

Background: Osteocyte apoptosis precedes bone loss induced by reduced mechanical forces, and unloading increases RANKL expression.
Results: Inhibition of osteocyte apoptosis prevents increased osteocytic RANKL but not bone loss induced by tail suspension.
Conclusion: Prevention of apoptosis and reduction of osteocytic RANKL are not sufficient to stop unloading-induced bone loss.
Significance: RANKL from non-osteocytic sources contributes to bone loss induced by reduced mechanical forces.
Keywords: apoptosis, bone, osteoblast, osteoclast, osteocyte, RANKL, immobilization, resorption
Abstract
Apoptosis of osteocytes and osteoblasts precedes bone resorption and bone loss with reduced mechanical stimulation, and receptor activator of NF-κB ligand (RANKL) expression is increased with unloading in mice. Because osteocytes are major RANKL producers, we hypothesized that apoptotic osteocytes signal to neighboring osteocytes to increase RANKL expression, which, in turn, increases osteoclastogenesis and bone resorption. The traditional bisphosphonate (BP) alendronate (Aln) or IG9402, a BP analog that does not inhibit resorption, prevented the increase in osteocyte apoptosis and osteocytic RANKL expression. The BPs also inhibited osteoblast apoptosis but did not prevent the increase in osteoblastic RANKL. Unloaded mice exhibited high serum levels of the bone resorption marker C-telopeptide fragments of type I collagen (CTX), elevated osteoclastogenesis, and increased osteoclasts in bone. Aln, but not IG9402, prevented all of these effects. In addition, Aln prevented the reduction in spinal and femoral bone mineral density, spinal bone volume/tissue volume, trabecular thickness, mechanical strength, and material strength induced by unloading. Although IG9402 did not prevent the loss of bone mass, it partially prevented the loss of strength, suggesting a contribution of osteocyte viability to strength independent of bone mass. These results demonstrate that osteocyte apoptosis leads to increased osteocytic RANKL. However, blockade of these events is not sufficient to restrain osteoclast formation, inhibit resorption, or stop bone loss induced by skeletal unloading.
Introduction
Mechanical forces have a positive effect on the skeleton and, in contrast, conditions under which decreased strain is imposed on bone lead to loss of bone mass and an increased risk of fractures (1). Such conditions include prolonged bed rest, physical inactivity, motor paralysis because of denervation, and reduced gravity. With the rise in the aging population, the predominance of a sedentary life style, and the prospective of commercialization of space flights, there is an increased need for understanding the mechanisms underlying the loss of bone with skeletal unloading and for new approaches to combat it.
Osteocytes are recognized mechanosensory cells of bone that detect the need for bone augmentation or reduction in response to changes in mechanical stimulation (2). Recent advances in osteocyte biology demonstrate that osteocytes produce molecules that regulate the generation and activity of both osteoblasts and osteoclasts (3). The protein sclerostin encoded by the gene Sost is secreted by osteocytes and inhibits bone formation by osteoblasts, providing the first example of a molecular mediator produced by matrix-embedded osteocytes that regulate the activity of cells on the bone surface (4). Sost/sclerostin expression is down-regulated by mechanical stimulation, an event that is required for the anabolic response of the skeleton to loading (5, 6). Further, sclerostin expression is increased with unloading, potentially explaining the decreased bone formation that ensues with disuse osteoporosis (6). Osteocytes also produce pro- and anti-osteoclastogenic cytokines that regulate bone resorption, including osteoprotegerin (OPG)6 and receptor activator of NF-κB ligand (RANKL) (7–11). In particular, mice lacking the RANKL gene primarily from osteocytes exhibit a progressive increase in bone mass because of a reduced number of osteoclasts, demonstrating that osteocytic RANKL is critical for bone remodeling (10, 11). However, the mechanisms that regulate RANKL and OPG expression in osteocytes in response to mechanical forces (or lack thereof) are not known.
Earlier work demonstrated that physiological levels of loading maintain osteocyte viability in vitro and in vivo (12–15). On the other hand, unloaded bone or bone subjected to excessive loading exhibit an increased prevalence of osteocyte apoptosis (14, 16). Furthermore, apoptosis of osteocytes precedes temporally and spatially osteoclast-mediated resorption because apoptotic osteocytes are found in bone before any detectable increase in osteoclasts and accumulate in areas that will be subsequently resorbed (14, 16). Moreover, the targeted ablation of osteocytes by genetic means is sufficient to increase RANKL and to induce osteoclast recruitment and bone resorption (17). However, whether the increase in osteocyte apoptosis and RANKL expression induced by unloading are mechanistically linked remains unknown. In addition, whether preserving osteocyte viability alters RANKL expression, osteoclast generation, and/or bone resorption in unloaded bones has not been explored before.
In this study, we addressed these questions using two bisphosphonate (BP) analogs that, as we showed previously, effectively block osteocyte and osteoblast apoptosis in vitro and apoptosis induced by glucocorticoid excess in vivo (18–21). The mechanism by which these agents prevent apoptosis of osteoblastic cells involves opening of connexin 43 hemichannels and activation of the prosurvival Src/ERK signaling pathway (21, 22). Traditional BPs such as alendronate also inhibit the mevalonate pathway and intoxicate osteoclasts, thereby inhibiting resorption (23). In contrast, the unique BP analog IG9402 only acts on osteocytes and osteoblasts and does not inhibit the mevalonate pathway or bone resorption (19, 21, 24). Furthermore, daily injections with alendronate decreases bone formation as a consequence of its antiresorptive activity (21). This was evidenced by reduced plasma levels of CTX and osteocalcin and mineralizing surface/bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate/bone surface (BFR/BS) and osteoblast number in cancellous bone. On the other hand, IG9402 did not affect any of these parameters in vehicle-treated mice. Moreover, IG9402 prevented the decrease in bone formation and osteoblast number in glucocorticoid-treated mice. Using these two pharmacologic tools, we dissected the contribution of osteocyte and osteoblast apoptosis to the changes in RANKL and OPG expression and to the increased resorption triggered by skeletal unloading.
We found that both BPs inhibited osteocyte and osteoblast apoptosis and decreased RANKL expression in osteocytes. In contrast, even when osteoblast apoptosis induced by unloading was inhibited by the bisphosphonates, the increased RANKL expression in osteoblasts was not reversed by the drugs. Alendronate also prevented the elevation in osteoclasts, bone resorption, and bone loss induced by unloading. In contrast, IG9402 did not prevent the increase in osteoclasts or bone resorption or the decrease in bone mass. These findings show that osteocyte apoptosis does indeed control osteocytic RANKL expression and that maintaining osteocyte viability is not sufficient to restrain resorption or prevent the loss of bone induced by unloading. Therefore, RANKL derived from non-osteocytic sources (likely osteoblasts) mediates osteoclastogenesis and the bone loss resulting from lack of mechanical forces.
Experimental Procedures
Tail Suspension
Female 4-month-old C57BL/6 mice (Harlan) were used. Mice were kept in cages under standard laboratory conditions with a 12-h dark, 12-h light cycle, a constant temperature of 20 °C, and a humidity of 48% for 2 weeks for acclimation. Mice were fed a standard rodent diet (Purina FormulaLab Diet 5008) with water ad libitum. Skeletal unloading was achieved using the tail suspension model described previously (14). The height was adjusted to maintain the mice in an ∼30° head-down tilt. Two tail-suspended animals were housed per cage. Fully ambulatory control mice were caged in groups of five and pair-fed with the tail-suspended mice. All protocols were approved by the Indiana University School of Medicine.
Bone Resorption Marker
CTX were measured using an enzyme-linked immunoadsorbent assay (RatLaps, Immunodiagnostic Systems Inc., Fountain Hills, AZ), as published previously (21).
Ex Vivo Osteoclastogenesis
Bone marrow cells collected from tibiae and femora of six mice of each group were combined and seeded in triplicates at a density of 50 × 103 cells/cm2. Cells were cultured in the presence of 30 ng/ml macrophage colony-stimulating factor (M-CSF) and 30 ng/ml soluble RANKL for 4 days. Cells when then fixed and stained for tartrate-resistant acid phosphatase (TRAP) as published previously (19).
TUNEL, Immunohistochemistry, and TRAP Staining
Vertebrae (L3-L4) were decalcified and paraffin-embedded as described previously (25). Consecutive 5-μm-thick sections were used for osteocyte apoptosis and for analysis of protein expression by immunohistochemistry. For apoptosis, a modified version of a commercial TUNEL kit (EMD Millipore, Billerica, MA) was used, and sections were counterstained with 2% methyl green, as described previously (20). Immunohistochemistry was performed in consecutive sections using goat anti-RANKL and anti-OPG antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA) with prior antigen retrieval (DeCal Retrieval Solution, BioGenex, San Ramon, CA) and followed by signal amplification (ABC kit, Vector Laboratories, Burlingame, CA). Non-immune IgG was used as a negative control. Cells were score as either positive (brown staining) or negative (blue/green staining) for each antigen. To visualize osteoclasts on the cancellous bone surface, sections were stained for TRAP and counterstained with toluidine blue, as described previously (25). Only TRAP-positive cells containing more than two nuclei were counted.
MLO-Y4 Osteocytic and Ob-6 Osteoblastic Cell Culture
Cells were cultured as published previously (18, 26). For gene analysis and trypan blue uptake, cells were treated with vehicle or 10−7 m alendronate or IG9402 for 1 h, followed by addition of vehicle, dexamethasone (10−6 m), etoposide (20 μM), or H2O2 (100 μM) in growing medium or with medium without serum for the indicated time points. Cells were then trypsinized to determine the percentage of dead cells by trypan blue uptake or lysed to isolate mRNA, as published previously (26). For the experiments using conditioned medium, cells were treated with vehicle or bisphosphonate for 1 h, followed by addition of H2O2 or by changing the medium to remove the serum. 6, 24, and 48 h later, culture supernatants were collected and added to cells plated the day before (together with growing medium, 1:1). 6 and 24 h later, cells were lysed to collect mRNA.
RNA Preparation and Real-time PCR
RNA was isolated from MLO-Y4 cells using TRIzol reagent (Invitrogen), as described previously (27). Quantitative PCR was performed using the housekeeping gene ribosomal protein S2 (ChoB) and the ΔCt method. Primers and probes were designed using the Assay Design Center (Roche Applied Science) or were available commercially (Applied Biosystems, Foster City, CA).
Bone Mineral Density (BMD) and Microcomputed Tomography (MicroCT) Analysis
BMD of the total body (excluding the head), spine (L1-L6), and femur was determined by dual energy x-ray absorptiometry using a PIXImus II densitometer (GE Medical Systems, Lunar Division, Madison, WI), as described previously (28). For microCT analysis, L5 vertebrae were dissected, cleaned of soft tissue, fixed, and stored in 70% ethanol until analyzed at 6-μm resolution using a Skyscan 1172 microCT (SkyScan, Kontich, Belgium) (28). Analyses were conducted on cancellous bone to measure bone volume/tissue volume (BV/TV), trabecular number (Tb.N), spacing (Tb.Sp), and thickness (Tb.Th); on cortical bone to measure the thickness of the posterior shell (cortical thickness); and on cortical and cancellous combined normalized to total area (total BV/TV).
Biomechanical Testing
Compression strength was measured in lumbar vertebrae (L6) using a single-column material testing machine and a calibrated tension/compression load cell (model 5542, Instron Corp., Norwood, MA) (21). Bone dimensions were measured with a digital caliper at a resolution of 0.01 mm (Mitutoyo, catalog no. 500-196, Ace Tools, Wantagh, NY).
Statistical Analysis
Statistical analysis was performed using SigmaStat (SPSS Science, Chicago, IL). Data were analyzed by two-way ANOVA, and, if a significant main effect or interaction was found, we examined it closely by performing pairwise multiple comparisons with Bonferroni correction (29). All values are reported as the mean ± S.D.
Results
Increased Osteocyte and Osteoblast Apoptosis and Accumulation of RANKL-positive Osteocytes Induced by Lack of Mechanical Stimulation Is Prevented by Bisphosphonates Independently of Their Antiresorptive Potential
Our previous work has shown that tail suspension consistently decreases bone mass and strength and increases osteoblast and osteocyte apoptosis in lumbar vertebral bone of Swiss Webster and C57BL/6 mice (14). We therefore examined the effect of BP administration on vertebral bone of unloaded mice. Tail suspension induced an increase in the prevalence of apoptotic osteocytes and osteoblasts in cancellous bone of the lumbar vertebra (Fig. 1). As found previously for glucocorticoid-induced apoptosis (21), daily injections of alendronate or IG9402 preserved both osteoblast and osteocyte viability. The prevalence of RANKL-positive osteocytes was increased significantly in vertebral bone from ∼7% in controls to 14% in mice subjected to tail suspension (Fig. 2A). This increase was completely prevented by daily administration of alendronate or IG9402. Tail suspension also increased the prevalence of RANKL-positive osteoblasts from ∼13% in controls to 23% in mice subjected to tail suspension, but neither alendronate nor IG9402 administration prevented this effect. The prevalence of OPG-positive osteocytes, which ranged from 19–25% in ambulatory control mice, was decreased by tail suspension (10–12%), independent of whether the animals received saline, alendronate, or IG9402 (Fig. 2B). On the other hand, the prevalence of OPG-positive osteoblasts was decreased in mice subjected to tail suspension and receiving vehicle or IG9402 (∼37 and 17% OPG-positive osteoblasts for control and tail-suspended mice, respectively), whereas alendronate administration reversed the decrease in OPG-expressing osteoblasts (∼25 and 38% OPG-positive osteoblasts for alendronate-treated control and tail-suspended mice, respectively). On the other hand, OPG expression in the bone marrow was not altered by any of the treatments. Taken together, this evidence suggests that signals from apoptotic osteocytes increase RANKL expression in osteocytes without modifying OPG expression and that the regulation of RANKL and OPG expression in osteoblasts is independent of changes in cell survival.
FIGURE 1.
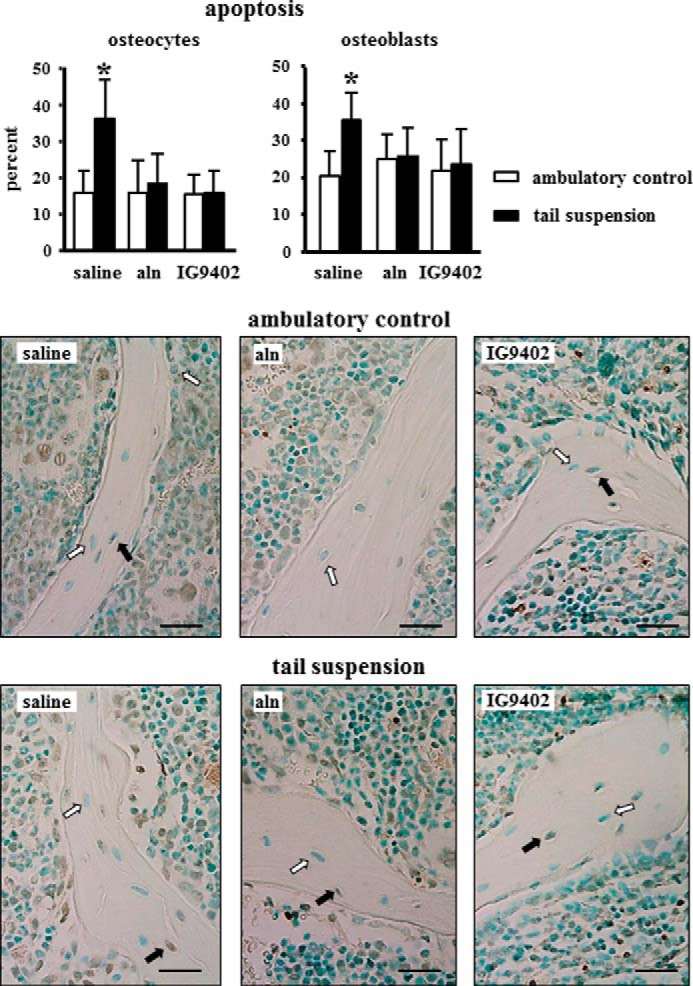
Alendronate and IG9402 prevent immobilization-induced osteocyte and osteoblast apoptosis. The prevalence of osteocyte and osteoblast apoptosis was determined in vertebral bone sections stained for TUNEL. Error bars represent the mean ± S.D. of five to nine replicas. *, p < 0.05 versus the respective ambulatory control by two-way ANOVA. Images are representative of sections with alive (white arrows) or apoptotic (black arrows) osteocytes and osteoblasts. Scale bars = 25 μm.
FIGURE 2.

Prevention of osteocyte apoptosis is associated with reduced prevalence of RANKL-positive osteocytes and no change in OPG-positive osteocytes in immobilized mice. A and B, the prevalence of RANKL- (A) and OPG-positive (B) osteocytes and osteoblasts was determined in vertebral bone sections stained with the respective antibodies. Error bars represent the mean ± S.D. of three to six replicas. *, p < 0.05 versus the respective ambulatory control mice; #, p < 0.05 versus saline-treated tail-suspended mice; two-way ANOVA. Images are representative of sections with osteocytes negative (white arrows) or positive (black arrows) for the indicated protein. Scale bars = 25 μm. Insets correspond to sections stained with non-immune IgG.
To determine whether inhibition of RANKL expression by alendronate or IG9402 is reproduced in vitro, we treated MLO-Y4 osteocytic cells and Ob-6 osteoblastic cells with different proapoptotic agents (dexamethasone, etoposide, H2O2, and serum starvation) in the presence or absence of bisphosphonates. Cells were cultured for 2, 6, 24, or 48 h with the proapoptotic agents, and RNA was extracted. We found that the proapoptotic agents induced apoptosis of osteocytic cells and that the bisphosphonates prevented apoptosis (data not shown), as published previously (18, 20, 22). However, RANKL expression was not affected either by the proapoptotic agents or by the bisphosphonates. In addition, and to determine whether soluble factors derived from apoptotic cells were able to increase RANKL expression, conditioned medium from MLO-Y4 osteocytic cells or Ob-6 osteoblastic cells treated with H2O2 or undergoing serum starvation was placed on a new set of cells, and RANKL expression was measured after 6 or 24 h. Conditioned media from none of the treatments/times increased RANKL expression in MLO-Y4 cells or Ob-6 cells. Taken together, these findings suggest that bisphosphonates do not regulate RANKL expression directly on osteocytes or osteoblasts and that soluble factors derived from osteocytes or osteoblasts are not sufficient to increase RANKL expression in vitro.
Osteoclastogenesis and Bone Resorption Induced by Tail Suspension Are Prevented by Alendronate but Not by IG9402
The levels of CTX in the circulation were increased in mice subjected to tail suspension (Fig. 3A). Daily alendronate administration reduced CTX levels under ambulatory conditions and prevented the increase observed in tail-suspended mice. On the other hand, IG9402 did not affect circulating CTX levels in either group of mice. Consistent with this, osteoclastogenesis induced ex vivo using non-adherent bone marrow cells isolated from the treated mice was increased in tail-suspended animals, inhibited by alendronate, and not affected by IG9402 (Fig. 2B). Moreover, osteoclast number and surface in the vertebra followed a similar pattern (Fig. 3C). However, there was a small but significant decrease in the number of osteoclast/bone surface in tail-suspended mice treated with IG9402 compared to saline-treated tail-suspended animals. Alendronate administration induced the appearance of giant osteoclasts and small, darkly stained TRAP-positive cells that appeared to be encapsulated in the bone marrow (Fig. 4). These were present independent of whether the mice were subjected to tail suspension or not and are consistent with previous evidence of giant inactive osteoclasts found in humans treated with BPs (30, 31).
FIGURE 3.

Alendronate, but not IG9402, inhibits osteoclastogenesis ex vivo and decreases osteoclast number and function in vivo. A, levels of the resorption marker CTX were measured in serum at the end of the experiment. Error bars represent the mean ± S.D. of 11–13 replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA. B, osteoclasts were generated ex vivo by treating non-adherent bone marrow cells with M-CSF and sRANKL, as indicated under “Experimental Procedures.” Error bars represent the mean ± S.D. of three replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA. Representative images of osteoclasts for each treatment are shown. Scale bars = 100 μm. C, number of osteoclasts per bone perimeter (NOc/BPm) and osteoclast surface per bone surface (OcS/BS) were quantified in vertebral bone sections stained for TRAP. Error bars represent mean ± S.D. of five to six replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA. Representative images of cancellous bone are shown. Scale bars = 50 μm.
FIGURE 4.
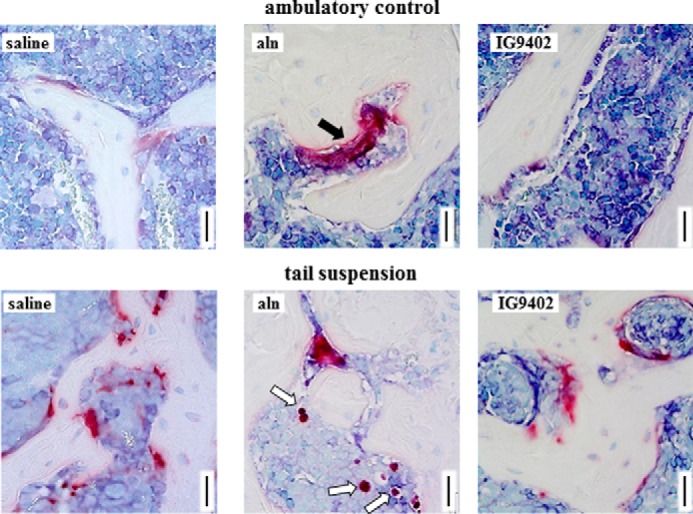
Abnormal osteoclasts accumulate in cancellous bone of alendronate-treated mice. Representative images of vertebral cancellous bone stained for TRAP show the presence of a giant osteoclast (white arrows) in the ambulatory control mice and potential osteoclast remnants (black arrow) in the tail-suspended mice receiving alendronate. Scale bars = 25 μm.
IG9402 Does Not Preserve Bone Mass but Partially Prevents the Decrease in Bone Strength Induced by Tail Suspension in Mice
Tail suspension for 28 days induced a decrease in total, spinal, and femoral BMD (Fig. 5). As described previously (20), daily administration of alendronate increased BMD in all sites in ambulatory controls and prevented bone loss induced by unloading. On the other hand, IG9402 did not have any effect either under ambulatory or tail-suspended conditions. Similar changes were observed by microCT because tail suspension decreased cancellous BV/TV and trabecular number and thickness without changing trabecular spacing (Fig. 6A). Alendronate increased BV/TV and trabecular number and thickness in ambulatory controls and prevented the decrease in tail-suspended mice. IG9402 did not affect the decrease in BV/TV and trabecular thickness observed in tail-suspended mice, but the trabecular number was not different in IG9402-treated, tail-suspended mice compared with ambulatory mice treated with the same BP. Similar changes were observed in vertebral cortical thickness (Fig. 6B) and in total BV/TV (Fig. 6C), which includes both cancellous and cortical bone.
FIGURE 5.
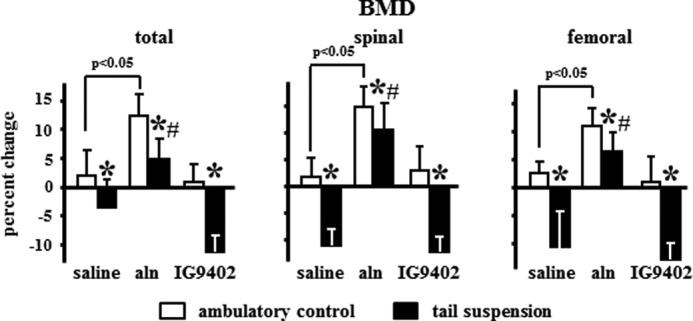
Alendronate, but not IG9402, prevented bone loss induced by immobilization. Shown is the percent change of BMD measured in the total body, spine, and femur. Error bars represent the mean ± S.D. of 8–13 replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA.
FIGURE 6.

IG9402 did not prevent the loss of bone volume and the changes in cancellous bone architecture induced by tail suspension. MicroCT analysis showing percent cancellous BV/TV and trabecular number (TbN), thickness (TbTh), and separation (TbSp) (A); cortical thickness (B), and total (cancellous + cortical) BV/TV in lumbar vertebrae (C). Error bars represent mean ± S.D. of three to seven replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA. Representative microCT images of the cancellous vertebral bone are shown.
Immobilization induced a decrease in vertebral strength at both the whole bone level (maximum load) and estimated material level (ultimate stress), and alendronate prevented these effects (Fig. 7). Therefore, there was no difference in ultimate load, and the difference in ultimate stress was greatly reduced in alendronate-treated, tail-suspended mice compared with ambulatory controls. Despite the lack of effect on bone mass and architecture, IG9402 administration partially prevented the decrease in ultimate load and ultimate stress in tail-suspended mice.
FIGURE 7.
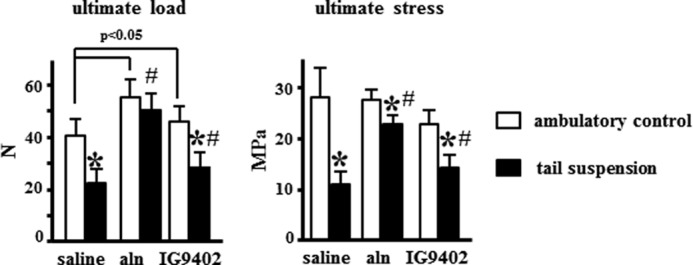
Alendronate and IG9402 prevented the decrease in bone strength induced by tail suspension. Shown are the ultimate load and ultimate stress of the vertebrae as determined by compression testing. Error bars represent mean ± S.D. of 10–13 replicas. *, p < 0.05 versus the respective ambulatory control; #, p < 0.05 versus saline-treated, tail-suspended mice; two-way ANOVA.
Discussion
Accumulation of apoptotic osteocytes precedes osteoclast-mediated resorption and the loss of bone induced by reduced mechanical forces, and RANKL expression is increased with unloading in mice (10, 14). Osteocytes are major producers of RANKL (3). However, the role of these cells in pathological bone resorption is unknown. Here we tested the hypothesis that apoptotic osteocytes signal to neighboring osteocytes to increase RANKL expression, which, in turn, increases osteoclast formation and bone resorption in disuse osteoporosis. Using the murine model of unloading by tail suspension, we found that, indeed, prevention of osteocyte apoptosis blocks the increase in RANKL expression in osteocytes. However, inhibition of apoptosis and reduction of osteocytic RANKL was not sufficient to prevent the increase in osteoclasts or bone resorption or the decrease in bone mass induced by unloading. These findings are consistent with previous studies showing that inhibition of apoptosis induced by fatigue loading results in decreased RANKL expression in osteocytes (32) and add additional support to the notion that there is a cause-effect relationship between osteocyte apoptosis and osteocytic RANKL. In contrast to osteocyte apoptosis induced by fatigue loading, however, the results of this study show that maintaining osteocyte viability is not sufficient to restrain resorption or prevent the loss of bone induced by unloading. These findings suggest that RANKL derived from cells other than osteocytes is critical for the increased osteoclastogenesis and the bone loss resulting from reduced mechanical forces. Because RANKL expression in osteoblasts is still high even when apoptosis is inhibited, our data suggest that osteoblasts contribute to the enhanced resorption in unloaded mice. Furthermore, on the basis of the lack of effect of bisphosphonates on RANKL expression in vitro, either directly or through soluble factors released to the media, we conclude that interactions between apoptotic osteocytes and other cells of the bone/bone marrow microenvironment that cannot be reproduced in vitro might be involved in the regulation of RANKL downstream of osteocyte apoptosis. In this regard, several molecules have been proposed as mediators for the regulation of RANKL expression in osteocytes following apoptosis, including VEGF and HMGB1 (3, 33). However, the identity of the molecules that mediate the increase in resorption in unloaded mice is not known. Future studies are warranted to investigate the cellular and molecular mechanism underlying this phenomenon.
As shown in previous studies (14), unloading increased the prevalence of apoptosis of osteocytes and osteoblasts. We found that BPs (a traditional compound as well as a novel analog that does not affect osteoclasts directly) prevented apoptosis of both osteocytes and osteoblasts. In addition, both BPs prevented the increase in RANKL expression in osteocytes, demonstrating that elevated expression of this cytokine is a consequence of apoptosis in these cells. However, neither agent prevented the increase in RANKL expression induced by unloading in osteoblasts, demonstrating that unloading regulates RANKL expression by different mechanisms in osteocytes versus osteoblasts. The decrease in OPG induced by unloading in osteocytes was not reversed by the BPs, demonstrating that this gene is not regulated by apoptosis. Similarly, IG9402 did not reverse the decrease in OPG induced by unloading in osteoblasts. The increase in osteoblastic OPG observed in alendronate-treated animals suggests that, besides its direct action on osteoclasts, part of the antiresorptive effects of this BP might result from effects on osteoblasts leading to up-regulation of this antiosteoclastogenic cytokine. Indeed, increased OPG levels induced by BPs have been shown previously in patients (34–36) and in bone marrow/mesenchymal stem cells (37–39).
An association between osteocyte apoptosis and osteocytic RANKL has also been reported for fatigue loading (32). In contrast to our study, in which inhibition of apoptosis with BPs did not prevent the increase in bone resorption nor the loss of bone mass, blocking osteocyte apoptosis induced by fatigue loading with the pan caspase inhibitor quinolyl-valyl-O-methylaspartyl-[2,6-difluorophenoxy]-methyl ketone (Q-VD-OPh) completely inhibited intracortical remodeling (40). The same caspase inhibitor also prevented endocortical remodeling induced by ovariectomy (41). In these latter cases, however, resorption was evaluated only at the local level in cortical bone, and systemic effects on circulating CTX or bone mass throughout the skeleton were not reported.
Our work showing that restraining the increase in osteocytic RANKL does not prevent the loss of bone with unloading contrasts with earlier studies demonstrating that mice lacking RANKL in osteocytes are protected from bone loss induced by unloading (10). A potential source for this difference is that the increase in osteoclasts and the decrease in cancellous bone volume induced by tail suspension in the study by O'Brien and colleagues (10) were much smaller compared with our study, likely because of the fact that they used mice from both genders and of mixed background. Another potential explanation is that, in their study, RANKL was completely removed from osteocytes instead of just preventing the increase in osteocytic RANKL induced by tail suspension, as in our study. It is then possible that a basal tonic expression of RANKL in osteocytes is required for the bone loss induced by tail suspension by acting in concert with RANKL expressed by other cells. In addition, the DMP1–10kb-Cre mice used to delete the RANKL gene in the study by O'Brien and colleagues (10) is also expressed in a population of mature osteoblasts. Therefore, RANKL could have been deleted from both osteocytes and some osteoblasts, which, as we show in this study, exhibit increased RANKL expression in unloaded mice. Then, the absence of RANKL from both cell types is what is needed to halt osteoclastogenesis and the loss of bone mass induced by unloading.
Even when IG9402 was not able to block the bone loss induced by unloading, mice receiving this BP analog exhibited more bone strength than vehicle-treated mice. Indeed, the decrease in ultimate load by 46%, and, in maximum stress, by 62% induced by tail suspension in vehicle-treated mice was reduced to a 38% decrease for both parameters in unloaded mice treated with IG9402. Taken together with the survival effect of IG9402 on osteoblasts and osteocytes, this evidence suggests that preservation of osteocyte and osteoblast viability by IG9402 contributes to maintain bone strength independent of changes in bone mass.
Author Contributions
L. I. P., A. R. G., and T. B. conceived and designed the experiments and coordinated the study. L. I. P. and T. B. wrote the paper. L. I. P., A. R. G., H. M. D., K. W. C., H. G., and M. M. performed the experiments. M. R. A. contributed with the analysis and interpretation of the data shown in Figs. 5 and 6. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Emily Atkinson and David Lopez for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK076007 and ARRA supplement S10-RR023710 (to T. B.) and R01AR053643 (to L. I. P.). This work was also supported by Veterans Affairs Merit Award I01BX002104 (to T. B.) and an IUSM Biomedical Research grant (to L. I. P.) and by scholarships from the Conchita Rábago Foundation (to A. R. G. and M. M.), the European Molecular Biology Organization (to M. M.), and Universidad Autónoma de Madrid (to H. G). The authors declare that they have no conflicts of interest with the contents of this article.
- OPG
- osteoprotegerin
- RANKL
- receptor activator of NF-κB ligand
- BP
- bisphosphonate
- CTX
- C-telopeptide fragments of type I collagen
- BMD
- bone mineral density
- microCT
- microcomputed tomography
- BV/TV
- bone volume/tissue volume
- ANOVA
- analysis of variance.
References
- 1. Turner C. H., Warden S. J., Bellido T., Plotkin L. I., Kumar N., Jasiuk I., Danzig J., Robling A. G. (2009) Mechanobiology of the skeleton. Sci. Signal. 2, pt3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonewald L. F. (2011) The amazing osteocyte. J. Bone Miner. Res. 26, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bellido T. (2014) Osteocyte-driven bone remodeling. Calcif. Tissue Int. 94, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baron R., Kneissel M. (2013) WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192 [DOI] [PubMed] [Google Scholar]
- 5. Tu X., Rhee Y., Condon K. W., Bivi N., Allen M. R., Dwyer D., Stolina M., Turner C. H., Robling A. G., Plotkin L. I., Bellido T. (2012) Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robling A. G., Niziolek P. J., Baldridge L. A., Condon K. W., Allen M. R., Alam I., Mantila S. M., Gluhak-Heinrich J., Bellido T. M., Harris S. E., Turner C. H. (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 283, 5866–5875 [DOI] [PubMed] [Google Scholar]
- 7. Kramer I., Halleux C., Keller H., Pegurri M., Gooi J. H., Weber P. B., Feng J. Q., Bonewald L. F., Kneissel M. (2010) Osteocyte Wnt/β-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 30, 3071–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You L., Temiyasathit S., Lee P., Kim C. H., Tummala P., Yao W., Kingery W., Malone A. M., Kwon R. Y., Jacobs C. R. (2008) Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone 42, 172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao S., Zhang Y. K., Harris S., Ahuja S. S., Bonewald L. F. (2002) MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res. 17, 2068–2079 [DOI] [PubMed] [Google Scholar]
- 10. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 12. Bakker A., Klein-Nulend J., Burger E. (2004) Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem. Biophys. Res. Commun. 320, 1163–1168 [DOI] [PubMed] [Google Scholar]
- 13. Plotkin L. I., Mathov I., Aguirre J. I., Parfitt A. M., Manolagas S. C., Bellido T. (2005) Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases and ERKs. Am. J. Physiol. Cell Physiol. 289, C633-C643 [DOI] [PubMed] [Google Scholar]
- 14. Aguirre J. I., Plotkin L. I., Stewart S. A., Weinstein R. S., Parfitt A. M., Manolagas S. C., Bellido T. (2006) Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 21, 605–615 [DOI] [PubMed] [Google Scholar]
- 15. Bellido T. (2010) Antagonistic interplay between mechanical forces and glucocorticoids in bone: a tale of kinases. J. Cell. Biochem. 111, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verborgt O., Gibson G. J., Schaffler M. B. (2000) Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J. Bone Miner. Res. 15, 60–67 [DOI] [PubMed] [Google Scholar]
- 17. Tatsumi S., Ishii K., Amizuka N., Li M., Kobayashi T., Kohno K., Ito M., Takeshita S., Ikeda K. (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464–475 [DOI] [PubMed] [Google Scholar]
- 18. Plotkin L. I., Weinstein R. S., Parfitt A. M., Roberson P. K., Manolagas S. C., Bellido T. (1999) Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J. Clin. Invest. 104, 1363–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plotkin L. I., Manolagas S. C., Bellido T. (2006) Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone 39, 443–452 [DOI] [PubMed] [Google Scholar]
- 20. Plotkin L. I., Lezcano V., Thostenson J., Weinstein R. S., Manolagas S. C., Bellido T. (2008) Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J. Bone Miner. Res. 23, 1712–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plotkin L. I., Bivi N., Bellido T. (2011) A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 49, 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plotkin L. I., Manolagas S. C., Bellido T. (2002) Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 277, 8648–8657 [DOI] [PubMed] [Google Scholar]
- 23. Rogers M. J., Crockett J. C., Coxon F. P., Mönkkönen J. (2011) Biochemical and molecular mechanisms of action of bisphosphonates. Bone 49, 34–41 [DOI] [PubMed] [Google Scholar]
- 24. Sanders J. M., Ghosh S., Chan J. M., Meints G., Wang H., Raker A. M., Song Y., Colantino A., Burzynska A., Kafarski P., Morita C. T., Oldfield E. (2004) Quantitative structure-activity relationships for γδ T cell activation by bisphosphonates. J. Med. Chem. 47, 375–384 [DOI] [PubMed] [Google Scholar]
- 25. Bivi N., Condon K. W., Allen M. R., Farlow N., Passeri G., Brun L. R., Rhee Y., Bellido T., Plotkin L. I. (2012) Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 27, 374–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bivi N., Lezcano V., Romanello M., Bellido T., Plotkin L. I. (2011) Connexin43 interacts with barrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J. Cell. Biochem. 112, 2920–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bivi N., Pacheco-Costa R., Brun L. R., Murphy T. R., Farlow N. R., Robling A. G., Bellido T., Plotkin L. I. (2013) Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J. Orthop. Res. 31, 1075–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Brien C. A., Plotkin L. I., Galli C., Goellner J. J., Gortazar A. R., Allen M. R., Robling A. G., Bouxsein M., Schipani E., Turner C. H., Jilka R. L., Weinstein R. S., Manolagas S. C., Bellido T. (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3, e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riffenburgh R. H. (2006) Statistics in Medicine, 2nd Ed., Academic Press, Burlington, MA [Google Scholar]
- 30. Weinstein R. S., Roberson P. K., Manolagas S. C. (2009) Giant osteoclast formation and long-term oral bisphosphonate therapy. N. Engl. J. Med. 360, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheung M. S., Glorieux F. H., Rauch F. (2009) Large osteoclasts in pediatric osteogenesis imperfecta patients receiving intravenous pamidronate. J. Bone Miner. Res. 24, 669–674 [DOI] [PubMed] [Google Scholar]
- 32. Kennedy O. D., Herman B. C., Laudier D. M., Majeska R. J., Sun H. B., Schaffler M. B. (2012) Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50, 1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Plotkin L. I. (2014) Apoptotic osteocytes and the control of targeted bone resorption. Curr. Osteoporos. Rep. 12, 121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szymczak J., Bohdanowicz-Pawlak A. (2013) Osteoprotegerin, RANKL, and bone turnover in primary hyperparathyroidism: the effect of parathyroidectomy and treatment with alendronate. Horm. Metab Res. 45, 759–764 [DOI] [PubMed] [Google Scholar]
- 35. Martini G., Gozzetti A., Gennari L., Avanzati A., Nuti R., Lauria F. (2006) The effect of zoledronic acid on serum osteoprotegerin in early stage multiple myeloma. Haematologica 91, 1720–1721 [PubMed] [Google Scholar]
- 36. Martini G., Gennari L., Merlotti D., Salvadori S., Franci M. B., Campagna S., Avanzati A., De Paola V., Valleggi F., Nuti R. (2007) Serum OPG and RANKL levels before and after intravenous bisphosphonate treatment in Paget's disease of bone. Bone 40, 457–463 [DOI] [PubMed] [Google Scholar]
- 37. Tsubaki M., Satou T., Itoh T., Imano M., Yanae M., Kato C., Takagoshi R., Komai M., Nishida S. (2012) Bisphosphonate- and statin-induced enhancement of OPG expression and inhibition of CD9, M-CSF, and RANKL expressions via inhibition of the Ras/MEK/ERK pathway and activation of p38MAPK in mouse bone marrow stromal cell line ST2. Mol. Cell. Endocrinol. 361, 219–231 [DOI] [PubMed] [Google Scholar]
- 38. Ohe J. Y., Kwon Y. D., Lee H. W. (2012) Bisphosphonates modulate the expression of OPG and M-CSF in hMSC-derived osteoblasts. Clin. Oral Investig. 16, 1153–1159 [DOI] [PubMed] [Google Scholar]
- 39. Eslami B., Zhou S., Van Eekeren I., LeBoff M. S., Glowacki J. (2011) Reduced osteoclastogenesis and RANKL expression in marrow from women taking alendronate. Calcif. Tissue Int. 88, 272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cardoso L., Herman B. C., Verborgt O., Laudier D., Majeska R. J., Schaffler M. B. (2009) Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner. Res. 24, 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emerton K. B., Hu B., Woo A. A., Sinofsky A., Hernandez C., Majeska R. J., Jepsen K. J., Schaffler M. B. (2010) Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46, 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]