 0.3 × 106 TSLPR CAR T cells.
Figure 6.

TSLPR CAR demonstrates activity comparable with CD19 CAR. (A) Schematic diagram of second-generation CD19 and the CD22 CAR constructs. Both CARs have identical transmembrane and signaling domains to the TSLPR CAR. (B) Flow cytometric surface expression of protein L on TSLPR, CD19, and CD22 CAR T cells at 5 days postviral transduction, demonstrating efficacy of CAR transduction. (C) TSLPR, CD19, and CD22 CAR-redirected T cells were injected IV into NSG mice (3 × 106/mouse) previously engrafted with the patient xenograft cell line JH331-LUC 29 days earlier and followed by bioluminescent imaging. T cells transduced with GFP were used as a negative control. ACT, adoptive cell transfer; D, day. (D) JH331-LUC–bearing NSG mice were treated as in Figure 6C but including a group receiving a log lower dose of CAR T cell. Control mice received 3 × 106 GD2-targeted CAR T cells.
Discussion
Much recent progress has been made in immunotherapeutic approaches to treat cancer, particularly in the area of adoptive cell therapy.40 For B-ALL, the use of T cells modified with CARs that fuse T-cell signaling constructs with antibody recognition domains targeting the B cell–associated antigen CD19 has resulted in remarkable complete remission rates in heavily pretreated, chemotherapy-refractory patients, including those who have relapsed after allogeneic hematopoietic stem cell transplantation.9-11 These studies have provided important proof-of-principle of the potency of adoptive cell therapy using CAR-expressing T cells. However, leukemia relapses with loss of expression of the CD19 target has been observed.14 CD19 loss has also been observed after treatment of ALL patients with blinatumomab, a CD19/CD3 bispecific T cell–engaging reagent.15 These observations indicate that, although CD19 is an excellent target, it is likely not required for B-ALL survival. Thus, identification of additional ALL targets is needed, particularly those less likely to be lost from the surface of the leukemic blasts and, theoretically, more likely to result in durable responses when targeted.
Recent gene expression profiling studies have revealed overexpression of the CRLF2 gene in 5% to 15% of children and adults with B-ALL, which results in high levels of TSLPR cell-surface expression that is easily measurable by flow cytometry assays.17,18,31 Detailed analyses of these leukemias indicated that TSLPR overexpression was associated with translocations or upstream deletions, resulting in alternative promoter regions in the majority of patients, then resulting in overexpression of a nonmutated receptor.20 Importantly, although survival and relapse data have been somewhat discrepant in standard-risk vs high-risk patients,23,26,41,42 CRLF2 rearrangements and other Ph-like kinase alterations are associated with a greater risk of relapse and inferior outcomes in high-risk patients.18 More recent genetic studies have demonstrated that Ph-like ALL (including those with CRLF2 rearrangements) are highly associated with IKZF1 deletions, which are also prevalent in Ph+ ALL.17,23,24,43 TSLPR is thus an excellent target for molecularly targeted therapeutic approaches, because its overexpression occurs in a sizable subset of B-ALL patients who have high risks of recurrence and generally poor outcomes.
The CRLF2 chain forms the heterodimeric TSLPLR complex with the IL-7Rα chain with inducible signaling by binding of the TSLP ligand.44 Important intracellular mediators of TSLP signaling include members of the JAK/STAT pathway, such as JAK1, JAK2, and STAT5. Physiologically, TSLP is important in allergic inflammation and T-cell development and can contribute to B-cell development. Recently, targeting of the TSLP/TSLPR axis has been shown to be therapeutically beneficial in patients with allergic asthma.45 In B-ALL, recurring mutations in the IL7RA gene (encoding the IL7Rα chain) have been identified that result in homodimerization and autophosphorylation of the TSLPR with ligand-independent signaling in contrast to the overexpression of nonmutated receptor in the majority of CRLF2-overexpressing ALL.16 Nonetheless, these data provide further evidence that IL-7/TSLP signaling pathways are functionally important for B-ALL. JAK2 mutations have also been identified in B-ALL and are frequently associated with CRLF2 rearrangements,17,20,46 although the specific mutations are different from those found in myeloproliferative neoplasms.47 Although JAK inhibition is under study for CRLF2-rearranged and/or JAK-mutant B-ALL, and preclinical efficacy has been demonstrated in PDX models,33 it remains unclear whether such agents will be effective in patients with JAK-activated Ph-like ALL or whether multiple inhibitors will be necessary given the diversity of mutations. Targeting the TSLPR protein on the cell surface thus provides another universal option for these high-risk patients. Collectively, these data indicate that B-ALL biology is firmly linked to this cytokine receptor family and suggest that targeting of these pathways could provide a therapeutic option.
In these studies, we demonstrate that T cells engineered with a CAR targeting the TSLPR protein are potent therapeutically and can eradicate TSLPR-overexpressing leukemia in murine xenograft models of human Ph-like ALL. Importantly, we observed these phenomena in multiple TSLPR-overexpressing B-ALL models. Interestingly, although other CAR constructs have successfully used a spacer region between the cell membrane and the scFv, T cells transduced with the long version of our anti-TSLPR CAR did not demonstrate in vivo activity despite comparable activity in vitro. We demonstrate that this lack of efficacy of the long-construct T cells is associated with lack of persistence in vivo. Similar observations have been recently reported for other CAR constructs.48,49 Finally, at suboptimal CAR T-cell dosing, residual or recurrent B-ALL did not lose surface expression of TSLPR.
In summary, we demonstrate that targeting of TSLPR using engineered chimeric antigen receptor T cells is efficacious against CRLF2-rearranged B-ALL and may be a promising therapeutic option for patients with these high-risk leukemias. To our knowledge, these data are the first demonstration that targeting of a cytokine receptor and oncoprotein can induce robust leukemia cytotoxicity. Although CAR-based immunotherapies have demonstrated potent activity in B-ALL, such approaches are currently under study only in patients with recurrent or chemorefractory ALL. Our findings also create opportunities to use TSLPR targeting with other treatment approaches, such as scFv-toxin conjugates or bispecific-antibody reagents, for patients with CRLF2-rearranged ALL as an intervention earlier in therapy given the high risk of relapse in these patients.
Acknowledgments
The authors thank Crystal L. Mackall for helpful discussions.
These studies were supported in part by the Stand Up To Cancer St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113) (S.K.T., M.D., D.M.B., R.J.O., P.H.B.S., S.A.G., T.J.F.); a grant from the William Lawrence and Blanche Hughes Foundation (T.J.F.); National Institutes of Health, National Cancer Institute–supported CHTN (UMICA1837390); the Alex’s Lemonade Stand Foundation (ALSF), the University of Pennsylvania Institute for Translational Medicine and Therapeutics (ITMAT) Transdisciplinary Program in Translational Medicine and Therapeutics (UL1RR024134 from the National Center For Research Resources), and the National Cancer Institute (K08CA184418) (S.K.T.).
S.K.T. was an ALSF Scholar in Developmental Therapeutics. D.M.B. is a St. Baldrick’s Foundation Scholar. S.A.G. is the Yetta Deitch Novotny Endowed Chair in Pediatric Oncology.
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.Q. performed experiments and wrote the manuscript; W.H., M.C., and L.Z. performed experiments and analyzed data; S.K.T. provided key patient-derived xenografts and edited the manuscript; H.Z.O., G.L.N., M.D., and P.H.B.S. performed and interpreted the TSLPR tissue microarray; J.Z., Y.L., and B.S.M. provided important normal tissue lines for potential off-tumor reactivity; S.M., D.T.T., D.M.B., and S.A.G. contributed key patient-derived xenografts; R.J.O. provided important reagents and intellectual contributions to the construction of the chimeric antigen receptors; and T.J.F. conceived of, designed, and supervised the research; analyzed data; and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Terry J. Fry, Pediatric Oncology Branch, Center for Cancer Research, NCI, NIH, Bethesda, MD 20816; e-mail: fryt@mail.nih.gov.
References
- 1.Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(3):185–196. doi: 10.1053/j.seminhematol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunger SP, Loh ML, Whitlock JA, et al. COG Acute Lymphoblastic Leukemia Committee. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(6):957–963. doi: 10.1002/pbc.24420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943–1955. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205–e217. doi: 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed] [Google Scholar]
- 5.Raetz EA, Bhatla T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematol Am Soc Hematol Educ Program. 2012;2012:129–136. doi: 10.1182/asheducation-2012.1.129. [DOI] [PubMed] [Google Scholar]
- 6.Oeffinger KC, Mertens AC, Sklar CA, et al. Childhood Cancer Survivor Study. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355(15):1572–1582. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 7.Mathisen MS, Kantarjian H, Thomas D, O’Brien S, Jabbour E. Acute lymphoblastic leukemia in adults: encouraging developments on the way to higher cure rates. Leuk Lymphoma. 2013;54(12):2592–2600. doi: 10.3109/10428194.2013.789509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schafer ES, Hunger SP. Optimal therapy for acute lymphoblastic leukemia in adolescents and young adults. Nat Rev Clin Oncol. 2011;8(7):417–424. doi: 10.1038/nrclinonc.2011.77. [DOI] [PubMed] [Google Scholar]
- 9.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224–225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fry TJ, Mackall CL. T-cell adoptive immunotherapy for acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. 2013;2013:348–353. doi: 10.1182/asheducation-2013.1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topp MS, Gökbuget N, Zugmaier G, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–4140. doi: 10.1200/JCO.2014.56.3247. [DOI] [PubMed] [Google Scholar]
- 16.Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901–908. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Blood. 2013;121(3):485–488. doi: 10.1182/blood-2012-04-422691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115(5):1006–1017. doi: 10.1182/blood-2009-08-235408. [DOI] [PubMed] [Google Scholar]
- 20.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 22.Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107(1):252–257. doi: 10.1073/pnas.0911726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119(15):3512–3522. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115(26):5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010;115(26):5393–5397. doi: 10.1182/blood-2009-11-256131. [DOI] [PubMed] [Google Scholar]
- 26.Moorman AV, Schwab C, Ensor HM, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30(25):3100–3108. doi: 10.1200/JCO.2011.40.3907. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita Y, Shimada A, Yamada T, et al. IKZF1 and CRLF2 gene alterations correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(10):1587–1592. doi: 10.1002/pbc.24571. [DOI] [PubMed] [Google Scholar]
- 28.Attarbaschi A, Morak M, Cario G, et al. Associazione Italiana di Ematologia ed Oncologia Pediatrica (AIEOP)-Berlin-Frankfurt-Münster (BFM) Study Group and National Cancer Research Institute (NCRI)-Children’s Cancer and Leukaemia (CCLG) Study Group. Treatment outcome of CRLF2-rearranged childhood acute lymphoblastic leukaemia: a comparative analysis of the AIEOP-BFM and UK NCRI-CCLG study groups. Br J Haematol. 2012;158(6):772–777. doi: 10.1111/j.1365-2141.2012.09221.x. [DOI] [PubMed] [Google Scholar]
- 29.Palmi C, Vendramini E, Silvestri D, et al. Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia. 2012;26(10):2245–2253. doi: 10.1038/leu.2012.101. [DOI] [PubMed] [Google Scholar]
- 30.van Bodegom D, Zhong J, Kopp N, et al. Differences in signaling through the B-cell leukemia oncoprotein CRLF2 in response to TSLP and through mutant JAK2. Blood. 2012;120(14):2853–2863. doi: 10.1182/blood-2012-02-413252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tasian SK, Doral MY, Borowitz MJ, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833–842. doi: 10.1182/blood-2011-12-389932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tasian SK, Loh ML. Understanding the biology of CRLF2-overexpressing acute lymphoblastic leukemia. Crit Rev Oncog. 2011;16(1-2):13–24. doi: 10.1615/critrevoncog.v16.i1-2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510–3518. doi: 10.1182/blood-2012-03-415448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006;107(3):1149–1155. doi: 10.1182/blood-2005-05-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 37.Ziegler SF, Liu YJ. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat Immunol. 2006;7(7):709–714. doi: 10.1038/ni1360. [DOI] [PubMed] [Google Scholar]
- 38.Lo Kuan E, Ziegler SF. Thymic stromal lymphopoietin and cancer. J Immunol. 2014;193(9):4283–4288. doi: 10.4049/jimmunol.1400864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haso W, Lee DW, Shah NN, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol. 2014;32(27):3012–3020. doi: 10.1200/JCO.2014.55.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ensor HM, Schwab C, Russell LJ, et al. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood. 2011;117(7):2129–2136. doi: 10.1182/blood-2010-07-297135. [DOI] [PubMed] [Google Scholar]
- 43.Mullighan CG, Su X, Zhang J, et al. Children’s Oncology Group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tal N, Shochat C, Geron I, Bercovich D, Izraeli S. Interleukin 7 and thymic stromal lymphopoietin: from immunity to leukemia. Cell Mol Life Sci. 2014;71(3):365–378. doi: 10.1007/s00018-013-1337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gauvreau GM, O’Byrne PM, Boulet LP, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370(22):2102–2110. doi: 10.1056/NEJMoa1402895. [DOI] [PubMed] [Google Scholar]
- 46.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106(23):9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 48.Guest RD, Hawkins RE, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 49.Hudecek M, Sommermeyer D, Kosasih PL, et al. The non-signaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]