

Abstract
Iron is a redox-active metal required as a cofactor in multiple metalloproteins essential for a host of life processes. The metal is highly toxic when present in excess and must be strictly regulated to prevent tissue and organ damage. Hepcidin, a molecule first characterized as an antimicrobial peptide, plays a critical role in the regulation of iron homeostasis. Multiple stimuli positively influence the expression of hepcidin, including iron, inflammation, and infection by pathogens. In this Minireview, I will discuss how inflammation regulates hepcidin transcription, allowing for sufficient concentrations of iron for organismal needs while sequestering the metal from infectious pathogens.
Keywords: bone morphogenetic protein (BMP), cell signaling, infectious disease, inflammation, iron metabolism, STAT3, hepcidin
Introduction
Iron is crucial for many life functions in both eukaryotic hosts and prokaryotic pathogens. Due to its ability to readily accept or donate electrons, iron is a valuable cofactor in proteins essential in metabolic processes. However, when left unsupervised, iron can also react with oxygen to generate radical oxygen species that can damage all facets of a cell, leading to tissue damage and eventual organ failure. Therefore, it is crucial for organisms to maintain strict control over iron uptake and distribution to assure appropriate amounts for life requirements, yet regulate and sequester it tightly to prevent oxidative stress or microbial proliferation during infection. Herein, I will present an overview of how iron balance is maintained in vertebrate organisms and discuss the role of hepcidin, the master regulator of iron metabolism, in iron regulation during inflammation and infection.
General Iron Homeostasis
Each day the average human must absorb 1–2 mg of iron from the diet to offset unregulated losses from general bleeding, menstruation, or the sloughing of epithelial cells. Iron is a critical cofactor required for DNA synthesis, mitochondrial respiration, and various signaling pathways. Most crucially, almost 25 mg of iron per day is required for hemoglobin synthesis and the replacement of an estimated 200 billion RBCs. The vast majority of this iron pool is acquired through the recycling of senescent erythrocytes by macrophages of the reticuloendothelial compartment. Whole body iron homeostasis is thought to occur completely at the level of iron absorption, as no physiologically regulated means of iron excretion has been elucidated. Hence, influx of iron from the diet, and recycling of iron from aged or damaged RBCs, must be closely regulated to prevent iron-restricted erythropoiesis resulting in anemia or excess iron loading and subsequent tissue damage caused by the generation of radical oxygen species. Proper distribution of circulating iron to tissues such as the brain, heart, and skeletal muscle is crucial for the prevention of human disease states.
Non-heme dietary reduced iron is admitted into the body through divalent metal transporter 1 (DMT1, Slc11a2) (1, 2), an iron transporter located on the apical membrane of duodenal enterocytes located in the first section of the small intestine (Fig. 1). After uptake into villus enterocytes, the iron may take two different paths. A fraction of the reduced iron may be oxidized and securely sequestered in ferritin. Alternatively, iron may be transferred across the basolateral membrane by ferroportin (3–5), where it is oxidized to the ferric state by the membrane-bound multi-copper ferroxidase hephaestin or the homologous soluble ferroxidase ceruloplasmin (6, 7). Oxidized iron is then loaded onto transferrin (TF),2 a soluble protein in the blood that securely transports and distributes iron to downstream tissues.
FIGURE 1.
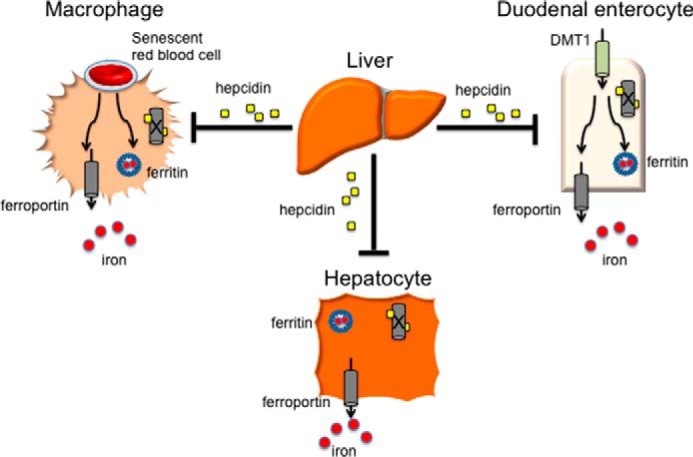
Role of the hepcidin/ferroportin axis in iron metabolism. Divalent metal transporter 1 is required for the uptake of dietary iron through duodenal epithelial cells. Hepcidin is predominately produced by hepatocytes of the liver. Stimulation of hepcidin production by elevated iron, inflammation, or infection causes ferroportin to be internalized and degraded in red blood cell-recycling macrophages, duodenal enterocytes of the small intestine, or hepatocytes themselves. In this manner, iron can be sequestered in ferritin within these cells, lowering iron concentration in the serum and preventing iron overload or theft of iron by invading pathogens. The opposite is also true. Iron-restricted erythropoiesis leads to a diminishment of hepcidin, elevated numbers of ferroportin transporters on iron-exporting cells, and increased serum iron availability for red blood cell maturation.
As previously noted, reticuloendothelial macrophages phagocytose senescent RBCs, harvesting the heme and returning almost 25 mg of iron into circulation each day. Like duodenal enterocytes and hepatocytes of the liver, macrophages of the RBC recycling compartment also employ ferroportin to export recovered iron into the general circulation (Fig. 1). Iron export, from both epithelial cells of the duodenum and macrophages within the RBC recycling compartment, is rate-controlling for total body iron flux; consequently, ferroportin expression must be closely regulated to assure sufficient iron for erythropoiesis and prevent excess tissue iron accumulation. Ferroportin expression is regulated post-translationally by its ligand hepcidin, the master regulator of iron metabolism.
Role of Hepcidin in Iron Metabolism
In 1994 Finch (8) postulated that soluble regulators of systemic iron metabolism, the “store regulators,” exist and that these hormones are essential for the maintenance of appropriate iron balance. However, it was not until 2000 that hepcidin, originally termed LEAP-1 (liver-expressed antimicrobial protein 1), was characterized as a defensin-like, liver-expressed, 25-residue antimicrobial peptide with four disulfide bonds (9). Simultaneous work in mice and humans demonstrated that hepcidin expression increases concomitantly with serum and tissue iron levels and is excreted in the urine (10, 11). Deletion of the hepcidin locus in mice demonstrated that animals lacking hepcidin have severe iron overload (12, 13); conversely, overexpression of hepcidin leads to severe iron deficiency (14, 15).
Hepcidin acts as a negative regulator of iron release from cells (Fig. 1), binding to ferroportin, the only known iron exporter, and causing the internalization and degradation of the transporter (16). Administration of synthetic exogenous hepcidin (17) leads to hypoferremia, a state of diminished TF-bound iron in the serum, and eventual iron deficiency. Interestingly, specific mutations in human ferroportin (18) prevent transporter binding to its ligand hepcidin, leading to iron overload and confirming the key function of this iron regulatory mechanism. In total, an increase in hepcidin expression leads to elevated iron storage in RBC recycling macrophages and hepatocytes and limits uptake from dietary sources. Conversely, diminished hepcidin expression permits more non-heme iron to be released from internal liver and macrophage stores and increases iron transfer through intestinal epithelial cells, effectively controlling the bioavailable iron supply.
Regulation of Hepcidin through the Bone Morphogenetic Protein (BMP)/SMAD Signaling Pathway
Hepcidin is only regulated at the transcriptional level, and expression is inhibited by anemia, hypoxia (19), and ineffective erythropoiesis (20), and stimulated by iron loading and inflammation. Multiple lines of inquiry have demonstrated that members of the TGF-β superfamily, including BMP receptors, associated BMP ligands, and the cytoplasmic SMAD transcription factors (homologs of the Caenorhabditis elegans protein SMA and the Drosophila protein mothers against decapentaplegic (MAD)) play a central role in transcriptionally regulating hepcidin expression (Fig. 2). Hemojuvelin (HJV) is a BMP co-receptor (21) required for appropriate iron metabolism that is expressed primarily in liver, heart, and skeletal muscle (22). Loss of HJV causes severe cases of iron loading in humans termed juvenile hemochromatosis (22), and mouse models confirmed that ablation of HJV (23, 24), specifically in hepatocytes (25, 26), leads to extreme iron overload due to depressed hepatic hepcidin expression. Furthermore, the ligand BMP-6 is essential for appropriate HJV-mediated hepcidin expression (27, 28), and serine/threonine type I (predominately ALK3 (29)) and type II (ActRIIA and BMPRII (30)) receptors are required for transmission of this signal. Stimulation of these receptors leads to phosphorylation of SMAD1/5/8 transcription factors. Earlier work demonstrated that loss of SMAD4 (31), the primary common mediator SMAD (co-SMAD) that binds to activated SMAD1/5/8, results in iron overload and decreased hepcidin expression. Finally, BMP-responsive elements are found in the hepcidin promoter and are critical for appropriate hepcidin regulation (32, 33). These data, in aggregate, establish that the BMP/SMAD signaling pathway plays a central role in the transcriptional regulation of hepcidin.
FIGURE 2.

Regulation of hepcidin expression by iron and inflammation. Increasing saturation of TF, and subsequent binding to TFR1, causes HFE to be released from a complex with TFR1. HFE is postulated to interact with TFR2 and HJV, a BMP co-receptor, to stimulate SMAD1/5/8 phosphorylation, dimerization with SMAD4, and elevation of hepcidin transcription. The ligand BMP6 is thought to play a key role in this process. HFE also interacts with ALK3, a type I BMP receptor, and stabilizes it on the cell membrane. During periods of inflammation or infection, the cytokine IL-6 is produced, activating the STAT3 signaling pathway to promote transcription of hepcidin through a gp130-, JAK1/2-mediated pathway. Both the phosphorylated SMAD1/5/8-SMAD4 heterodimer and the STAT3 transcription factors have known binding sites in the hepcidin promoter, and the two pathways are believed to work together in hepcidin regulation. Other inflammation-mediated stimulatory signals may act to positively stimulate hepcidin expression.
Regulation of BMP/SMAD pathway signaling by iron sensors is complex and not completely understood, and dysregulation of this system can lead to human disease. Although loss of multiple members of the iron-sensing mechanism, including HFE (34), the classic hereditary hemochromatosis gene, and TFR2 (transferrin receptor 2) (35), a homolog of the iron uptake receptor TFR1, is known to diminish hepcidin expression, leading to excess iron absorption, how they interact with the BMP/SMAD signaling complex is uncertain. HFE binds to transferrin receptor 1 (TFR1), sharing a binding site on the receptor with TF (36). One model asserts that increasing transferrin saturation displaces HFE from TFR1, leading to increased hepcidin expression (37). Although work in vitro has demonstrated that HFE, TFR2, and HJV may form a stable complex that functions to regulate hepcidin expression through HJV (38), other data suggest that this interaction is dispensable or works through a different mechanism (39, 40). To this end, recent work has demonstrated that HFE directly interacts with ALK3, stabilizing the receptor on the cell surface and helping transduce a signal for hepcidin transcriptional regulation (41). Further research will be required to fully comprehend how these proteins, as well as other effectors of BMP/SMAD signaling, work together to regulate hepcidin expression.
Regulation of Hepcidin by Inflammation
A large number of plant and animal tissues contain antimicrobial peptides involved in host defense. Due to its eight cysteine residues and defensin-like structure, hepcidin was originally postulated to be a liver-generated member of this large protein family. In fact, the peptide has both antifungal and antimicrobial activities (11). Importantly, it was noted that murine hepcidin transcription surges upon treatment with LPS (10) or turpentine (19). LPS is an endotoxin and the major component of the outer membrane of Gram-negative bacteria, and as such, is an extremely potent pathogen-derived inflammatory signal. Toll-like receptor 4 (TLR4), a member of the TLR family, which plays a fundamental role in pathogen recognition and activation of innate immunity, is the receptor for LPS (42). Terpenes are thought to play a protective role in conifers, and turpentine has long been known to initiate inflammatory responses in mammals. Taken together, these investigations suggested that the hepcidin peptide was likely regulated by both inflammation and infection.
Inflammatory cytokines are generated in response to infection by iron-dependent invading pathogens. Particular molecular patterns are recognized by specific receptor families (TLRs), and cytokines are released to instigate an immune response. This response can stimulate an acute hypoferremia, inhibiting pathogen growth and proliferation. Several cytokines, including primarily IL-6 (43), but also IL-1 (44), IL-22 (45), and interferon α (46), have been shown to positively up-regulate hepcidin expression. This is mediated by STAT3 signaling (47, 48), and loss of STAT3 specifically in the liver prevents hepcidin regulation by cytokine stimulation (49). In the current model, IL-6 binds to the gp130 protein receptor complex (50), instigating a JAK1/2 tyrosine kinase-mediated phosphorylation of the transcription factor STAT3. Activated STAT3 then translocates to the nucleus and binds to the STAT3-responsive element on the proximal hepcidin promoter, inducing hepcidin transcription (Fig. 2).
More recently, ALK3, the primary type I receptor BMP receptor involved in hepcidin regulation, was shown to be crucial for IL-6-mediated hepcidin induction (51). Furthermore, activin B, a member of the TGF-β superfamily, is involved in response to inflammation in an IL-6-independent manner (52). Upon treatment with LPS, expression of the activin β (B)-subunit is significantly increased, and this leads to a rise in SMAD1/5/8 phosphorylation and subsequent hepcidin induction. Importantly, IL-6 is known to be crucial for the response to common bacterial or viral infections or to pathogen-derived molecules in mice (53). Interestingly, cytokine induction, and the resulting increase in hepcidin expression by inflammation, also leads to decreased numbers of erythroid progenitors (54), possibly helping to match the diminished amount of iron available for erythropoiesis. Finally, upon infection, activated inflammatory cells undergo an oxidative burst that results in the release of large amounts of reactive oxygen species, which help to kill invading microbes. Neutrophils generate H2O2 when activated, and work in cell culture has shown that low levels of H2O2 stimulation contribute to hepatic hepcidin induction through STAT3 (55).
There is also some suggestion that inflammatory signals may modulate iron metabolism without the need for hepcidin induction. Animals with complete genetic ablation of hepcidin and treated with LPS have diminished ferroportin expression in the duodenum and spleen, leading to slightly decreased plasma iron (56). Furthermore, TLRs are key components in the innate immune system and are required for the induction of the adaptive immunity response. They are normally expressed in sentinel cells such as tissue macrophages and recognize pathogen-derived molecules. Stimulation of TLR2 and -6 receptors (Fig. 3) reduces ferroportin expression in mouse bone marrow-derived macrophages, liver, and spleen independently of hepcidin (57). Ferroportin containing a C326C mutation is known to be resistant to hepcidin-mediated degradation (18). Injection with two separate TLR2/6 ligands down-regulated ferroportin and induced hypoferremia in mice containing the C326C mutation. These data suggest that there may be multiple pathways by which organisms attempt to withhold iron from invading pathogens during periods of inflammation, but further research is essential to better understand these additional inflammatory responses leading to hypoferremia.
FIGURE 3.
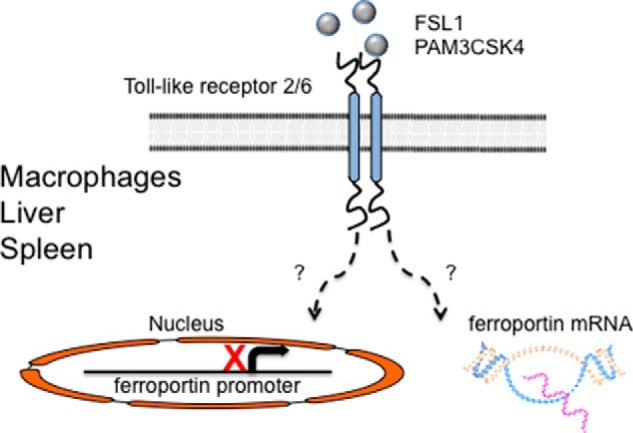
Hepcidin-independent, inflammation-mediated regulation of ferroportin. Stimulation of TLRs 2 or 6 by the ligands FSL1 or PAM3CSK4 causes diminished ferroportin mRNA and protein expression in bone marrow-derived macrophages, liver, or spleen. This occurs through TLR2/6 heterodimers or TLR2 homodimers. Decreased ferroportin expression leads to an acute hypoferremia that may precede or complement the hepcidin-mediated decrease in bioavailable iron during inflammatory conditions. Further research is necessary to determine whether this pathway suppresses ferroportin mRNA expression by diminishing transcription or through increased mRNA degradation.
Cooperation between BMP/SMAD and JAK1/2-STAT3 Inflammatory Signaling Pathway
Increasing weight of evidence suggests that essential crosstalk exists between the BMP/SMAD and JAK1/2-STAT3 inflammatory signaling pathway (Fig. 2). SMAD4 is the co-SMAD necessary for dimerization with phosphorylated SMAD1/5/8 and subsequent hepcidin induction. Early studies suggested that mice lacking Smad4 are unable to induce hepcidin expression after treatment with LPS (31). Pharmacological intervention with dorsomorphin, LDN-193189, or other specific small-molecule inhibitors of the BMP pathway (58–60) is able to attenuate hepcidin expression in rodents treated with inflammatory agents. Loss of HFE and TFR2, either alone or in combination, leads to inappropriately phosphorylated SMAD1/5/8 and suppressed hepcidin expression (39, 61–63). Mice lacking HFE (64) or both HFE and TFR2 (65) are able to mount an appropriate immune response to LPS, but do not elevate hepcidin production nor develop hypoferremia. Furthermore, concomitant stimulation of both the BMP/SMAD and the JAK1/2-STAT3 pathways in rodents causes, at minimum, additive, and also in some experiments synergistic, effects upon up-regulation of hepcidin (59, 66, 67). In toto, this research suggests that the BMP/SMAD and JAK1/2-STAT3 signaling pathways impinge on one other, leading to increased hepcidin expression under conditions of infection or inflammation.
Role of Hepcidin in the Anemia of Inflammation
The anemia of chronic disease (68), now commonly termed the anemia of inflammation, is known to occur in settings of infection by microbial pathogens, in inflammatory, autoimmune conditions such as arthritis or lupus, in chronic kidney disease, or as a result of cancer. The mild to moderate anemia is normocytic and normochromic with a reduced number of erythrocytes; however, patients may progress to a more serious condition with microcytic and hypochromic red blood cells over the course of a long, serious illness. In most cases, iron is retained within macrophages of the reticuloendothelial system, leading to inappropriately low availability of iron-bound transferrin required for erythropoiesis. In these inflammatory states, release of cytokines leads to elevated hepcidin expression, diminishing ferroportin on the surface of enterocytes, recycling macrophages and hepatocytes, sequestering iron in storage sites, and diminishing iron uptake from the diet. One of the first studies to demonstrate the involvement of hepcidin in this condition showed that patients with glycogen storage disease type 1a, a population that spontaneously develops large adenomas in the liver and the anemia of inflammation, have elevated hepcidin expression. The observed anemia was ameliorated upon resection of the adenomas (69).
Anemia of Inflammation and Infection
Historically, the anemia of inflammation in humans was most readily observed and understood in the context of infection with pathogenic organisms. The hypoferremia associated with infection was first noted over 70 years ago by Cartwright et al. (101) and was hypothesized to sequester iron in tissues to prevent transfer to invading microbes. Based on the overwhelming weight of evidence, this response is primarily due to the up-regulation of hepcidin. Interestingly, hepcidin regulation by infection was first noted in sea bass (70) where a massive induction of hepcidin occurs after bacterial infection. Subsequently, it was demonstrated that patients with various causes of anemia of inflammation or infection had elevated urinary hepcidin excretion (71). Furthermore, humans treated with Il-6 (43) or with LPS (72) have elevated hepcidin expression leading to an acute hypoferremia.
Significant data demonstrate that hepcidin-mediated iron regulation plays a crucial role in the interaction between human hosts and their microbial pathogens. For example, hepcidin is induced in malarial infection (73), a disease that is estimated to kill 600,000 people every year. The relevance of hepcidin, and its role in the treatment and outcome of infection by this human scourge, has been reviewed extensively elsewhere (74). Furthermore, the crucially important nature of hepcidin in host defense was starkly illustrated by a laboratory accident. Attenuated strains of Yersinia pestis lacking a high-pathogenicity island involved in iron uptake are commonly employed in vaccine research. A researcher with an undiagnosed case of hereditary hemochromatosis caused by a mutation in HFE, a disease where inappropriately low hepcidin expression leads to elevated total body iron body burden, died after developing a case of septicemic plague (75). Of note, the up-regulation of hepcidin and resulting acute hypoferremia may not be a universal phenomenon in pathogen infection. For example, hepcidin is induced upon HIV-1 infection, but not by infection with hepatitis B or C, in humans (76). This suggests that the hepcidin-mediated iron sequestration in infection may be pathogen-, tissue-, and inflammation response-specific.
The vast majority of hepcidin is expressed by hepatocytes of the liver. However, immune cells at the site of infection produce small concentrations of the peptide (77), likely through a TLR4-dependent mechanism (78). Furthermore, treatment of monocytes from anemia of inflammation patients with IL-6 or LPS induced a more robust hepcidin induction than in controls, and this leads to diminished ferroportin expression and a decrease in iron export in an autocrine manner (79). Taken together, this implies that hepcidin production at the site of infection may lead to localized iron sequestration and prevent iron theft by pathogens; however, further research is required to understand this response to microbial invasion.
It appears that a regulatory pathway facilitates the amelioration of inflammation-mediated anemia once infection has resolved (Fig. 4). Recent investigation has uncovered erythroferrone (ERFE), a hormone produced by erythroblasts in response to erythropoietin, which mediates hepcidin suppression during the early stages of stress erythropoiesis (80). Significantly, ERFE also appears to play a role in the recovery of organisms from the anemia of inflammation (81). Heat-killed Brucella abortus (HKBA) is known to induce an anemia of inflammation response in rodents (82, 83). Mice lacking ERFE have both a more severe and a more prolonged anemia, and more greatly elevated hepcidin expression, as compared with wild type animals upon exposure to HKBA. Elevated hepcidin expression causes depressed serum iron available for erythropoiesis in HKBA-treated mice. Increased ERFE expression appears to suppress the deleterious effects of iron sequestration during infection or the anemia of inflammation, and as such, may be a possible target for therapeutic intervention in disorders with chronic inflammation.
FIGURE 4.
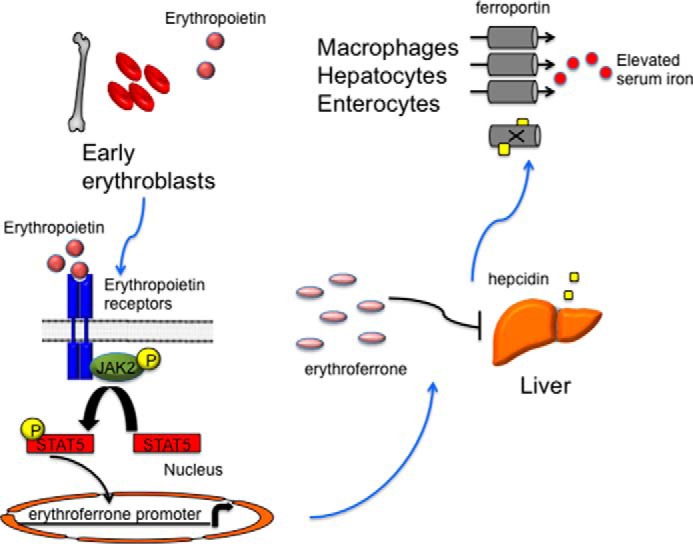
A model for erythroferrone-mediated recovery from the anemia of inflammation. Hypoxia is sensed in the kidney, and erythropoietin levels are increased through an IRP1-HIF2α signaling pathway. Elevated erythropoietin causes a JAK2/STAT5 phosphorylation cascade, leading to the production of ERFE in early erythroblasts. Circulating erythroferrone suppresses hepcidin production in hepatocytes, leading to elevated ferroportin expression and augmenting serum iron availability for red blood cell maturation.
Other Causes of the Anemia of Inflammation
The anemia of inflammation has also been linked with chronic kidney disease (CKD), a disorder most commonly caused by diabetes and high blood pressure, and cancer. Patients with CKD are known to present with anemia during the course of their illness and have elevated hepcidin expression (84). Hepcidin is also greatly increased in multiple myeloma, a plasma cell malignancy that is thought to account for a large percentage of hematologic cancers (85). Hepcidin induction in this cancer requires BMP2, another BMP/SMAD pathway ligand, and the inflammatory cytokine IL-6 (67). Furthermore, recent work demonstrated that hepcidin is elevated in breast cancer patients and that diminished tumor expression of ferroportin promotes breast cancer growth (86, 87). Finally, patients with the most advanced cancers have the lowest RBC hemoglobin concentration, and hemoglobin measurements are conversely correlated with inflammatory markers and hepcidin (88). This research indicates that efficacious treatment of CKD, and a number of distinct malignancies, requires not only directed therapy toward each disease, but also appropriate management of patient iron status.
Interestingly, anemia is widespread in elderly populations although iron is not normally diet-limited. This anemia has been attributed to low grade inflammation; however, the cause of this inflammation is often not explained by infection, CKD, or cancer and has been termed the unexplained anemia of the elderly. Studies in aged mice demonstrate that IL-6 and hepcidin are not directly required for aging-related anemia; however, mice lacking these genes have improved erythropoiesis later in life (89). More recent work has demonstrated that patients with unexplained anemia of the elderly, all of whom have no known history of chronic inflammatory disease, do in fact have features of low grade inflammation including elevated IL-6 expression (90).
Finally, endoplasmic reticulum (ER) stress induces multiple pathways that are collectively known as the unfolded protein response. Toxins, misfolded proteins, disruption of ER homeostasis, and inflammation are all known to generate ER stress. ER stress was directly linked to the acute inflammatory response when it was demonstrated that the ER stress-activated transcription factor CREBH (cyclic AMP-response element-binding protein H) responds to induction by both LPS and IL-6 (91). Importantly, hepcidin expression can be modulated by the transcription factors CHOP (CCAAT-enhancer-binding protein homologous protein), C/EBPα (CCAAT-enhancer-binding protein α), and CREBH, which bind to specific binding sites on the hepcidin promoter (92, 93).
Conclusions
Hepcidin is the master regulator of vertebrate iron metabolism and homeostasis. Expression of hepcidin is modulated by multiple signaling pathways, and up-regulation of the anti-microbial peptide is triggered by elevated iron status, inflammation, and infection. Inflammation-mediated induction of hepcidin is thought to occur through the combined efforts of the BMP/SMAD and JAK1/2-STAT3 signaling pathways. Stimulation of hepcidin expression during episodes of inflammation and infection greatly decreases access of bioavailable iron to invading pathogens however, this may cause iron-restricted erythropoiesis in the host. Accordingly, there is a constant struggle within a host to meet organismal iron demands for heme production while preventing iron theft by invading infectious agents. This balance is primarily maintained by the attenuation of hepcidin production.
Moving forward, much of the research concerning hepcidin and its link to various human disease states will need to be completed in animal models. Accordingly, new rodent models of cancer (94) and infection (82, 83) have been recently generated. These model systems will be essential for dissecting the more intricate interactions between hepcidin regulation and changes in iron homeostasis induced by inflammation or infection.
Furthermore, the intimate link between hepcidin regulation, iron metabolism, and human health suggests that therapeutic manipulation of hepcidin is an essential future goal. To that end, neutralizing hepcidin antibodies (95), small-molecule inhibitors of the BMP/SMAD pathway (59), siRNA (96) and antisense oligonucleotide (97) technology, and hepcidin mimetics (minihepcidins) (98) have been employed to inhibit or induce hepcidin expression, respectively. Additional work will be necessary to determine the most efficacious methods of hepcidin modulation in clinical settings.
The direct measurement of hepcidin protein, both in rodent models of human disease and in patients themselves, is vital for further investigation and treatment of the anemia of inflammation. Moreover, determination of the therapeutic efficacy of hepcidin modulation requires specific, non-invasive measurements of the ligand, which can be ascertained over the course of treatment. Several methods have been proposed to directly measure the peptide in easily accessible body fluids. Human serum or urine hepcidin can be measured by ELISA (84) or time-of-flight mass spectroscopy (99). Recently, a highly specific ELISA assay has been generated that quantitatively measures hepcidin in mouse serum or urine under conditions where hepcidin is greatly elevated or repressed (100).
Although our understanding of how inflammation regulates hepcidin expression, and by extension modulates vertebrate iron metabolism, is rapidly increasing, many pertinent questions yet remain. For example, do other regulatory pathways exist that influence hepcidin expression during inflammatory events? Are there additional mechanisms that modify iron flux under acute inflammatory conditions without the need for hepcidin? Finally, and perhaps most importantly, how do we best modify hepcidin expression in patients with the anemia of inflammation? Significant further research will be required to answer these questions and identify novel therapeutic approaches for the safe and effective treatment of affected individuals.
Acknowledgments
I thank Mark Fleming for careful reading of the manuscript.
This is the fifth article in the Thematic Minireview series “Metals at the Host-Pathogen Interface.” The author declares that he has no conflicts of interest with the contents of this article.
- TF
- transferrin
- BMP
- bone morphogenetic protein
- TLR
- Toll-like receptor
- ERFE
- erythroferrone
- CKD
- chronic kidney disease
- HKBA
- Heat-killed Brucella abortus
- ER
- endoplasmic reticulum.
References
- 1. Gunshin H., Mackenzie B., Berger U. V., Gunshin Y., Romero M. F., Boron W. F., Nussberger S., Gollan J. L., Hediger M. A. (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488 [DOI] [PubMed] [Google Scholar]
- 2. Fleming M. D., Trenor C. C., 3rd, Su M. A., Foernzler D., Beier D. R., Dietrich W. F., Andrews N. C. (1997) Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 16, 383–386 [DOI] [PubMed] [Google Scholar]
- 3. Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., Paw B. H., Drejer A., Barut B., Zapata A., Law T. C., Brugnara C., Lux S. E., Pinkus G. S., Pinkus J. L., Kingsley P. D., Palis J., Fleming M. D., Andrews N. C., Zon L. I. (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 [DOI] [PubMed] [Google Scholar]
- 4. McKie A. T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T. J., Farzaneh F., Hediger M. A., Hentze M. W., Simpson R. J. (2000) A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 5, 299–309 [DOI] [PubMed] [Google Scholar]
- 5. Abboud S., Haile D. J. (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 275, 19906–19912 [DOI] [PubMed] [Google Scholar]
- 6. Vulpe C. D., Kuo Y. M., Murphy T. L., Cowley L., Askwith C., Libina N., Gitschier J., Anderson G. J. (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 21, 195–199 [DOI] [PubMed] [Google Scholar]
- 7. Harris Z. L., Durley A. P., Man T. K., Gitlin J. D. (1999) Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. U.S.A. 96, 10812–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finch C. (1994) Regulators of iron balance in humans. Blood 84, 1697–1702 [PubMed] [Google Scholar]
- 9. Krause A., Neitz S., Mägert H. J., Schulz A., Forssmann W. G., Schulz-Knappe P., Adermann K. (2000) LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 480, 147–150 [DOI] [PubMed] [Google Scholar]
- 10. Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., Loréal O. (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819 [DOI] [PubMed] [Google Scholar]
- 11. Park C. H., Valore E. V., Waring A. J., Ganz T. (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 276, 7806–7810 [DOI] [PubMed] [Google Scholar]
- 12. Nicolas G., Bennoun M., Devaux I., Beaumont C., Grandchamp B., Kahn A., Vaulont S. (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lesbordes-Brion J. C., Viatte L., Bennoun M., Lou D. Q., Ramey G., Houbron C., Hamard G., Kahn A., Vaulont S. (2006) Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108, 1402–1405 [DOI] [PubMed] [Google Scholar]
- 14. Nicolas G., Bennoun M., Porteu A., Mativet S., Beaumont C., Grandchamp B., Sirito M., Sawadogo M., Kahn A., Vaulont S. (2002) Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. U.S.A. 99, 4596–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roy C. N., Mak H. H., Akpan I., Losyev G., Zurakowski D., Andrews N. C. (2007) Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood 109, 4038–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., Kaplan J. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 [DOI] [PubMed] [Google Scholar]
- 17. Rivera S., Nemeth E., Gabayan V., Lopez M. A., Farshidi D., Ganz T. (2005) Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood 106, 2196–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drakesmith H., Schimanski L. M., Ormerod E., Merryweather-Clarke A. T., Viprakasit V., Edwards J. P., Sweetland E., Bastin J. M., Cowley D., Chinthammitr Y., Robson K. J., Townsend A. R. (2005) Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 106, 1092–1097 [DOI] [PubMed] [Google Scholar]
- 19. Nicolas G., Chauvet C., Viatte L., Danan J. L., Bigard X., Devaux I., Beaumont C., Kahn A., Vaulont S. (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 110, 1037–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adamsky K., Weizer O., Amariglio N., Breda L., Harmelin A., Rivella S., Rachmilewitz E., Rechavi G. (2004) Decreased hepcidin mRNA expression in thalassemic mice. Br. J. Haematol. 124, 123–124 [DOI] [PubMed] [Google Scholar]
- 21. Babitt J. L., Huang F. W., Wrighting D. M., Xia Y., Sidis Y., Samad T. A., Campagna J. A., Chung R. T., Schneyer A. L., Woolf C. J., Andrews N. C., Lin H. Y. (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531–539 [DOI] [PubMed] [Google Scholar]
- 22. Papanikolaou G., Samuels M. E., Ludwig E. H., MacDonald M. L., Franchini P. L., Dubé M. P., Andres L., MacFarlane J., Sakellaropoulos N., Politou M., Nemeth E., Thompson J., Risler J. K., Zaborowska C., Babakaiff R., Radomski C. C., Pape T. D., Davidas O., Christakis J., Brissot P., Lockitch G., Ganz T., Hayden M. R., Goldberg Y. P. (2004) Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 36, 77–82 [DOI] [PubMed] [Google Scholar]
- 23. Huang F. W., Pinkus J. L., Pinkus G. S., Fleming M. D., Andrews N. C. (2005) A mouse model of juvenile hemochromatosis. J. Clin. Invest. 115, 2187–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niederkofler V., Salie R., Arber S. (2005) Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Invest. 115, 2180–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen W., Huang F. W., de Renshaw T. B., Andrews N. C. (2011) Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis. Blood 117, 6319–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gkouvatsos K., Wagner J., Papanikolaou G., Sebastiani G., Pantopoulos K. (2011) Conditional disruption of mouse HFE2 gene: maintenance of systemic iron homeostasis requires hepatic but not skeletal muscle hemojuvelin. Hepatology 54, 1800–1807 [DOI] [PubMed] [Google Scholar]
- 27. Andriopoulos B., Jr., Corradini E., Xia Y., Faasse S. A., Chen S., Grgurevic L., Knutson M. D., Pietrangelo A., Vukicevic S., Lin H. Y., Babitt J. L. (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 41, 482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meynard D., Kautz L., Darnaud V., Canonne-Hergaux F., Coppin H., Roth M. P. (2009) Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 41, 478–481 [DOI] [PubMed] [Google Scholar]
- 29. Steinbicker A. U., Bartnikas T. B., Lohmeyer L. K., Leyton P., Mayeur C., Kao S. M., Pappas A. E., Peterson R. T., Bloch D. B., Yu P. B., Fleming M. D., Bloch K. D. (2011) Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118, 4224–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia Y., Babitt J. L., Sidis Y., Chung R. T., Lin H. Y. (2008) Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 111, 5195–5204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang R. H., Li C., Xu X., Zheng Y., Xiao C., Zerfas P., Cooperman S., Eckhaus M., Rouault T., Mishra L., Deng C. X. (2005) A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2, 399–409 [DOI] [PubMed] [Google Scholar]
- 32. Verga Falzacappa M. V., Casanovas G., Hentze M. W., Muckenthaler M. U. (2008) A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. (Berl.) 86, 531–540, 10.1007/s00109-008-0313-7 [DOI] [PubMed] [Google Scholar]
- 33. Truksa J., Lee P., Beutler E. (2009) Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 113, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feder J. N., Gnirke A., Thomas W., Tsuchihashi Z., Ruddy D. A., Basava A., Dormishian F., Domingo R., Jr., Ellis M. C., Fullan A., Hinton L. M., Jones N. L., Kimmel B. E., Kronmal G. S., Lauer P., Lee V. K., Loeb D. B., Mapa F. A., McClelland E., Meyer N. C., Mintier G. A., Moeller N., Moore T., Morikang E., Prass C. E., Quintana L., Starnes S. M., Schatzman R. C., Brunke K. J., Drayna D. T., Risch N. J., Bacon B. R., Wolff R. K. (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13, 399–408 [DOI] [PubMed] [Google Scholar]
- 35. Camaschella C., Roetto A., Calì A., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., Gasparini P. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15 [DOI] [PubMed] [Google Scholar]
- 36. West A. P., Jr., Giannetti A. M., Herr A. B., Bennett M. J., Nangiana J. S., Pierce J. R., Weiner L. P., Snow P. M., Bjorkman P. J. (2001) Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites. J. Mol. Biol. 313, 385–397 [DOI] [PubMed] [Google Scholar]
- 37. Schmidt P. J., Toran P. T., Giannetti A. M., Bjorkman P. J., Andrews N. C. (2008) The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 7, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D'Alessio F., Hentze M. W., Muckenthaler M. U. (2012) The hemochromatosis proteins HFE, TfR2 and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 57, 1052–1060 [DOI] [PubMed] [Google Scholar]
- 39. Wallace D. F., Summerville L., Crampton E. M., Frazer D. M., Anderson G. J., Subramaniam V. N. (2009) Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 50, 1992–2000 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt P. J., Fleming M. D. (2012) Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am. J. Hematol. 87, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu X. G., Wang Y., Wu Q., Cheng W. H., Liu W., Zhao Y., Mayeur C., Schmidt P. J., Yu P. B., Wang F., Xia Y. (2014) HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 124, 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 43. Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B. K., Ganz T. (2004) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113, 1271–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee P., Peng H., Gelbart T., Wang L., Beutler E. (2005) Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. U.S.A. 102, 1906–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Armitage A. E., Eddowes L. A., Gileadi U., Cole S., Spottiswoode N., Selvakumar T. A., Ho L. P., Townsend A. R., Drakesmith H. (2011) Hepcidin regulation by innate immune and infectious stimuli. Blood 118, 4129–4139 [DOI] [PubMed] [Google Scholar]
- 46. Ryan J. D., Altamura S., Devitt E., Mullins S., Lawless M. W., Muckenthaler M. U., Crowe J. (2012) Pegylated interferon-α induced hypoferremia is associated with the immediate response to treatment in hepatitis C. Hepatology 56, 492–500 [DOI] [PubMed] [Google Scholar]
- 47. Wrighting D. M., Andrews N. C. (2006) Interleukin-6 induces hepcidin expression through STAT3. Blood 108, 3204–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Verga Falzacappa M. V., Vujic Spasic M., Kessler R., Stolte J., Hentze M. W., Muckenthaler M. U. (2007) STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109, 353–358 [DOI] [PubMed] [Google Scholar]
- 49. Sakamori R., Takehara T., Tatsumi T., Shigekawa M., Hikita H., Hiramatsu N., Kanto T., Hayashi N. (2010) STAT3 signaling within hepatocytes is required for anemia of inflammation in vivo. J. Gastroenterol. 45, 244–248 [DOI] [PubMed] [Google Scholar]
- 50. Pietrangelo A., Dierssen U., Valli L., Garuti C., Rump A., Corradini E., Ernst M., Klein C., Trautwein C. (2007) STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology 132, 294–300 [DOI] [PubMed] [Google Scholar]
- 51. Mayeur C., Lohmeyer L. K., Leyton P., Kao S. M., Pappas A. E., Kolodziej S. A., Spagnolli E., Yu B., Galdos R. L., Yu P. B., Peterson R. T., Bloch D. B., Bloch K. D., Steinbicker A. U. (2014) The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 123, 2261–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Besson-Fournier C., Latour C., Kautz L., Bertrand J., Ganz T., Roth M. P., Coppin H. (2012) Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 120, 431–439 [DOI] [PubMed] [Google Scholar]
- 53. Rodriguez R., Jung C. L., Gabayan V., Deng J. C., Ganz T., Nemeth E., Bulut Y. (2014) Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infect. Immun. 82, 745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Langdon J. M., Yates S. C., Femnou L. K., McCranor B. J., Cheadle C., Xue Q. L., Vaulont S., Civin C. I., Walston J. D., Roy C. N. (2014) Hepcidin-dependent and hepcidin-independent regulation of erythropoiesis in a mouse model of anemia of chronic inflammation. Am. J. Hematol. 89, 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Millonig G., Ganzleben I., Peccerella T., Casanovas G., Brodziak-Jarosz L., Breitkopf-Heinlein K., Dick T. P., Seitz H. K., Muckenthaler M. U., Mueller S. (2012) Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3). J. Biol. Chem. 287, 37472–37482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deschemin J. C., Vaulont S. (2013) Role of hepcidin in the setting of hypoferremia during acute inflammation. PLoS One 8, e61050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guida C., Altamura S., Klein F. A., Galy B., Boutros M., Ulmer A. J., Hentze M. W., Muckenthaler M. U. (2015) A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 125, 2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu P. B., Hong C. C., Sachidanandan C., Babitt J. L., Deng D. Y., Hoyng S. A., Lin H. Y., Bloch K. D., Peterson R. T. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Steinbicker A. U., Sachidanandan C., Vonner A. J., Yusuf R. Z., Deng D. Y., Lai C. S., Rauwerdink K. M., Winn J. C., Saez B., Cook C. M., Szekely B. A., Roy C. N., Seehra J. S., Cuny G. D., Scadden D. T., Peterson R. T., Bloch K. D., Yu P. B. (2011) Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 117, 4915–4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Theurl I., Schroll A., Sonnweber T., Nairz M., Theurl M., Willenbacher W., Eller K., Wolf D., Seifert M., Sun C. C., Babitt J. L., Hong C. C., Menhall T., Gearing P., Lin H. Y., Weiss G. (2011) Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood 118, 4977–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ahmad K. A., Ahmann J. R., Migas M. C., Waheed A., Britton R. S., Bacon B. R., Sly W. S., Fleming R. E. (2002) Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol. Dis. 29, 361–366 [DOI] [PubMed] [Google Scholar]
- 62. Muckenthaler M., Roy C. N., Custodio A. O., Miñana B., deGraaf J., Montross L. K., Andrews N. C., Hentze M. W. (2003) Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat. Genet. 34, 102–107 [DOI] [PubMed] [Google Scholar]
- 63. Kawabata H., Fleming R. E., Gui D., Moon S. Y., Saitoh T., O'Kelly J., Umehara Y., Wano Y., Said J. W., Koeffler H. P. (2005) Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 105, 376–381 [DOI] [PubMed] [Google Scholar]
- 64. Roy C. N., Custodio A. O., de Graaf J., Schneider S., Akpan I., Montross L. K., Sanchez M., Gaudino A., Hentze M. W., Andrews N. C., Muckenthaler M. U. (2004) An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat. Genet. 36, 481–485 [DOI] [PubMed] [Google Scholar]
- 65. Wallace D. F., McDonald C. J., Ostini L., Subramaniam V. N. (2011) Blunted hepcidin response to inflammation in the absence of Hfe and transferrin receptor 2. Blood 117, 2960–2966 [DOI] [PubMed] [Google Scholar]
- 66. Kartikasari A. E., Roelofs R., Schaeps R. M., Kemna E. H., Peters W. H., Swinkels D. W., Tjalsma H. (2008) Secretion of bioactive hepcidin-25 by liver cells correlates with its gene transcription and points towards synergism between iron and inflammation signaling pathways. Biochim. Biophys. Acta 1784, 2029–2037 [DOI] [PubMed] [Google Scholar]
- 67. Maes K., Nemeth E., Roodman G. D., Huston A., Esteve F., Freytes C., Callander N., Katodritou E., Tussing-Humphreys L., Rivera S., Vanderkerken K., Lichtenstein A., Ganz T. (2010) In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood 116, 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weiss G., Goodnough L. T. (2005) Anemia of chronic disease. N. Engl. J. Med. 352, 1011–1023 [DOI] [PubMed] [Google Scholar]
- 69. Weinstein D. A., Roy C. N., Fleming M. D., Loda M. F., Wolfsdorf J. I., Andrews N. C. (2002) Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 100, 3776–3781 [DOI] [PubMed] [Google Scholar]
- 70. Shike H., Lauth X., Westerman M. E., Ostland V. E., Carlberg J. M., Van Olst J. C., Shimizu C., Bulet P., Burns J. C. (2002) Bass hepcidin is a novel antimicrobial peptide induced by bacterial challenge. Eur. J. Biochem. 269, 2232–2237 [DOI] [PubMed] [Google Scholar]
- 71. Nemeth E., Valore E. V., Territo M., Schiller G., Lichtenstein A., Ganz T. (2003) Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 101, 2461–2463 [DOI] [PubMed] [Google Scholar]
- 72. Kemna E., Pickkers P., Nemeth E., van der Hoeven H., Swinkels D. (2005) Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 106, 1864–1866 [DOI] [PubMed] [Google Scholar]
- 73. Howard C. T., McKakpo U. S., Quakyi I. A., Bosompem K. M., Addison E. A., Sun K., Sullivan D., Semba R. D. (2007) Relationship of hepcidin with parasitemia and anemia among patients with uncomplicated Plasmodium falciparum malaria in Ghana. Am. J. Trop. Med. Hyg. 77, 623–626 [PubMed] [Google Scholar]
- 74. Spottiswoode N., Duffy P. E., Drakesmith H. (2014) Iron, anemia and hepcidin in malaria. Front. Pharmacol. 5, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Frank K. M., Schneewind O., Shieh W. J. (2011) Investigation of a researcher's death due to septicemic plague. N. Engl. J. Med. 364, 2563–2564 [DOI] [PubMed] [Google Scholar]
- 76. Armitage A. E., Stacey A. R., Giannoulatou E., Marshall E., Sturges P., Chatha K., Smith N. M., Huang X., Xu X., Pasricha S. R., Li N., Wu H., Webster C., Prentice A. M., Pellegrino P., Williams I., Norris P. J., Drakesmith H., Borrow P. (2014) Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc. Natl. Acad. Sci. U.S.A. 111, 12187–12192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Motley S. T., Morrow B. J., Liu X., Dodge I. L., Vitiello A., Ward C. K., Shaw K. J. (2004) Simultaneous analysis of host and pathogen interactions during an in vivo infection reveals local induction of host acute phase response proteins, a novel bacterial stress response, and evidence of a host-imposed metal ion limited environment. Cell. Microbiol. 6, 849–865 [DOI] [PubMed] [Google Scholar]
- 78. Peyssonnaux C., Zinkernagel A. S., Datta V., Lauth X., Johnson R. S., Nizet V. (2006) TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107, 3727–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Theurl I., Theurl M., Seifert M., Mair S., Nairz M., Rumpold H., Zoller H., Bellmann-Weiler R., Niederegger H., Talasz H., Weiss G. (2008) Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 111, 2392–2399 [DOI] [PubMed] [Google Scholar]
- 80. Kautz L., Jung G., Valore E. V., Rivella S., Nemeth E., Ganz T. (2014) Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 46, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kautz L., Jung G., Nemeth E., Ganz T. (2014) Erythroferrone contributes to recovery from anemia of inflammation. Blood 124, 2569–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kim A., Fung E., Parikh S. G., Valore E. V., Gabayan V., Nemeth E., Ganz T. (2014) A mouse model of anemia of inflammation: complex pathogenesis with partial dependence on hepcidin. Blood 123, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gardenghi S., Renaud T. M., Meloni A., Casu C., Crielaard B. J., Bystrom L. M., Greenberg-Kushnir N., Sasu B. J., Cooke K. S., Rivella S. (2014) Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood 123, 1137–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ganz T., Olbina G., Girelli D., Nemeth E., Westerman M. (2008) Immunoassay for human serum hepcidin. Blood 112, 4292–4297 [DOI] [PubMed] [Google Scholar]
- 85. Sharma S., Nemeth E., Chen Y. H., Goodnough J., Huston A., Roodman G. D., Ganz T., Lichtenstein A. (2008) Involvement of hepcidin in the anemia of multiple myeloma. Clin. Cancer Res. 14, 3262–3267 [DOI] [PubMed] [Google Scholar]
- 86. Zhang S., Chen Y., Guo W., Yuan L., Zhang D., Xu Y., Nemeth E., Ganz T., Liu S. (2014) Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell. Signal. 26, 2539–2550 [DOI] [PubMed] [Google Scholar]
- 87. Chen Y., Zhang S., Wang X., Guo W., Wang L., Zhang D., Yuan L., Zhang Z., Xu Y., Liu S. (2015) Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell. Signal. 27, 168–176 [DOI] [PubMed] [Google Scholar]
- 88. Macciò A., Madeddu C., Gramignano G., Mulas C., Tanca L., Cherchi M. C., Floris C., Omoto I., Barracca A., Ganz T. (2015) The role of inflammation, iron, and nutritional status in cancer-related anemia: results of a large, prospective, observational study. Haematologica 100, 124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. McCranor B. J., Langdon J. M., Prince O. D., Femnou L. K., Berger A. E., Cheadle C., Civin C. I., Kim A., Rivera S., Ganz T., Vaulont S., Xue Q. L., Walston J. D., Roy C. N. (2013) Investigation of the role of interleukin-6 and hepcidin antimicrobial peptide in the development of anemia with age. Haematologica 98, 1633–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Artz A. S., Xue Q. L., Wickrema A., Hesdorffer C., Ferrucci L., Langdon J. M., Walston J. D., Roy C. N. (2014) Unexplained anaemia in the elderly is characterised by features of low grade inflammation. Br. J. Haematol. 167, 286–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D. T., Back S. H., Kaufman R. J. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599 [DOI] [PubMed] [Google Scholar]
- 92. Vecchi C., Montosi G., Zhang K., Lamberti I., Duncan S. A., Kaufman R. J., Pietrangelo A. (2009) ER stress controls iron metabolism through induction of hepcidin. Science 325, 877–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Oliveira S. J., Pinto J. P., Picarote G., Costa V. M., Carvalho F., Rangel M., de Sousa M., de Almeida S. F. (2009) ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPα activity. PLoS One 4, e6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim A., Rivera S., Shprung D., Limbrick D., Gabayan V., Nemeth E., Ganz T. (2014) Mouse models of anemia of cancer. PLoS One 9, e93283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sasu B. J., Cooke K. S., Arvedson T. L., Plewa C., Ellison A. R., Sheng J., Winters A., Juan T., Li H., Begley C. G., Molineux G. (2010) Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 115, 3616–3624 [DOI] [PubMed] [Google Scholar]
- 96. Schmidt P. J., Toudjarska I., Sendamarai A. K., Racie T., Milstein S., Bettencourt B. R., Hettinger J., Bumcrot D., Fleming M. D. (2013) An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe−/− mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood 121, 1200–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guo S., Casu C., Gardenghi S., Booten S., Aghajan M., Peralta R., Watt A., Freier S., Monia B. P., Rivella S. (2013) Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Invest. 123, 1531–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ramos E., Ruchala P., Goodnough J. B., Kautz L., Preza G. C., Nemeth E., Ganz T. (2012) Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 120, 3829–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Anderson D. S., Heeney M. M., Roth U., Menzel C., Fleming M. D., Steen H. (2010) High-throughput matrix-assisted laser desorption ionization-time-of-flight mass spectrometry method for quantification of hepcidin in human urine. Anal. Chem. 82, 1551–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gutschow P., Schmidt P. J., Han H., Ostland V., Bartnikas T. B., Pettiglio M. A., Herrera C., Butler J. S., Nemeth E., Ganz T., Fleming M. D., Westerman M. (2015) A competitive enzyme-linked immunosorbent assay specific for murine hepcidin-1: correlation with hepatic mRNA expression in established and novel models of dysregulated iron homeostasis. Haematologica 100, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cartwright G. E., Lauritsen M. A., Jones P. J., Merrill I. M., Wintrobe M. M. (1946) The anemia of infection. I. Hypoferremia, hypercupremia, and alterations in porphyrin metabolism in patients. J. Clin. Invest. 25, 65–80, 10.1172/JCI101690 [DOI] [PMC free article] [PubMed] [Google Scholar]