

Background: Persulfide dioxygenases (PDOs), which belong to the metallo-β-lactamase (MBL) enzyme superfamily, oxidize glutathione persulfide (GSSH).
Results: Crystal structures and ITC data provide information on ligand binding by PDOs.
Conclusion: MBLs share conserved amino acid residues, but the functions of these residues vary by class.
Significance: These results provide criteria for distinguishing PDOs from other MBL superfamily members.
Keywords: bacterial metabolism, bioenergetics, dioxygenase, enzyme structure, sulfur, ETHE1, glutathione, glyoxalase II
Abstract
Persulfide dioxygenases (PDOs), also known as sulfur dioxygenases (SDOs), oxidize glutathione persulfide (GSSH) to sulfite and GSH. PDOs belong to the metallo-β-lactamase superfamily and play critical roles in animals, plants, and microorganisms, including sulfide detoxification. The structures of two PDOs from human and Arabidopsis thaliana have been reported; however, little is known about the substrate binding and catalytic mechanism. The crystal structures of two bacterial PDOs from Pseudomonas putida and Myxococcus xanthus were determined at 1.5- and 2.5-Å resolution, respectively. The structures of both PDOs were homodimers, and their metal centers and β-lactamase folds were superimposable with those of related enzymes, especially the glyoxalases II. The PDOs share similar Fe(II) coordination and a secondary coordination sphere-based hydrogen bond network that is absent in glyoxalases II, in which the corresponding residues are involved instead in coordinating a second metal ion. The crystal structure of the complex between the Pseudomonas PDO and GSH also reveals the similarity of substrate binding between it and glyoxalases II. Further analysis implicates an identical mode of substrate binding by known PDOs. Thus, the data not only reveal the differences in metal binding and coordination between the dioxygenases and the hydrolytic enzymes in the metallo-β-lactamase superfamily, but also provide detailed information on substrate binding by PDOs.
Introduction
Accumulating evidence indicates that hydrogen sulfide (H2S) plays significant roles as a signaling molecule in animals (1, 2). The cellular H2S concentration is maintained by equilibrium between its formation and oxidation. It is produced from sulfur-containing amino acids by cystathionine β-synthase, cystathionine γ-lyase, and mercaptopyruvate sulfur transferase (3). In eukaryotes, H2S is oxidized in the mitochondrion. First, sulfide:quinone oxidoreductase oxidizes it to sulfane sulfur that is likely present as a cysteinyl persulfide intermediate within the active site (4). Sulfide:quinone oxidoreductase passes the sulfur to a sulfane sulfur acceptor. Although sulfite is an effective acceptor, GSH is more likely to be the physiological acceptor to produce glutathione persulfide (GSSH) (5). Then, persulfide dioxygenase (PDO),4 which is also known as sulfur dioxygenase (EC 1.13.11.18), oxidizes GSSH to sulfite and GSH (6). Furthermore, a rhodanese or sulfurtransferase is involved in the metabolism (5, 7, 8). Mutations in the human PDO gene ethe1 are the cause of the recessive hereditary disease ethylmalonic encephalopathy, which can lead to unusually high excretion of short-chain carboxylic acids in the urine, brain defects, and early death (9). PDO activity is also important in plants; its gene inactivation in Arabidopsis thaliana has been reported to produce defects in seed development and embryonic arrest by the early heart stage (10). Thus, H2S oxidation plays a major physiological role in both plants and animals.
The activity of PDO was initially discovered in bacterial cell extracts, and GSSH was identified as a substrate (11). The human PDO (referred to as hPDO in this report, and is known as hETHE1) is the first characterized PDO (10, 12). Sequence analysis reveals that it belongs to the metallo-β-lactamase superfamily, which consists of mainly hydrolytic enzymes (13). Proteins in the family share the same structural fold of two stacked β-sheets surrounded by α-helices. The metal-binding residues are located at one edge of the β-sheets, and substrate binding is from the loops and α-helices surrounding the metal center. These proteins have six or seven conserved amino acid residues for binding one or two metal ions at the metal center. Because the other members of the superfamily are mainly hydrolytic enzymes, their reaction mechanisms cannot apply to explain the dioxygenase activity of PDOs.
Using the hPDO sequence, we have identified a wide distribution of PDO genes in sequenced bacterial genomes and have recently characterized 10 bacterial PDOs (14). On the basis of sequence analysis, we have proposed three subclasses of PDO: (i) ETHE1, which is present in animals, plants, and bacteria, (ii) persulfide dioxygenase A (PdoA), also known as sulfur dioxygenase A (SdoA), which is common in Proteobacteria, and (iii) Blh, which is an acronym for “β-lactamase-like hydrolase” (14). To be consistent with scientific names, we rename the bacterial ETHE1 type as PDO type I, and the PdoA type as PDO type II throughout this report. Our new structural data also support this classification. Apo-form crystal structures of hPDO and A. thaliana PDO (referred to as AtPDO in this report, and is also known as AtETHE1) have been determined (15, 16).
In this report, we present structural and biochemical analyses for a PDO from Pseudomonas putida (PpPDO2, also known as PpSdoA) and for a PDO from Myxococcus xanthus (MxPDO1b, also known as MxETHE1b because M. xanthus possesses three type I PDOs). The crystal structure of PpPDO2 with GSH in its binding pocket permits identification of the amino acid residues involved in substrate binding. In addition, structural comparison of PpPDO2 with MxPDO1b reveals that there are differences in the GS-moiety binding sites between them. Furthermore, the change of metal binding in the PDOs in comparison with other members of the MBL superfamily is discussed for the evolution of a dioxygenase from a hydrolase.
Experimental Procedures
Chemicals and Enzymes
Chemicals were obtained from Sigma or Fisher Scientific. Crystallization screens were obtained from Hampton Research and Qiagen.
Cloning and Enzyme Purification
Genes encoding PpPDO2 from P. putida (ABQ76243) and MxPDO1b from M. xanthus (WP_011554322, ex. YP_632494) were cloned into pET30 Ek/LIC with Escherichia coli BL21(DE3) as the host (14).
For expression and purification of C terminally His-tagged PpPDO2 or MxPDO1b, cultures were grown at 37 °C in LB broth containing 30 μg/ml of kanamycin. The cultures were allowed to reach an A600 of 0.6 prior to inducing protein expression, which was done by adding 0.5 mm isopropyl β-d-thiogalactopyranoside to the media and incubating for 22 h at 20 °C. Cells were harvested by centrifugation at 5000 × g, frozen, and then suspended in 50 mm Tris buffer, pH 8.0, supplemented with 300 mm NaCl and 20 mm imidazole. Cells were lysed by sonication and lysates were cleared by centrifugation at 15,000 × g. The supernatant was stirred into nickel-nitrilotriacetic acid-agarose resin (Qiagen), the column was washed with 2 volumes of the lysis buffer, and recombinant enzyme was eluted with buffer containing 50 mm Tris, pH 8.0, 300 mm NaCl, and 250 mm imidazole. The eluted sample was concentrated and exchanged into 20 mm Tris, pH 7.5, applied to a 6-ml Resource Q column (GE Healthcare), and the enzyme was eluted with a 50 mm stepwise NaCl gradient in the same buffer. PpPDO2 or MxPDO1b-containing fractions, which eluted at ∼200 and 100 mm NaCl, respectively, were pooled, buffer-exchanged, and concentrated into the appropriate buffer for crystallization or biochemical experiments. To ensure higher occupancy of ferrous iron in the active sites, all enzyme preparations were incubated with ferrous ammonium sulfate and ascorbic acid on ice for 2 h prior to use, and at concentrations equal to those of the enzymes. Purity was monitored for all protein preparations by SDS-PAGE and protein concentrations were determined with the method of Bradford, using BSA as a standard.
Protein Crystallization and Structure Determination
Crystals of PpPDO2 and MxPDO1b were grown using the hanging-drop, vapor-diffusion method. For PpPDO2 crystallization, purified protein at 30 mg/ml in 20 mm Tris, pH 8.0, was mixed with an equal volume of reservoir solution and equilibrated against the same solution at 4 °C. The reservoir solution was 100 mm HEPES, pH 7.5, 200 mm ammonium acetate, and 25% (w/v) PEG 3350. Crystals of this enzyme typically appeared within 3 days. To obtain structural data for PpPDO2 in complex with GSH, crystals were soaked in 5 mm GSH for 1 h prior to harvest. Adequate cryoprotection was achieved by passing crystals through a small drop of storage buffer/mother liquor mixture (50% of each component by volume) that was brought to 18% glycerol. For MxPDO1b crystallization, purified protein at 11 mg/ml in 20 mm MOPS, pH 7.1, was mixed with an equal volume of reservoir solution and equilibrated against the same solution at 4 °C. The reservoir solution was 200 mm calcium chloride, 100 mm HEPES, pH 7.5, 15% (w/v) PEG 400, and 15% glycerol (w/v). Because the mother liquor was a sufficient cryoprotectant, no additional glycerol was required to prevent freezing.
The space groups of PpPDO2 and MxPDO1b were P43 and P61, respectively; the asymmetric units of the unit cells contained both molecules of the respective homodimers. Diffraction data were collected up to 1.5-Å resolution for PpPDO2 in complex with GSH and 2.5 Å for the apo-forms of both MxPDO1b and PpPDO2 at the Berkeley Advanced Light Source (ALS, beamline 8.2.1). The diffraction data were processed with the HKL2000 package (17). The statistics for the diffraction data are listed in Table 1. Initial phasing of apo-form PpPDO2 diffraction data were conducted by molecular replacement with the PDB coordinates of model 4EFZ using PHENIX Phaser (18). MxPDO1b initial phasing was conducted by molecular replacement as well, using the atomic coordinates of the unpublished structure for a PDO1 protein from Cupriavidus necator. Iterative model building and refinement was conducted using the programs COOT (19) and PHENIX. The coordinate and diffraction data have been deposited in the Protein Data Bank.
TABLE 1.
Crystallographic data for the PpPDO2 and MxPDO1b structures
| Data | PpPDO2 (apo-form) | PpPDO2 (complex) | MxPDO1b |
|---|---|---|---|
| PDB ID | 4YSK | 4YSL | 4YSB |
| Space group | P43 | P43 | P61 |
| Cell dimensions (a, b, c) (Å) | (79.505, 79.505, 94.523)a | (79.730, 79.730, 93.990) | (116.603, 116.603, 65.061) |
| (α, β, γ) (°) | (90, 90, 90) | (90, 90, 90) | (90, 90,120) |
| Resolution (Å) | 50.00-2.40 | 50.00-1.46 | 50-2.50 |
| Rsym | 0.165 (0.545) | 0.020 (0.042) | 0.075 (0.319) |
| I/σI | 42.143 (7.892) | 30.600 (2.957) | 33.625 (6.692) |
| Completeness (%) | 97.25 (65.0) | 98.18 (89.1) | 99.89 (99.4) |
| Redundancy | 5.5 (5.3) | 2.4 (2.2) | 11.2 (11.1) |
| Refinement | |||
| Resolution (Å) | 40.6-2.47 | 40.5-1.46 | 43.4-2.50 |
| Unique reflections | 19,717 | 99,194 | 17,540 |
| Rwork/Rfree | 0.213/0.262 | 0.180/0.204 | 0.175/0.226 |
| R.m.s deviations | |||
| R.m.s. deviation bonds (Å) | 0.002 | 0.009 | 0.003 |
| R.m.s. deviation angles (°) | 0.641 | 1.126 | 0.595 |
| Number of atoms | |||
| Protein | 4,717 | 4,871 | 3,531 |
| Ligand | 0 | 40 | 0 |
| Ion | 2 | 2 | 2 |
| Water | 131 | 637 | 127 |
| B-factors | |||
| Protein | 41.11 | 26.78 | 34.27 |
| Ligand | 67.16 | ||
| Ion | 38.24 | 23.22 | 32.58 |
| Water | 39.88 | 39.58 | 35.50 |
| Ramachandran | |||
| Favored | 94.28 | 97.38 | 96.70 |
| Outliers | 0.00 | 0.00 | 0.22 |
a Numbers in parentheses refer to the highest resolution shell.
Multiangle Light Scattering and Isothermal Titration Calorimetry (ITC)
To determine a predominant oligomeric state, multiangle laser light scattering was performed for PpPDO2 as previously described (20). Briefly, 200 μg of PpPDO2 were loaded onto a Yarra 3-μm SEC-200 column (Phenomenex) and eluted isocratically at 0.5 ml min−1. The elution buffer for PpPDO2 was 20 mm MOPS, pH 6.8, supplemented with 100 mm NaCl. The eluate was successively passed through a UV detector (Gilson), an Optilab DSP interferometric refractometer (Wyatt Technology), and a Dawn EOS laser light scattering detector (Wyatt Technology). Data analysis was performed using the Zimm fitting method with software (ASTRA) provided by the instrument manufacturer.
Isothermal calorimetry titrations were executed for PpPDO2 and several substrate and product analogs in an ITC200 instrument (Malvern Instruments). The protein was prepared by extensive buffer exchange into the titration buffer, which consisted of 20 mm MOPS, pH 7.1. The concentration of protein in the calorimetric titration cell was diluted to 200 μm. All titrations were performed at 25 °C with a stirring speed of 750 rpm and 27 injections (1.4 μl each). All ligands were brought to a concentration of 2 mm in the titration buffer and injected into the protein solution, and the heats of binding were recorded. Ligands were also titrated against buffer alone to account for the heats of dilution. Ligand concentrations were adjusted to obtain significant heats of binding, and the time intervals between injections were adjusted to ensure proper baseline equilibration. All samples were degassed prior to titration.
Preparation of GSSCH3
To a solution of GSH (11 mg, 0.036 mmol) in 2 ml of sodium phosphate buffer (50 mm, pH 7.4), 10 eq of methyl methanethiosulfonate in 1 m N,N-dimethylformamide were added. The resulting solution was vigorously stirred for 1.5 h at room temperature. The solution was frozen on a dry ice/acetone bath and then lyophilized. The residue was washed and filtered with cold diethyl ether (3 × 5 ml) to remove excess methyl methanethiosulfonate. The white solid was collected by filtration to afford a product in 90% yield and characterized by 1H NMR, which was compared with a previous report (21). 1H NMR (300 MHz, D2O) δ 4.80 (m, CHCH2S, 1H), 3.97 (s, CH2CO2, 2H), 3.82 (t, J = 6.3 Hz, CHNH2, 1H), 3.25 (dd, J = 14.6, 4.9 Hz, CH2S, 1H), 3.02 (m, CH2S, 1H), 2.53 (q, J = 6.9 Hz, CH2CH2CO, 2H), 2.43 (s, SSCH3, 3H), 2.17 (q, J = 7.6 Hz, CH2CH2CO, 2H).
Preparation of GSSO3−
The compound was synthesized following a reported procedure (22). Freshly prepared 2 ml of Na2SO3 solution (200 mm: sodium phosphate buffer, pH 7.1) was directly added to freshly prepared 2 ml of GSNO solution (80 mm: sodium phosphate buffer, pH 7.1), and the resulting mixture was stirred for 1 h at room temperature. The formation of GSSO3− was monitored by 1H NMR and the spectral characteristics of GSSO3− were compared with previously reported 1H NMR data (22). 1H NMR (300 MHz, D2O, pD 7.4, 2 m KCl, 80 mm sodium phosphate, 100 μm EDTA) δ 5.01 (dd, J = 8.9, 4.6 Hz, CHCH2S, 1H), 3.97–3.89 (overlapped-m, CHNH2, CH2CO2, 3H), 3.76 (dd, J = 14.6, 4.7 Hz, CH2S, 1H), 3.57 (dd, J = 14.7, 8.9 Hz, CH2S, 1H), 2.72–2.59 (m, CH2CH2CO, 2H), 2.32 (m, CH2CH2CO, 2H).
Results
Global Structure
Purified recombinant PpPDO2 and MxPDO1b were crystallized in the P43 and P61 space groups and solved at 1.5- and 2.5-Å resolution, respectively. The structures of both PpPDO2 and MxPDO1b molecules consisted of two similarly sized, six-stranded central β-sheets surrounded by α-helices (Fig. 1, A and B), a structural motif strongly conserved among the metallo-β-lactamase superfamily. Although there were several insertions and deletions between PpPDO2 and MxPDO1b, which were primarily located in loops and N/C termini, the Cα positions of the two molecules were superimposable with a r.m.s. deviation value of 1.18 Å (Fig. 1C). The C-terminal eight residues of MxPDO1b were not visible in the final 2Fo − Fc electron density map, indicating their disordered nature.
FIGURE 1.
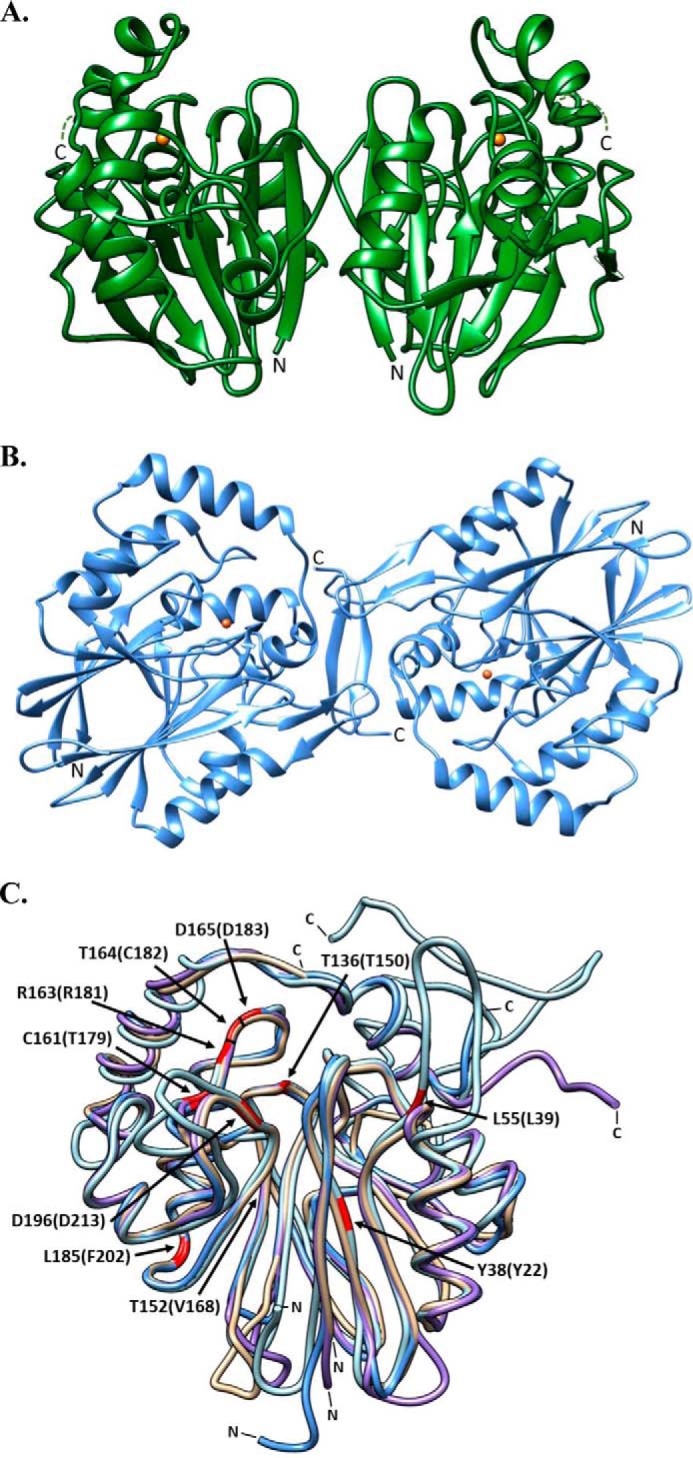
Ribbon diagrams representing the oligomeric and superimposed structures of PDOs. A, PpPDO2 and B, MxPDO1b are represented in their dimeric states. The orange spheres in both enzymes represent Fe(II). C, the superimposed structures of MxPDO1b (tan), PpPDO2 (light blue), hPDO (blue), and AtPDO (purple). The corresponding N and C termini are marked N and C, respectively. Residues corresponding to ethylmalonic encephalopathy-causing mutations are highlighted in red. The residues are numbered according to the hPDO sequence, with equivalent residues from PpPDO2 in parenthesis. This figure was generated using Chimera (UCSF) (32).
Throughout this report, the residue numbers for PpPDO2 were used, with those of MxPDO1b in parentheses unless stated otherwise. In both PpPDO2 and MxPDO1b structures, a single ferrous iron was coordinated by three residues occupying one face of the coordination sphere, including His74 (His57), His149 (His112), and Asp170 (Asp129) (Fig. 2A). This is a typical coordination pattern among mononuclear non-heme iron (II) oxygenases and is known as the 2-His 1-carboxylate facial triad (13, 23). The locations and orientations of participating residues observed in both PpPDO2 and MxPDO1b were identical and all three residues were from the same edge of the central β-sheets. The nitrogen atoms in Fe(II)-coordinating imidazole rings of His74 (His57) and His149 (His112) were within a hydrogen bond distance from the neighboring residues, Arg181 (Arg138) and Thr73 (Thr56), respectively (Fig. 2A). Thus both His residues could not only fix the orientation but also tune the redox potential of the Fe(II).
FIGURE 2.

GSH complex and ligand-free forms of PpPDO2. A, the stereo image of the Fe(II) coordinated by three water molecules in the apo-form of PpPDO2. The Fe(II)-coordinated water molecule W1 represents the water that is displaced upon binding of GSH, whereas water W2 is in the proposed site of O2 binding. The third water, W3, is engaged in hydrogen bonding with neighboring residues and may function as a proton relay. Notably, the lipophilic region of the Lys44 side chain in chain B interfaces with neighboring hydrophobic residues of chain A, likely contributing to binding pocket stability. B, the difference Fourier map clearly shows PpPDO2 in complex with GSH. Surrounding residues important for binding and catalysis are displayed and their residue numbers are indicated. The water molecules are shown as red spheres. C, the ribbon diagrams for Cα atoms of apo-form PpPDO2 (tan) and the same enzyme in complex with GSH (blue) are superimposable with a r.m.s. deviation of 0.3 Å. This figure was generated using Chimera (UCSF) (32).
Three water molecules (W1, W2, and W3) were noticed occupying the opposite face of the Fe(II) coordination sphere (Fig. 2A). W3 was hydrogen bonded to the side chains of His76 (His59), Asp78 (Asp61), and His212 (His170), closely mimicking the 2-His-1-carboxylate facial triad. W2 was also hydrogen-bonded to the backbone of Ala77 (Ala60). W1, which was displaced by GSH upon its binding, was connected only to the bulk solvent.
Oligomeric Structure of PpPDO2 and MxPDO1b
The asymmetric units of both PpPDO2 and MxPDO1b were composed of two tightly associated monomers in a non-crystallographic, 2-fold manner (Fig. 1, A and B). This dimeric status of PpPDO2 is maintained in solution and was verified by multiangle laser light scattering experiments (Fig. 3). The dimer interface had a symmetrically oriented inter-subunit β-sheet between two C-terminal peptides (Val255–Leu262), resulting in a large area of hydrophobic interaction. The observed dimer interface contributed to stabilizing one face of the substrate binding pocket, indicating the dimer as a functional unit (Fig. 2A).
FIGURE 3.
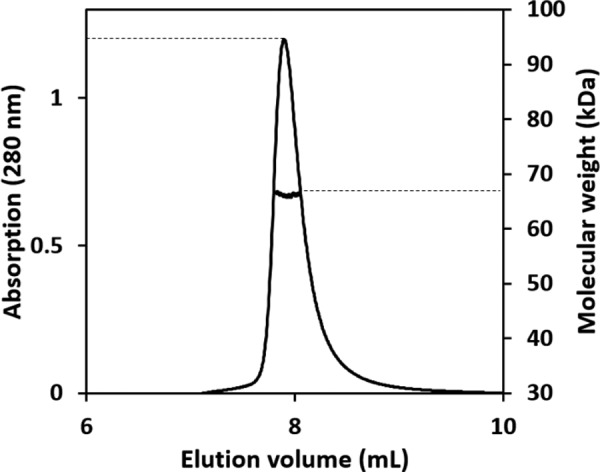
The oligomeric state of PpPDO2 in solution. The elution profile for PpPDO2 was monitored with multiangle laser light scattering and is shown as absorbance (left y axis) and molecular weight (right y axis) versus elution volume (ml). The solid line represents changes in absorption at 280 nm. The thick black cluster in the middle of the peak indicates the calculated molecular mass of 67.4 kDa from the light scattering, illustrating the dimeric nature of PpPDO2.
GSH Complex
The Fo − Fc map of GSH-soaked PpPDO2 crystals showed the corresponding electron density for the bound molecules located on the other side of the facial triad open to the bulk solvent (Fig. 2B). The r.m.s. deviation value for Cα atoms between apo-form and GSH complex PpPDO2 was 0.3 Å, indicating little change upon GSH binding (Fig. 2C). One of the metal-coordinating water molecules (W1) was replaced by GSH and the distance between the sulfur atom of GSH and Fe(II) was 2.5 Å (Fig. 2, A and B). A deep binding pocket was established by Asp78, His149, Asp170, Arg181, Tyr214, Arg250, Arg253, Val261, and Leu262. Noticeably, the apo-form structure of PpPDO2 had a few water molecules at equivalent positions of the GSH ligand (Fig. 2A). The backbone of Arg181 and the side chains of Arg181, Tyr214, Arg250, and Arg253 were in direct interaction with GSH (Fig. 2B). Specifically, a glycinyl carboxyl oxygen of GSH was electrostatically interacting with the guanidinium groups of the Arg250 and Arg253 side chains, and the two backbone carbonyl oxygen atoms of GSH were within hydrogen bond distance from the backbone nitrogen and guanidinium group of the Arg181 side chain. In addition, the cysteinyl amide hydrogen of GSH established a hydrogen bond with the phenolic hydroxyl group of Tyr214. The glutamyl carboxyl group of GSH displayed both direct and indirect interactions through a water molecule-mediated hydrogen bond with neighboring residues' backbone atoms. Contrary to the result of our efforts with GSH, our numerous attempts to diffuse GSSO3− into the apo-form PpPDO2 crystals were not successful, resulting in non-diffracting crystals or diffraction data of very low resolution.
ITC Data
We used ITC to confirm the differential binding affinities among the PpPDO2 product and two of its analogs. A small amount of heat was released when GSH or GSSO3− was titrated into PpPDO2-containing solutions (ΔH = −0.3 and −2.2 kcal mol−1, respectively) (Table 2, Fig. 4). Further analysis of the ITC data revealed favorable entropic contributions for both GSH and GSSO3− binding (ΔS = 25.5 and 15.1 cal mol−1 K−1), probably indicating that several solvent molecules were displaced from the pocket upon binding of either compound. Supporting this, there were several water molecules in the substrate-binding pocket of the apo-form crystal structure of PpPDO2. The calculated Kd values for GSH and GSSO3− (Table 2) were 1.6 and 12 μm, respectively. PpPDO2 did not appear to have affinity for GSSCH3 (Fig. 4).
TABLE 2.
Thermodynamic parameters for binding substrate analogs by PpPDO2
| Kd | ΔH | ΔS | |
|---|---|---|---|
| μm | kcal mol−1 | cal mol−1 K−1 | |
| GSH | 1.6 ± 0.6 | −0.31 ± 0.01 | 25.5 |
| GSSO3− | 12 ± 3 | −2.2 ± 0.2 | 15.1 |
| GSSCH3 | NDa | ND | ND |
a ND, not determined.
FIGURE 4.
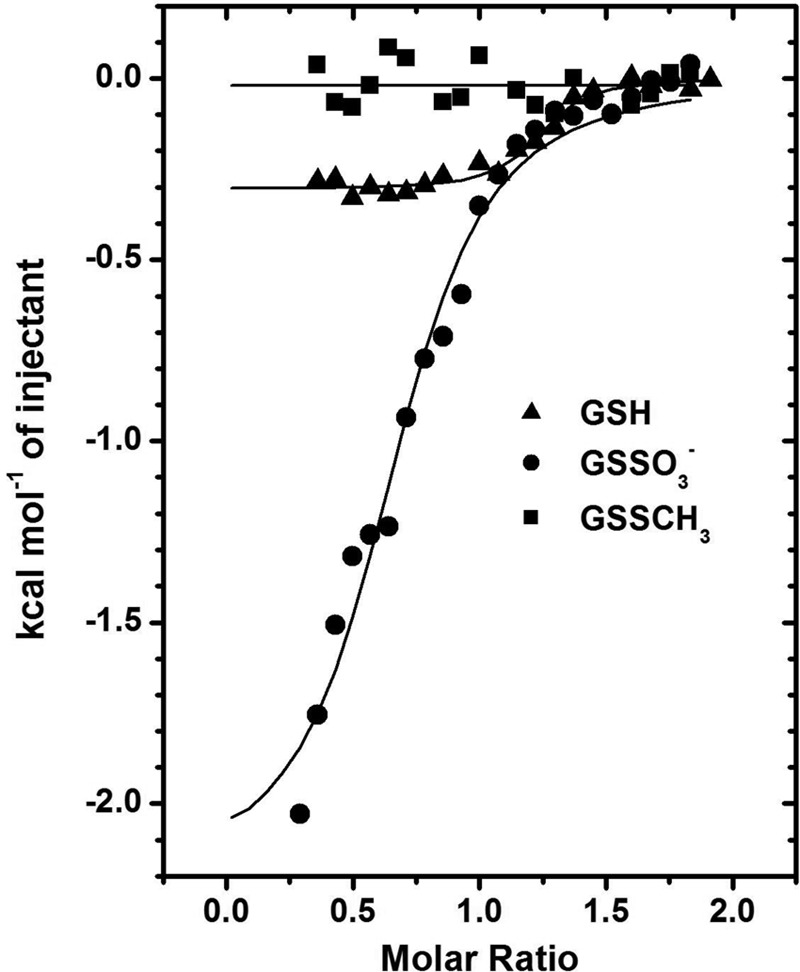
Measurements of PpPDO2 binding for the product and substrate analogs via ITC. The trends of heat released during serial injections of GSH (▴), GSSO3− (●), and GSSCH3 (■) into a solution of PpPDO2 are presented. The data reveal low micromolar affinity for GSH and GSSO3−, but no apparent binding of GSSCH3.
Discussion
To establish the proper classification of bacterial PDOs and to identify any unique signatures, comparisons with available structures in the Protein Data Bank (PDB) were executed using Dali (24) and BLAST (25) searches. The superimposed three-dimensional structures of PpPDO2, MxPDO1b, and the related enzymes displayed that most of the regions with high sequence similarity were located around the residues of the facial triad. Significantly, the Fe(II)-coordinating residues and critical residues in the second coordination sphere were completely conserved among those closely related PDOs, including AtPDO and hPDO. Those residues and their physical arrangements may be conserved to maintain the orientation of Fe(II)-coordinating residues and water molecules, as well as for facilitating rearrangements of electrons and protons of substrates.
As shown by our Dali and BLAST searches, both PpPDO2 and MxPDO1b share a high level of sequence identity with PDOs and glyoxalases II (Fig. 5). In addition, their β-lactamase folds are superimposable with those of PDOs and glyoxalases II from various species. Their metal-binding centers also closely resemble that of the first metal-binding site of the glyoxalases II (Fig. 6), which use di-metallic reaction centers to hydrolyze S-lactoylglutathione.
FIGURE 5.

Multiple sequence alignment of four PDOs and three glyoxalases II. Included in the alignment are PDOs from P. putida (PpPDO2), M. xanthus (MxPDO1b), human (hPDO), and Arabidopsis (AtPDO), as well as glyoxalase II enzymes from human (hGloB), A. thaliana (AtGloB), and Salmonella typhimurium (StGloB). The α-helices and β-strands are highlighted in red and yellow, respectively. Functionally significant residues are bolded, and residues involved in binding metals are underlined. Single conserved residues are marked with an asterisk, whereas conserved motifs are enclosed by a box. Multiple sequence alignment was performed with CLUSTALW2 using a BLOSUM weighting matrix. Secondary structure for each enzyme was calculated from PDB coordinates using DSSP (version 2.0)(29).
FIGURE 6.

Metal-binding and secondary coordination sphere architectures of PDOs and glyoxalases II. The two structures shown at the top panel correspond to PpPDO2 (A) and MxPDO1b (B). At the bottom panel are glyoxalase II enzymes from human (C) and S. typhimurium (D). The extensive hydrogen bond network that appears to be characteristic of PDOs is substituted with a conserved, one-turn α-helix (resides 56–60 of human and 55–59 of S. typhimurium) in the glyoxalases II that is involved in coordinating the second metal.
The secondary coordination sphere with hydrogen bond networks observed in both PpPDO2 and MxPDO1b are essentially the same among the PDOs (Fig. 6A, B). This similarity, however, does not extend to the glyoxalases II, because the three-dimensional structures of those enzymes do not show any secondary coordination sphere with hydrogen bond networks. The corresponding residues in the glyoxalases II are instead involved in coordinating a second metal ion (Fig. 6, C and D). Given the fact that most members of the metallo-β-lactamase superfamily are hydrolytic enzymes with binuclear metal centers (26), PDOs likely evolved from a hydrolytic enzyme that has two coordinated metal ions. Evolution has led to loss of affinity for a second metal and gain of the coordination for water molecules together with a hydrogen bond network in their second coordination sphere, which is likely critical to Fe(II) positioning and catalysis, as we proposed in the case of 2,6-dichloro-p-hydroquinone 1,2-dioxygenase (27). Consequently, the metal-binding center is highly conserved among PDOs.
Both primary and tertiary structures of MxPDO1b closely resemble hPDO and AtPDO in regard to the GS-moiety binding pocket and to Fe(II) coordination, thus supporting our classification scheme for type I and type II PDOs (14). On the contrary, the structure of PpPDO2 is more similar to those of the glyoxalases II than to hPDO and AtPDO in terms of its three specific arginine residues (Arg181, Arg250, and Arg253) electrostatically interacting with GSH (Figs. 2B and 7). The structure of PpPDO2 in complex with GSH showed that the two interior carbonyl oxygens of GSH were anchored by Arg181, and the glycinyl carboxyl group of GSH electrostatically interacted with two other arginine residues (Arg250 and Arg253) (Fig. 2B). Significantly, both location and orientation of the bound GSH in PpPDO2 were similar to those in the crystal structure of human glyoxalase II (PDB code 1QH5 (28)) (Fig. 7). Furthermore, the positions of the three specified arginine residues in PpPDO2 were conserved and superimposable when the three-dimensional structures of glyoxalases II and PpPDO2 were aligned. However, due to large insertions or deletions just before those basic residues among PDOs and glyoxalases II, all alignment programs used failed to capture that structural/functional conservation, which was only possible to grasp with the aid of tertiary structure alignment. In type I PDOs, including hPDO, AtPDO, and MxPDO1b, there is a single basic residue (Arg214 of hPDO or Arg189 of MxPDO1b) located in the set of conserved residues NPR(L/V), suggesting the possibility of convergent evolution to accommodate the binding of GSSH. As shown in Fig. 5, many sequence alignment programs aligned Arg253 of PpPDO2 with Arg221 of MxPDO1b and the Arg246 of hPDO, which turned out meaningless. The latter two residues are located in a different α-helix that is irrelevant to substrate binding, and their guanidinium groups instead point to the bulk solvent.
FIGURE 7.

Active site representations of PpPDO2 and human glyoxalase II in complex with GSH. A, the binding pocket of human glyoxalase II (Protein Data Bank code 1QH5) with a bound GSH molecule is shown. The two gray spheres represent the catalytic Zn(II) ions (28). B, for comparison, the GSH-binding modes and participating residues of PpPDO2 (green) and human glyoxalase II (gray) were superimposed. Residue labels that correspond to residues of the glyoxalase binding pocket are indicated in parentheses. Bond lengths are written near their respective bond representations. This figure was generated using PyMOL Molecular Graphics System, version 1.7.0.3 (Schrödinger, LLC).
Significantly, the His-rich region of the glyoxalases II includes a one-turn α-helix, and the 1st, 3rd, 5th, and 6th residues in its HHHXDH motif are involved in metal coordination (Figs. 5 and 6, C and D). On the contrary, in the corresponding region in PDOs, only the first histidine of HXHXDH is involved in Fe(II) coordination (Figs. 5 and 7, A and B). The remaining residues are involved in the hydrogen bond network, with the Fe(II)-coordinating water molecules and neighboring residues establishing the core of the secondary coordination sphere. Therefore, HHHXDH versus HXHXDH could serve as a signature sequence for distinguishing between glyoxalases II and PDOs.
Nonsense mutations of Gln12 and Gln63 and missense mutations of Tyr38, Leu55, Thr136, Thr152, Cys161, Arg163, Thr164, Asp165, Leu185, and Asp196 of hPDO have been implicated in ethylmalonic encephalopathy (6, 9, 30). These residues are conserved among PDOs (Fig. 5). The structure of PpPDO2 in complex with GSH suggests that the backbone amide and side chain of Arg163 in hPDO (Arg181 in PpPDO2) jointly anchor the flanking carbonyl oxygens of GSH (Fig. 2B). The residues near Arg163 of hPDO showed similarity with those of the glyoxalases II (Fig. 5). Tyr38 of hPDO (Fig. 1C), which is conserved among PDOs, establishes a side chain-mediated hydrogen bond to the backbone carbonyl oxygen of the residue located at the β-strand of the opposite side in the central β-sheet. Consequently, the observed Y38C mutation can impact the overall folding of hPDO and proper configuration of the Fe(II) site. Thr136, which is also conserved, is located next to the Fe(II)-coordinating histidine residue, forming a β-bulge structure through its side chain hydrogen bond. Thus, the T136A/T136G mutations will cause the loss of proper geometry for Fe(II) coordination. Leu185, which is conserved in PDOs and glyoxalases II, interacts with neighboring hydrophobic residues. Alteration to a charged residue such as arginine (L185R) will greatly reduce the stability of enzymes or result in misfolding. It may also be possible that conservation of hydrophobic residues in this region is necessary to shield iron-bound oxygen from bulk solvent, thus preventing formation of reactive oxygen species in non-productive side reactions.
Active Site of PpPDO2 and Plausible Reaction Mechanism
Both apo-form and GSH binary complex structures of PpPDO2 are suggestive of a likely mechanism. The resting state of PpPDO2 in its Fe(II) form is coordinated by two histidines, an aspartate, and three water molecules. The incoming substrate, GSSH, replaces the solvent-exposed water molecule (W1 in Fig. 2A) coordinated to Fe(II) and liberates several water molecules from the GSSH-binding pocket (Fig. 2, A and B). Upon docking into the binding pocket, the carboxyl groups of GSSH are anchored by hydrogen bonds and salt bridges to the backbones and side chains of the residues that constitute the pocket.
The pKa of the sulfhydryl group in GSSH has been estimated to be 7.2, which is lower than that of GSH (31). Thus, it is likely deprotonated or being deprotonated as it approaches Fe(II), which is facilitated by the Lewis acid nature of the metal, the entropic effect of the departing water molecules, and the lower dielectric constant upon departure of those waters. In addition, the Fe(II)-coordinating water molecule, W3 (Fig. 2A), could act as a proton relay to facilitate the deprotonation of GSSH.
Association of substrate into the active site should change the electronic properties of Fe(II) and the hydrogen bond network, which triggers replacement of a water molecule in the primary coordination sphere by O2. It is tempting to speculate that a coordinating water (W2), which is proximal to Phe184, is replaced by O2 (Fig. 2A). Phe184 could act as a gate, because two alternate conformations of its side chain were detected in the electron density maps of both apo-form PpPDO2 and the enzyme in complex with GSH. The two conformers lend plausibility to a mechanism for transient exposure of the hydrophobic environment to the bulk solvent area.
Conclusion
Our results for PpPDO2 and MxPDO1b indicate a close relationship among PDOs, especially around the Fe(II) binding site. Although the shape and location of their binding sites for the GS moiety are superimposable between the type I and type II of PDOs, the key amino acid residues for substrate binding originated from different parts of the proteins, supporting the idea for grouping of them into individual subclasses. Our structural characterization also helps to gain a comprehensive picture of their binding and catalytic mechanisms that is likely conserved among PDOs.
Author Contributions
C. K., L. X., and M. X. conceived and coordinated the study. C. K., L. X., S. A. S., K. M. L., M. X., and C. P. wrote the paper. X. W., Y. X., and H. L. generated clones of PDO genes. S. A. S., X. W., and P. J. D. expressed, purified, and crystallized the proteins. C. P. and M. X. performed the synthesis of GSSO3− and GSSCH3, which were used for ITC and protein crystal soaking. C. P. performed NMR to assess purity of the synthetic compounds. S. A. S. and K. M. L. obtained x-ray diffraction data for PpPDO2 and MxPDO1b. S. A. S. and P. J. D. performed ITC and multiangle light scattering. S. A. S. solved the molecular structures of PpPDO2 and MxPDO1b. All authors contributed to experimental design, reviewed the results, and approved the final version of the manuscript.
This work was supported in part by National Science Foundation Grants MCB 1021148 and DBI 0959778 and the M. J. Murdock Charitable Trust. The authors declare that they have no conflicts of interest with the contents of this article.
- PDO
- persulfide dioxygenase
- ITC
- isothermal titration calorimetry
- r.m.s.
- root mean square.
References
- 1. Coletta C., Papapetropoulos A., Erdelyi K., Olah G., Módis K., Panopoulos P., Asimakopoulou A., Gerö D., Sharina I., Martin E., Szabo C. (2012) Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. U.S.A. 109, 9161–9166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kolluru G. K., Shen X., Kevil C. G. (2013) A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 1, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh S., Banerjee R. (2011) PLP-dependent H2S biogenesis. Biochim. Biophys. Acta 1814, 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackson M. R., Melideo S. L., Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- 5. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kabil O., Banerjee R. (2012) Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Melideo S. L., Jackson M. R., Jorns M. S. (2014) Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53, 4739–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hildebrandt T. M., Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 9. Tiranti V., Briem E., Lamantea E., Mineri R., Papaleo E., De Gioia L., Forlani F., Rinaldo P., Dickson P., Abu-Libdeh B., Cindro-Heberle L., Owaidha M., Jack R. M., Christensen E., Burlina A., Zeviani M. (2006) ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 43, 340–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holdorf M. M., Owen H. A., Lieber S. R., Yuan L., Adams N., Dabney-Smith C., Makaroff C. A. (2012) Arabidopsis ETHE1 encodes a sulfur dioxygenase that is essential for embryo and endosperm development. Plant Physiol. 160, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rohwerder T., Sand W. (2003) The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology 149, 1699–1710 [DOI] [PubMed] [Google Scholar]
- 12. Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205 [DOI] [PubMed] [Google Scholar]
- 13. Bugg T. (2001) Oxygenases: mechanisms and structural motifs for O2 activation. Curr. Opin. Chem. Biol. 5, 550–555 [DOI] [PubMed] [Google Scholar]
- 14. Liu H., Xin Y., Xun L. (2014) Distribution, diversity, and activities of sulfur dioxygenases in heterotrophic bacteria. Appl. Environ. Microbiol. 80, 1799–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pettinati I., Brem J., McDonough M. A., Schofield C. J. (2015) Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Hum. Mol. Genet. 24, 2458–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCoy J. G., Bingman C. A., Bitto E., Holdorf M. M., Makaroff C. A., Phillips G. N., Jr. (2006) Structure of an ethe1-like protein from Arabidopsis thaliana. Acta Crystallogr. D Biol. Crystallogr. 62, 964–970 [DOI] [PubMed] [Google Scholar]
- 17. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18. Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 19. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Webb B. N., Ballinger J. W., Kim E., Belchik S. M., Lam K. S., Youn B., Nissen M. S., Xun L., Kang C. (2010) Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Biol. Chem. 285, 2014–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arisawa M., Suwa A., Yamaguchi M. (2006) RhCl3-catalyzed disulfide exchange reaction using water solvent in homogeneous and heterogeneous systems. J. Organomet. Chem. 691, 1159–1168 [Google Scholar]
- 22. Choi L.-S., Bayley H. (2012) S-Nitrosothiol chemistry at the single-molecule level. Angew. Chem. Int. Ed. Engl. 51, 7972–7976 [DOI] [PubMed] [Google Scholar]
- 23. Hegg E. L., Que L., Jr. (1997) The 2-His-1-carboxylate facial triad: an emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 250, 625–629 [DOI] [PubMed] [Google Scholar]
- 24. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 25. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bebrone C. (2007) Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 74, 1686–1701 [DOI] [PubMed] [Google Scholar]
- 27. Hayes R. P., Green A. R., Nissen M. S., Lewis K. M., Xun L., Kang C. (2013) Structural characterization of 2,6-dichloro-p-hydroquinone 1,2-dioxygenase (PcpA), a new type of aromatic ring-cleavage enzyme. Mol. Microbiol. 88, 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cameron A. D., Ridderström M., Olin B., Mannervik B. (1999) Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Structure 7, 1067–1078 [DOI] [PubMed] [Google Scholar]
- 29. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 30. Mineri R., Rimoldi M., Burlina A. B., Koskull S., Perletti C., Heese B., von Döbeln U., Mereghetti P., Di Meo I., Invernizzi F., Zeviani M., Uziel G., Tiranti V. (2008) Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy. J. Med. Genet. 45, 473–478 [DOI] [PubMed] [Google Scholar]
- 31. Stockdreher Y., Venceslau S. S., Josten M., Sahl H. G., Pereira I. A., Dahl C. (2012) Cytoplasmic sulfurtransferases in the purple sulfur bacterium Allochromatium vinosum: evidence for sulfur transfer from DsrEFH to DsrC. PLoS One 7, e40785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]