

Background: LKB1 is an important serine/threonine kinase with key roles in cancer and metabolism. How LKB1 is regulated remains unclear.
Results: LPS induced S-nitrosylation of LKB1, resulting in its proteasomal degradation, which aggravated LPS-induced mortality in mice in vivo.
Conclusion: LPS induces LKB1 degradation through S-nitrosylation in macrophages.
Significance: We have unveiled a novel mechanism for regulating LKB1 stability.
Keywords: liver kinase B1 (LKB1), macrophage, nitric oxide, proteasome, S-nitrosylation, shock
Abstract
LKB1 (liver kinase B1) plays important roles in tumor suppression, energy metabolism, and, recently, in innate immune responses. However, how LKB1 is regulated under physiological or pathological conditions is still unclear. Here, we report that LKB1 protein (but not mRNA) was decreased in both LPS-treated RAW 264.7 cells and peritoneal macrophages isolated from LPS-challenged mice. Additional LPS treatment promoted protein ubiquitination and degradation of LKB1. Pharmacological inhibition or gene silencing of inducible NOS abrogated LPS-induced LKB1 degradation, whereas exposure of RAW 264.7 cells to S-nitroso-l-glutathione, a NO donor, triggered LKB1 S-nitrosylation. Consistently, mutation of one cysteine (C430S) in LKB1 prevented LPS-induced S-nitrosylation, ubiquitination, and degradation. Moreover, S-nitrosylation and ubiquitination of LKB1 were confirmed in macrophages from LPS-challenged mice in vivo. Co-administration of the inducible NOS inhibitor S-methylisothiourea or the proteasome inhibitor MG132 prevented LPS-induced LKB1 degradation and improved the survival rate. Finally, mice lacking LKB1 in macrophages had significantly lower survival rates in response to LPS challenge compared with wild-type mice. Thus, we concluded that LKB1 is degraded by LPS treatment via S-nitrosylation-dependent proteasome pathways, and this had a protective role in LPS-induced septic shock.
Introduction
NO is an important gaseous signaling molecule involved in many physiological processes, including cognitive function, synaptic plasticity, hormone secretion, and neurotransmission in the central nervous system (1), as well as in maintaining vascular homeostasis (2). NO is also a principle bactericidal mediator, which is produced primarily by inducible NOS (iNOS)2 upon LPS stimulation in macrophage cells as part of the immune response. There is increasing evidence that NO can act by directly modifying cysteine residues on target proteins. Cysteine nitrosylation (S-nitrosylation) is now regarded as a selective and specific signal controlled by cellular NO and has been closely associated with protein localization, stability, and function (3–7).
LKB1 (liver kinase B1) is a serine/threonine kinase that was first identified as a tumor suppressor responsible for Peutz-Jeghers syndrome (8, 9). It has been implicated in a wide range of processes such as cardiac function, angiogenesis, and glucose homeostasis (10–12). In a previous study, we found that LKB1 inhibited LPS-induced NF-κB activation in macrophages (13), which revealed a novel function of LKB1 in innate immune responses. However, how LKB1 is regulated during physiological and pathological conditions remains unknown.
LKB1 can undergo many different post-translational modifications, which influence its structure and activity. LKB1 can be phosphorylated on at least eight residues (14), and its phosphorylation at Ser-428 and Ser-307 was found to regulate its nucleocytoplasmic transport and activation (15, 16). LKB1 can also be acetylated, and its acetylation status is closely associated with its cytosolic localization and activity (17, 18). Recently, the reactive lipid species 4-hydroxy-trans-2-nonenal was also found to covalently modify LKB1 in its activation loop and thereby inhibit its activity (19). However, other post-translational modifications, especially those associated with LKB1 stability, have been less investigated. Here, we report that LPS-induced S-nitrosylation of LKB1 at Cys-430 promoted its ubiquitination and degradation.
Experimental Procedures
Reagents
LPS (Escherichia coli 0111:B4) and cycloheximide were obtained from Sigma-Aldrich. Protein A-Sepharose CL-4B beads were form GE Healthcare. S-Nitroso-l-glutathione (GSNO), S-methylisothiourea sulfate (SMT), and (S)-MG132 were from Cayman Chemical Co. (Ann Arbor, MI). Antibodies against iNOS were purchased from Abcam (Cambridge, MA). Antibodies against LKB1, ubiquitin, GAPDH, and actin were from Santa Cruz Biotechnology (Dallas, TX). Recombinant LKB1 protein was provided by Dr. Dietbert Neumann (Maastricht University, Maastricht, The Netherlands).
Cell Culture
RAW 264.7 macrophages were cultured in Dulbecco's modified Eagle's medium containing 10% heat-inactivated FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine in a humidified atmosphere of 5% CO2 at 37 °C. Resident peritoneal macrophages were isolated as described previously (20). Peritoneal cells were collected and cultured in RPMI 1640 medium supplemented with 10% FBS for 1 h at 37 °C. Non-adherent cells were then removed by gently washing three times with warm PBS.
Cell Transfections
For transfection of siRNAs, cells at 30–50% confluency were transfected using Lipofectamine RNAiMAX reagent (Invitrogen) following the manufacturer's protocol. For transfection of plasmids, the Amaxa Nucleofection system (Lonza, Köln, Germany) was used.
Animals
Myeloid-specific LKB1 knock-out (KO) mice were generated as we described previously (13). All mice were housed in a controlled environment (20 ± 2 °C, 12-h/12-h light/dark cycle) and maintained on a standard chow diet with free access to water. The animal protocol was reviewed and approved by the Animal Care and Use Committee of the University of Oklahoma Health Sciences Center.
Immunoprecipitation and Immunoblotting
Cells were lysed with radioimmune precipitation assay buffer (Santa Cruz Biotechnology) containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm glycerophosphate, 1 mm phenylmethylsulfonyl fluoride, 10 g/ml leupeptin, 10 g/ml aprotinin, 1 mm Na3VO4, and 10 mm NaF. Lysates were centrifuged at 10,000 × g for 10 min at 4 °C. Protein concentration was measured using the BCA protein assay (Bio-Rad). Cleared lysates were incubated overnight with the indicated antibodies and for 1 h with protein A-Sepharose beads. The pellets were then washed five times with ice-cold lysis buffer and resuspended in SDS sample buffer. Eluted immunoprecipitates or whole cell lysates were separated by SDS-PAGE and analyzed by immunoblotting.
Biotin Switch Assay
S-Nitrosylated proteins were detected using an S-nitrosylated protein detection assay kit (Cayman Chemical Co.). Briefly, free thiols were first blocked by incubation with thiol-specific methylthiolation reagent. Nitrosothiols were then selectively decomposed and labeled with biotin maleimide. Biotinylated proteins were detected using avidin-coupled reagents.
Plasmid Construction and Transfection
The LKB1 C430S mutant was generated using a QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The specific primers for site-directed mutagenesis were as follows: forward, 5′-CGGCTGTCGGCCTCCAAGCAGCAGTGA-3′; and reverse, 5′-TCACTGCTGCTTGGAGGCCGACAGCCG-3′. The mutation was verified by DNA sequencing. The Amaxa Nucleofection system was used for transfection of RAW 264.7 cells.
Statistical Analysis
Data are presented as the means ± S.E. of at least three independent experiments. The statistical significance of differences between two groups was analyzed by Student's t tests. Values of p < 0.05 were considered statistically significant.
Results
LPS Induces LKB1 Loss in Macrophages
To determine the effect of LPS on LKB1 expression, RAW 264.7 macrophages were treated with LPS for different time periods. As shown in Fig. 1 (A and B), LKB1 protein expression was decreased to 73 and 32% of control levels at 12 and 24 h, respectively, after LPS treatment. This expression was further decreased to 20 and 15% of control levels at 36 and 48 h, respectively, after LPS treatment. These results suggested that LPS induced LKB1 loss in a time-dependent manner. We further treated RAW 264.7 cells with different doses of LPS for 24 h. As shown in Fig. 1 (C and D), LPS also down-regulated LKB1 expression in a dose-dependent manner. Concentrations of LPS as low as 1 ng/ml were found to significantly induce LKB1 loss after 24 h. These results suggested that LPS suppressed LKB1 levels in macrophages.
FIGURE 1.

LPS induces LKB1 loss in macrophages. A, RAW 264.7 cells were treated with LPS (100 ng/ml) for the indicated times. B, quantification of LKB1 protein levels as in A (n = 4). *, p < 0.05 versus control. C, RAW 264.7 cells were treated with different doses of LPS for 24 h. D, quantification of LKB1 protein levels as in C (n = 3). #, p < 0.01 versus control.
LPS Induces LKB1 Loss via Proteasome-dependent Degradation
To further determine the times at which LPS down-regulates LKB1 expression, the mRNA levels of LKB1 were measured by real-time PCR. As shown in Fig. 2A, LKB1 mRNA expression was mildly decreased after 4 h of LPS treatment; however, it returned to normal levels after 8 h of treatment and remained unchanged thereafter. These results suggested that LPS-mediated LKB1 suppression was unrelated to LKB1 mRNA levels. Therefore, we further examined the protein stability of LKB1. To this end, RAW 264.7 cells were treated with the protein synthesis inhibitor cycloheximide in the absence or presence of LPS. In cycloheximide-treated cells, LKB1 protein was gradually reduced over 12 h (Fig. 2, B and C). At 12 h of cycloheximide treatment, LKB1 was decreased to 20% (Fig. 2, B and C). In the absence of LPS, the half-life of LKB1 was 5.2 h. As shown in Fig. 2 (B and C), LPS significantly accelerated LKB1 reduction, and the half-life of LKB1 was decreased to 2.6 h in the presence of LPS.
FIGURE 2.

LPS-induced LKB1 loss via proteasome-dependent degradation. A, real-time PCR quantification of LKB1 mRNA levels in RAW 264.7 cells treated with LPS (100 ng/ml) for different time periods (n = 3). B, RAW 264.7 cells were treated with cycloheximide (CHX; 10 μm) in the presence or absence of LPS (100 ng/ml) for the indicated time periods. C, quantification of LKB1 protein levels as in B (n = 3). D, RAW 264.7 cells were treated with or without LPS for 16 h, and whole cell lysates were immunoprecipitated (IP) with anti-LKB1 antibodies and stained with anti-ubiquitin (Ub) or anti-LKB1 antibodies. The blot is representative of three blots from three individual experiments. IB, immunoblot. E, RAW 264.7 cells were treated with or without LPS (100 ng/ml) and different concentrations of MG132 for 24 h. The blot is representative of three blots from three individual experiments.
Because LKB1 was reported to be ubiquitinated (21), we further investigated whether LPS promotes LKB1 ubiquitination. RAW 264.7 cells treated with or without LPS were analyzed for ubiquitination. As shown in Fig. 2D, LPS treatment dramatically enhanced the ubiquitination of LKB1. In addition, treatment with MG132 (0.5 μm), a potent proteasome inhibitor, reversed LPS-induced LKB1 loss (Fig. 2E). Taken together, these data suggested that LPS promoted LKB1 ubiquitination, resulting in its proteasome-dependent degradation.
LPS Induces LKB1 Degradation and Is NO-dependent
As shown in Fig. 3A, the expression of LKB1 was negatively associated with the levels of iNOS protein present in response to LPS. We thus hypothesized that iNOS-derived NO plays a role in mediating LKB1 degradation. To test this hypothesis, SMT was used to inhibit iNOS activity in cells. As shown in Fig. 3B, SMT abrogated LPS-induced nitrite production. Consistent with these results, SMT ablated LPS-induced LKB1 degradation (Fig. 3D). To rule out the possibility of nonspecific effects of SMT, RAW 264.7 cells were transfected with scrambled or iNOS-specific siRNA. As shown in Fig. 3 (E and F), iNOS siRNA dramatically decreased LPS-induced iNOS expression and cellular nitrite production compared with control siRNA. Consistent with these results, iNOS siRNA significantly increased LKB1 protein levels after 24 h of LPS treatment compared with control siRNA (Fig. 3, F and G). In contrast, treatment with the physiological NO donor GSNO induced LKB1 loss in a dose-dependent manner (Fig. 3, H and I). Overall, these data suggested that the LPS-induced LKB1 loss was NO-dependent.
FIGURE 3.

LPS-induced LKB1 degradation is NO-dependent. A, RAW 264.7 cells were treated with LPS for the indicated time periods. The blot is representative of four blots from four individual experiments. B, RAW 264.7 cells were treated with or without LPS and the iNOS inhibitor SMT (200 μm) for 24 h. Nitrite concentrations in the culture supernatants were measured with the Griess test (n = 6). ***, p < 0.001 versus control. NS, not significant. C, RAW 264.7 cells were treated as described for B, and cell lysates were analyzed for LKB1 expression. The blot is representative of three blots from three individual experiments. D, quantification of LKB1 protein levels as in C (n = 6). ***, p < 0.001 versus control. E, RAW 264.7 cells were transfected with control (si-control) or iNOS (si-iNOS) siRNA (100 nm) for 48 h and treated with LPS for 0, 12, and 24 h. Nitrite concentrations in the culture supernatants were measured with the Griess test (n = 3). *, p < 0.05 versus siRNA control; #, p < 0.01 versus siRNA control. F, RAW 264.7 cells were treated as described for E, and cell lysates were analyzed for LKB1 expression. The blot is representative of three blots from three individual experiments. G, quantification of LKB1 protein levels as in F (n = 3). *, p < 0.05 versus siRNA control. H, RAW 264.7 cells were treated with different doses of GSNO for 24 h. I, quantification of LKB1 protein levels as in H (n = 3). #, p < 0.01 versus control.
LKB1 Can Be S-Nitrosylated
NO has been reported to regulate protein stability via S-nitrosylation of targets (22). As our results indicated that the LPS-induced LKB1 loss was NO-dependent, we next investigated whether LKB1 can be S-nitrosylated. Recombinant LKB1 proteins were incubated with light-exposed GSNO (degraded GSNO) or fresh GSNO, and S-nitrosylation of LKB1 was examined using the biotin switch assay. As shown in Fig. 4 (A and B), fresh GSNO treatment significantly increased S-nitrosylation of LKB1 compared with degraded GSNO used as a control. Furthermore, RAW 264.7 cells were treated with GSNO and examined for S-nitrosylated LKB1. As shown in Fig. 4 (C and D), GSNO significantly increased S-nitrosylated LKB1 levels in macrophage cells. Next, we investigated S-nitrosylation of LKB1 in LPS-treated cells. As shown in Fig. 4 (E and F), LPS significantly increased the ratio of S-nitrosylated LKB1 and total LKB1 protein levels. These results indicated that LKB1 could be S-nitrosylated by LPS-induced NO effects.
FIGURE 4.
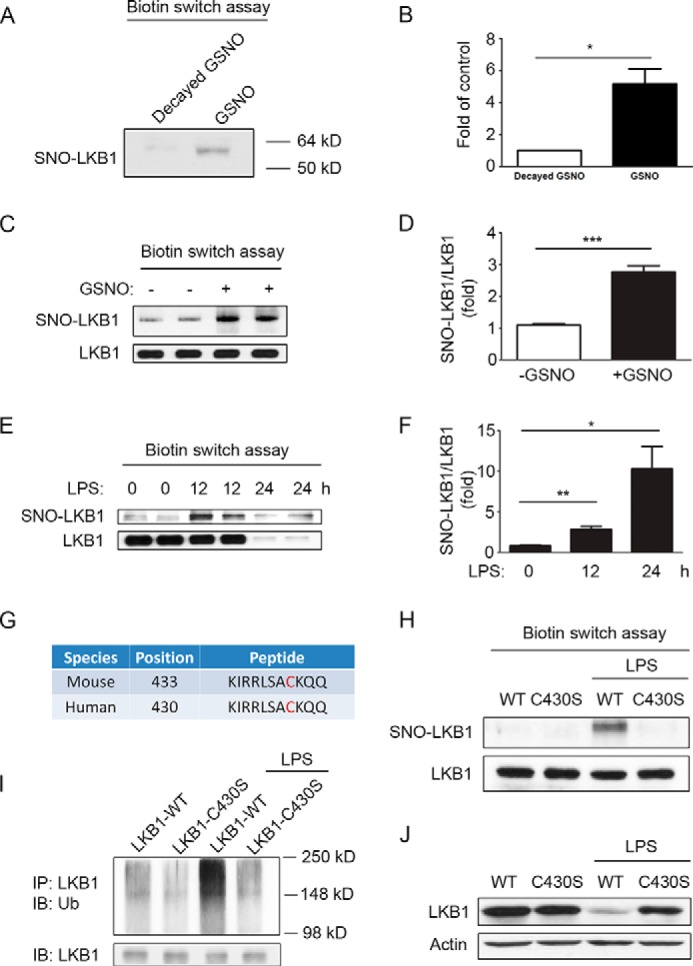
LKB1 can be S-nitrosylated, and S-nitrosylation of LKB1 regulates its ubiquitination and degradation. A, recombinant LKB1 protein was incubated with degraded or fresh GSNO (100 nm) for 30 min. After removal of the residual GSNO, protein samples were subjected to biotin switch assays, further immunoprecipitated with anti-LKB1 antibodies, and detected with anti-biotin antibodies. SNO-LKB1, S-nitrosylated LKB1. B, quantification of LKB1 protein levels as in A (n = 3). *, p < 0.05 versus degraded GSNO control. C, RAW 264.7 cells were treated with or without GSNO (500 nm) for 12 h and then subjected to biotin switch assays. Dissolved protein samples were immunoprecipitated with anti-LKB1 antibodies and detected with anti-biotin antibodies. The total levels of LKB1 in dissolved protein samples were also detected. D, quantification of S-nitrosylated LKB1/LKB1 ratio as in C (n = 6). ***, p < 0.001 versus control. E, RAW 264.7 cells were treated with LPS for different time periods and then subjected to biotin switch assays. F, quantification of S-nitrosylated LKB1/LKB1 ratio as in E (n = 6). *, p < 0.05 versus 0-h control; #, p < 0.01 versus 0-h control. G, predictions of S-nitrosylated sites in mouse and human LKB1 protein sequences using GPS-SNO 1.0 software. H, RAW 264.7 cells were transfected with WT or C430S mutant plasmid for 24 h and treated with or without LPS for 12 h. Cells were then subjected to biotin switch assays and further examined for S-nitrosylated and total LKB1 levels. The blot is representative of three blots from three individual experiments. I, RAW 264.7 cells transfected with WT or C430S mutant plasmid for 24 h were treated with or without LPS for 16 h. Whole cell lysates were immunoprecipitated (IP) with anti-LKB1 antibodies and stained with anti-ubiquitin (Ub) or anti-LKB1 antibodies. The blot is representative of two blots from two individual experiments. IB, immunoblot. J, RAW 264.7 cells transfected with WT or C430S mutant plasmid for 24 h were treated with or without LPS for 24 h. Cell lysates were then subjected to Western blot analysis for LKB1 expression. The blot is representative of three blots from three individual experiments.
To further identify which cysteine residue are responsible for S-nitrosylation of LKB1, the LKB1 protein sequence was analyzed using the S-nitrosylation site prediction software GPS-SNO 1.0 (23). As shown in Fig. 4G, a single site (Cys-430) in the human LKB1 sequence (Cys-433 in the mouse sequence) was identified. To determine whether Cys-430 is the target for S-nitrosylation, we constructed an LKB1 mutant plasmid with Cys-430 replaced by Ser (C430S). The WT and mutant LKB1 plasmids were introduced into RAW 264.7 cells by electrotransfection. Transfected cells were treated with or without LPS, and cell lysates were prepared and analyzed for S-nitrosylated LKB1. As shown in Fig. 4H, LPS dramatically increased the level of S-nitrosylated LKB1 in RAW 264.7 cells transfected with WT LKB1, but not in cells transfected with the C430S mutant, indicating that Cys-430 in LKB1 is the site for S-nitrosylation.
S-Nitrosylation of LKB1 Regulates Its Ubiquitination and Degradation
As shown in Fig. 4E, LPS-increased S-nitrosylation preceded the degradation of LKB1, suggesting a role for S-nitrosylation in regulating LKB1 protein stability. Therefore, we next investigated the ubiquitination of WT LKB1 and the C430S mutant in response to LPS. As shown in Fig. 4I, LPS increased the level of detectable ubiquitinated LKB1 in cells transfected with WT LKB1. In contrast, LPS did not alter LKB1 ubiquitination in cells transfected with the C430S mutant. Consistently, LPS induced degradation of WT LKB1, whereas mutation of Cys-430 prevented this degradation. Taken together, these results indicated that S-nitrosylation regulated LKB1 protein stability in macrophage cells.
LPS Induces S-Nitrosylation and Degradation of LKB1 in Macrophages in Vivo
We next investigated whether LPS promotes LKB1 degradation via S-nitrosylation in vivo. To this end, LKB1 protein levels were monitored in peritoneal macrophages isolated from mice challenged by injection of PBS or LPS. As shown in Fig. 5 (A and B), LPS challenge significantly decreased LKB1 protein levels in peritoneal macrophages. Additionally, LPS challenge significantly increased S-nitrosylation and ubiquitination of LKB1 in peritoneal macrophages in mice (Fig. 5, C and D, respectively). Pretreatment of mice with the iNOS inhibitor SMT (Fig. 5, E and F) and the proteasome inhibitor MG132 (Fig. 5, G and H) dramatically prevented LPS-induced LKB1 degradation in vivo. These data suggested an important role of S-nitrosylation in regulating LKB1 levels in vivo.
FIGURE 5.

LPS induces S-nitrosylation and degradation of LKB1 in macrophages in vivo. A, C57BL/6 mice were intraperitoneally injected with PBS or LPS (20 mg/kg) for 24 h, and peritoneal macrophages were then isolated and lysed for Western blotting. Three mice were used in each group, and two individual experiments were performed. B, quantification of LKB1 protein levels as in A (n = 6). **, p < 0.01 versus PBS control. C, C57BL/6 mice were injected with thioglycollate for 3 days and then challenged with PBS or LPS for 12 h. Peritoneal macrophages from five mice were pooled together for biotin switch assays. The blot is representative of two individual experiments. SNO-LKB1, S-nitrosylated LKB1. D, C57BL/6 mice were injected with thioglycollate for 3 days and then challenged with PBS or LPS for 16 h. Cell lysates of peritoneal macrophages were immunoprecipitated (IP) with anti-LKB1 antibodies and stained with anti-ubiquitin (Ub) antibodies. The blot is representative of three blots from three individual experiments. IB, immunoblot. E, mice were pretreated with PBS or SMT (10 mg/kg) for 1 h and then challenged with LPS (10 mg/kg) for 12 h. Peritoneal macrophages were isolated and analyzed for LKB1 expression. F, quantification of LKB1 protein levels as in E (n = 3). ***, p < 0.001 versus control. NS, not significant. G, mice were pretreated with dimethyl sulfoxide or MG132 (10 μm/kg) for 1 h and then challenged with LPS (10 mg/kg) for 12 h. Peritoneal macrophages were isolated and analyzed for LKB1 expression. H, quantification of LKB1 protein levels as in G and E (n = 3). ***, p < 0.001 versus control.
Preventing LKB1 Degradation Protects Mice from LPS-induced Death
We investigated if LKB1 loss in macrophages affects mouse mortality induced by LPS. To this end, WT and macrophage-specific LKB1 KO mice were challenged with lethal doses of LPS. As shown in Fig. 6A, LKB1 KO mice had a significantly lower survival rate than WT mice, suggesting that macrophage LKB1 protects mice from LPS-induced death.
FIGURE 6.
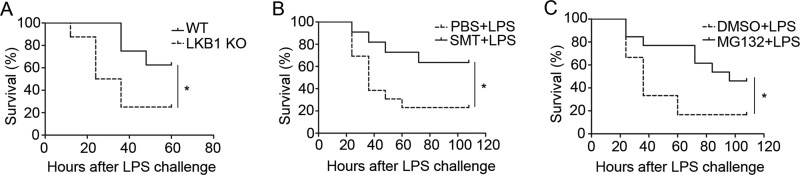
Preventing LKB1 degradation protects mice from LPS-induced death. A, WT and macrophage-specific LKB1 KO mice were challenged with LPS (25 mg/kg intraperitoneally), and their mortality was monitored every 12 h. The differences between WT and LKB1 KO mice were analyzed by log-rank tests (n = 8). *, p < 0.05 versus WT control. B, survival rates of mice pretreated with PBS or SMT (10 mg/kg) for 1 h and then challenged with LPS (25 mg/kg intraperitoneally; n = 11–13). *, p < 0.05 versus PBS control. C, survival rates of mice pretreated with dimethyl sulfoxide (DMSO) or MG132 (10 μm/kg) for 1 h and then challenged with LPS (25 mg/kg intraperitoneally; n = 12–13). *, p < 0.05 versus dimethyl sulfoxide control.
As LKB1 was degraded by the S-nitrosylation-dependent proteasome pathway, we next examined whether preventing its degradation improves mouse survival during LPS challenge. To achieve this, mice were pretreated with PBS or the iNOS inhibitor SMT and then challenged with LPS. As shown in Fig. 6B, mice pretreated with SMT showed a significantly improved survival rate when challenged with LPS. We further pretreated mice with dimethyl sulfoxide or the proteasome inhibitor MG132, followed by LPS challenge. As shown in Fig. 6C, mice pretreated with MG132 also showed a significantly improved survival rate when challenged with LPS. These data indicated that preventing LKB1 degradation protected the mice from LPS-induced death.
Discussion
LKB1 is an important kinase regulating tumor growth, energy metabolism, cardiovascular functions, and immune responses. Here, we have reported a novel modification of LKB1 protein that regulates its stability. We observed that LKB1 was degraded by LPS and that this degradation was NO-dependent. Mechanistically, we found that NO induced S-nitrosylation of LKB1 at Cys-430. Mutation of Cys-430 abrogated LPS-induced S-nitrosylation, ubiquitination, and degradation. Finally, we found that mice lacking LKB1 in macrophages had a higher death rate in response to LPS challenge, whereas preventing LKB1 degradation using a proteasome inhibitor significantly improved survival. Overall, our results demonstrate for the first time that LKB1 is degraded under pathological conditions through components of the S-nitrosylation-dependent proteasome pathway.
LKB1 is ubiquitously expressed in embryonic and adult tissues and is thought to be constitutively expressed, with its function carried out exclusively through regulation of its kinase activity. However, our results indicated that LKB1 can be regulated at the protein level, which plays a critical role in affecting LKB1 function. This was consistent with previous studies suggesting that LKB1 protein expression is regulated. For example, sex hormones such as androgen and estrogen were reported to regulate LKB1 gene expression through affecting LKB1 promoter activity (24, 25). SIRT1, a conserved NAD+-dependent deacetylase implicated in delayed aging, was found to promote deacetylation, ubiquitination, and proteasome-mediated degradation of LKB1 (26).
In this study, we demonstrated that LKB1 was degraded by LPS via components of the S-nitrosylation-dependent proteasome pathway. S-Nitrosylation is a NO-mediated modification of protein cysteine residues. NO production has been found to be increased in many infectious diseases (27–29) and in a variety of other human disorders such as ischemia/stroke (30) and tumor growth (31). In addition, oxidants such as oxidized LDL and hydrogen peroxide were found to either increase iNOS expression or up-regulate iNOS enzyme activity and NO levels (32–34). Therefore, our study suggests that NO-mediated S-nitrosylation of LKB1 may represent a general mechanism in various diseases.
Importantly, we found that mice lacking LKB1 in macrophages were much more sensitive to LPS challenge and had a higher rate of death, suggesting a protective role of LKB1 in LPS-induced septic shock. We reported previously that LKB1 suppresses LPS-induced NF-κB activation and pro-inflammatory responses (13). LKB1 deficiency in macrophages results in increased production of pro-inflammatory cytokines, enzymes, and other mediators (13). Excessive production of these mediators leads to tissue injury, organ failure, and even death (35). Therefore, the increased death of LKB1-deficient mice may be due to the excessive production of proinflammatory mediators through overactivation of the NF-κB signaling pathway. More importantly, preventing LKB1 degradation by iNOS inhibitors and proteasome inhibitors was found to protect mice from LPS-induced death. This result suggests that therapeutic agents that modulate LKB1 stability may potentially contribute to future clinical treatments.
In summary, we have demonstrated for the first time that LKB1 is S-nitrosylated by NO, which promotes its proteasomal degradation. Preventing LKB1 loss through inhibiting S-nitrosylation or proteasome activity may constitute a viable strategy for treating LPS-induced septic shock and possibly other disorders associated with increased NO production.
Author Contributions
Z. L. performed most of experiments, analyzed the data, and wrote the draft of the manuscript. X. D., H. Z., and M. Z. performed some of the experiments. M.-H. Z. designed the experiments, obtained the funding, and wrote the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL079584, HL080499, HL074399, HL089920, HL096032, HL105157, HL110488, and AG047776 (to M.-H. Z.) and HL128014 (to Z. Xie). The authors declare that they have no conflicts of interest with the contents of this article.
- iNOS
- inducible NOS
- GSNO
- S-nitroso-l-glutathione
- SMT
- S-methylisothiourea sulfate
- KO
- knock-out.
References
- 1. Guix F. X., Uribesalgo I., Coma M., Muñoz F. J. (2005) The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 76, 126–152 [DOI] [PubMed] [Google Scholar]
- 2. Tousoulis D., Kampoli A. M., Tentolouris C., Papageorgiou N., Stefanadis C. (2012) The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 10, 4–18 [DOI] [PubMed] [Google Scholar]
- 3. Azad N., Vallyathan V., Wang L., Tantishaiyakul V., Stehlik C., Leonard S. S., Rojanasakul Y. (2006) S-Nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 281, 34124–34134 [DOI] [PubMed] [Google Scholar]
- 4. Qu J., Liu G. H., Wu K., Han P., Wang P., Li J., Zhang X., Chen C. (2007) Nitric oxide destabilizes Pias3 and regulates sumoylation. PLoS ONE 2, e1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nott A., Watson P. M., Robinson J. D., Crepaldi L., Riccio A. (2008) S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455, 411–415 [DOI] [PubMed] [Google Scholar]
- 6. Numajiri N., Takasawa K., Nishiya T., Tanaka H., Ohno K., Hayakawa W., Asada M., Matsuda H., Azumi K., Kamata H., Nakamura T., Hara H., Minami M., Lipton S. A., Uehara T. (2011) On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc. Natl. Acad. Sci. U.S.A. 108, 10349–10354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Z. Q., Sunico C. R., McKercher S. R., Cui J., Feng G. S., Nakamura T., Lipton S. A. (2013) S-Nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc. Natl. Acad. Sci. U.S.A. 110, 3137–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., Toro T., Bodmer W., Olschwang S., Olsen A. S., Stratton M. R., de la Chapelle A., Aaltonen L. A. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 [DOI] [PubMed] [Google Scholar]
- 9. Jenne D. E., Reimann H., Nezu J., Friedel W., Loff S., Jeschke R., Müller O., Back W., Zimmer M. (1998) Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 18, 38–43 [DOI] [PubMed] [Google Scholar]
- 10. Ikeda Y., Sato K., Pimentel D. R., Sam F., Shaw R. J., Dyck J. R., Walsh K. (2009) Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 284, 35839–35849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohashi K., Ouchi N., Higuchi A., Shaw R. J., Walsh K. (2010) LKB1 deficiency in Tie2-Cre-expressing cells impairs ischemia-induced angiogenesis. J. Biol. Chem. 285, 22291–22298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z., Zhang W., Zhang M., Zhu H., Moriasi C., Zou M.-H. (2015) Liver kinase B1 suppresses lipopolysaccharides-induced NF-κB activation in macrophages. J. Biol. Chem. 290, 2312–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alessi D. R., Sakamoto K., Bayascas J. R. (2006) LKB1-dependent signaling pathways. Annu. Rev. Biochem. 75, 137–163 [DOI] [PubMed] [Google Scholar]
- 15. Xie Z., Dong Y., Scholz R., Neumann D., Zou M.-H. (2008) Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117, 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie Z., Dong Y., Zhang J., Scholz R., Neumann D., Zou M.-H. (2009) Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 29, 3582–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lan F., Cacicedo J. M., Ruderman N., Ido Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pillai V. B., Sundaresan N. R., Kim G., Gupta M., Rajamohan S. B., Pillai J. B., Samant S., Ravindra P. V., Isbatan A., Gupta M. P. (2010) Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 285, 3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Calamaras T. D., Lee C., Lan F., Ido Y., Siwik D. A., Colucci W. S. (2012) Post-translational modification of serine/threonine kinase LKB1 via adduction of the reactive lipid species 4-hydroxy-trans-2-nonenal (HNE) at lysine residue 97 directly inhibits kinase activity. J. Biol. Chem. 287, 42400–42406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X., Goncalves R., Mosser D. M. (2008) The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nony P., Gaude H., Rossel M., Fournier L., Rouault J. P., Billaud M. (2003) Stability of the Peutz-Jeghers syndrome kinase LKB1 requires its binding to the molecular chaperones Hsp90/Cdc37. Oncogene 22, 9165–9175 [DOI] [PubMed] [Google Scholar]
- 22. Kim S., Wing S. S., Ponka P. (2004) S-Nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 24, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xue Y., Liu Z., Gao X., Jin C., Wen L., Yao X., Ren J. (2010) GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS ONE 5, e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown K. A., McInnes K. J., Takagi K., Ono K., Hunger N. I., Wang L., Sasano H., Simpson E. R. (2011) LKB1 expression is inhibited by estradiol-17β in MCF-7 cells. J. Steroid Biochem. Mol. Biol. 127, 439–443 [DOI] [PubMed] [Google Scholar]
- 25. McInnes K. J., Brown K. A., Hunger N. I., Simpson E. R. (2012) Regulation of LKB1 expression by sex hormones in adipocytes. Int. J. Obes. 36, 982–985 [DOI] [PubMed] [Google Scholar]
- 26. Zu Y., Liu L., Lee M. Y., Xu C., Liang Y., Man R. Y., Vanhoutte P. M., Wang Y. (2010) SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 106, 1384–1393 [DOI] [PubMed] [Google Scholar]
- 27. Adamson D. C., Wildemann B., Sasaki M., Glass J. D., McArthur J. C., Christov V. I., Dawson T. M., Dawson V. L. (1996) Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science 274, 1917–1921 [DOI] [PubMed] [Google Scholar]
- 28. Stachura J., Konturek J. W., Karczewska A., Domschke W., Popiela T., Konturek S. J. (1996) Helicobacter pylori from duodenal ulcer patients expresses inducible nitric oxide synthase immunoreactivity in vivo and in vitro. J. Physiol. Pharmacol. 47, 131–135 [PubMed] [Google Scholar]
- 29. Nicholson S., Bonecini-Almeida Mda G., Lapa e Silva J. R., Nathan C., Xie Q. W., Mumford R., Weidner J. R., Calaycay J., Geng J., Boechat N., Linhares C., Rom W., Ho J. L. (1996) Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183, 2293–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haywood G. A., Tsao P. S., von der Leyen H. E., Mann M. J., Keeling P. J., Trindade P. T., Lewis N. P., Byrne C. D., Rickenbacher P. R., Bishopric N. H., Cooke J. P., McKenna W. J., Fowler M. B. (1996) Expression of inducible nitric oxide synthase in human heart failure. Circulation 93, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 31. Jenkins D. C., Charles I. G., Thomsen L. L., Moss D. W., Holmes L. S., Baylis S. A., Rhodes P., Westmore K., Emson P. C., Moncada S. (1995) Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. U.S.A. 92, 4392–4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Romeo G. R., Moulton K. S., Kazlauskas A. (2007) Attenuated expression of profilin-1 confers protection from atherosclerosis in the LDL receptor null mouse. Circ. Res. 101, 357–367 [DOI] [PubMed] [Google Scholar]
- 33. Huang H., Koelle P., Fendler M., Schröttle A., Czihal M., Hoffmann U., Conrad M., Kuhlencordt P. J. (2014) Induction of inducible nitric oxide synthase (iNOS) expression by oxLDL inhibits macrophage derived foam cell migration. Atherosclerosis 235, 213–222 [DOI] [PubMed] [Google Scholar]
- 34. Banan A., Fields J. Z., Zhang Y., Keshavarzian A. (2001) iNOS upregulation mediates oxidant-induced disruption of F-actin and barrier of intestinal monolayers. Am. J. Physiol. Gastrointest. Liver Physiol. 280, G1234–G1246 [DOI] [PubMed] [Google Scholar]
- 35. Laskin D. L., Pendino K. J. (1995) Macrophages and inflammatory mediators in tissue injury. Annu. Rev. Pharmacol. Toxicol. 35, 655–677 [DOI] [PubMed] [Google Scholar]