

Background: GPS2 is a multifunctional protein controlling cellular homeostasis, inflammation, and lipid metabolism.
Results: Arginine methylation modulates GPS2 interaction with TBL1 and prevents its degradation upon Siah2 ubiquitination.
Conclusion: A tightly regulated balance between stabilization and degradation determines GPS2 levels.
Significance: Understanding the molecular mechanisms controlling GPS2 expression and localization is critical for dissecting its multiple roles in the cell.
Keywords: gene expression, protein degradation, protein methylation, transcription corepressor, ubiquitin, GPS2, PRMT6, Siah2, TBL1
Abstract
G protein pathway suppressor 2 (GPS2) is a multifunctional protein involved in the regulation of a number of metabolic organs. First identified as part of the NCoR-SMRT corepressor complex, GPS2 is known to play an important role in the nucleus in the regulation of gene transcription and meiotic recombination. In addition, we recently reported a non-transcriptional role of GPS2 as an inhibitor of the proinflammatory TNFα pathway in the cytosol. Although this suggests that the control of GPS2 localization may be an important determinant of its molecular functions, a clear understanding of GPS2 differential targeting to specific cellular locations is still lacking. Here we show that a fine balance between protein stabilization and degradation tightly regulates GPS2 nuclear function. Our findings indicate that GPS2 is degraded upon polyubiquitination by the E3 ubiquitin ligase Siah2. Unexpectedly, interaction with the exchange factor TBL1 is required to protect GPS2 from degradation, with methylation of GPS2 by arginine methyltransferase PRMT6 regulating the interaction with TBL1 and inhibiting proteasome-dependent degradation. Overall, our findings indicate that regulation of GPS2 by posttranslational modifications provides an effective strategy for modulating its molecular function within the nuclear compartment.
Introduction
GPS2 is a small transcriptional cofactor that was originally identified while screening for suppressors of Ras activation in the yeast pheromone response pathway (1). The nuclear role of GPS2 has been reported in numerous studies describing functional interactions between GPS2 and several transcriptional regulators, including the NCoR-SMRT corepressor complex, the histone acetyltransferase p300, the DNA repair proteins MSH4/MSH5, and DNA-binding transcription factors p53, RFX4 (regulatory factor X4), FXR (farnesoid X receptor), SHP (small heterodimer partner), HNF4 (hepatocyte nuclear factor 4), LXR (liver X receptor), and PPARγ (peroxisome proliferator-activated receptor γ) (2–9). This body of work implicates GPS2 in a number of important nuclear functions, including transcriptional repression and activation, the cell cycle, and meiosis (2–4, 6–8, 10–13). In addition, our recent work has identified an unexpected, non-transcriptional role for GPS2 in the cytoplasm, specifically linking GPS2 with the modulation of TNFα signaling and JNK activity (8). Interestingly, our findings reveal that GPS2 complementary transcriptional and non-transcriptional functions rely on a conserved regulatory strategy on the basis of the inhibition of ubiquitin-conjugating complexes that are responsible for the formation of non-degradative Lys-63 ubiquitin chains (TRAF2/Ubc13 in the cytosol and RNF8/Ubc13 in the nucleus) (8, 9).
Although this suggests that regulatory mechanisms must exist to dictate GPS2 intracellular localization and to control the inhibitory activity of GPS2 on different ubiquitin complexes, not much is known about the pathways and posttranslational modifications that can regulate GPS2 in vitro and/or in vivo or about the physiological strategies governing GPS2 expression, stabilization, and/or degradation in different cellular compartments, cell types, or tissues. Other components of the NCoR-SMRT complex, including NCoR, SMRT, TBL1/TBLR1, and HDAC3, have been reported to be modified in response to different signaling pathways via phosphorylation, sumoylation, and ubiquitination events that contribute to the regulation of both their function and cellular localization (14–18). Recent reports of posttranslational modification by arginine methylation and sumoylation suggest that a similarly complex picture is likely to emerge for GPS2 as well (19–21). Interestingly, GPS2 was linked to cutaneous cancers and T cell lymphomas caused by human papilloma viruses and human T cell leukemia virus type 1, with the interaction between GPS2 and the corresponding viral interacting protein promoting the degradation of GPS2 (3, 22, 23). This suggests that unrelated viruses may have evolved similar mechanisms to get rid of an undesired host protein, possibly by hijacking an existing regulatory system.
On the basis of our previous work, which characterized an active derepression mechanism for the dismissal of the NCoR-SMRT corepressor complex on the basis of TBL1/TBLR1-dependent recruitment of the Siah/UbcH5 ubiquitin machinery (24–26), we explored the possibility that the GPS2 protein level would be modulated similarly. Our results reveal the existence of a similar but distinct strategy, with nuclear GPS2 levels being regulated by a fine balance between protein degradation and stabilization. Unexpectedly, under these circumstances, TBL1 plays a protective role against Siah-dependent degradation, with GPS2 interaction with TBL1 being modulated by the protein arginine methyltransferase 6 (PRMT6).
Experimental Procedures
Cells, Antibodies, siRNA, and Other Reagents
The HeLa and 293T human cell lines were grown in DMEM supplemented with 10% FBS. TBL1 KO ES cells have been generated and described previously (24). To inhibit proteasomal degradation, cells were treated with MG132 10 nm (InSolution 474791, Calbiochem-EMD) for 4 h. Commercial antibodies used were as follows: anti-ubiquitin (P4D1 clone, Cell Signaling Technology), anti-βtubulin (TUB 2.1 clone, Sigma), anti-HDAC2 (Santa Cruz Biotechnology, catalog no. sc-9959), anti-PRMT6 (catalog no. A300–929A, Bethyl Labs), anti-H3R17me2a (catalog no. ab8284, Abcam), anti-HA-HRP (Roche), and anti-FLAG-M2 and anti-FLAG-HRP (Sigma). Guinea pig antibody against TBL1 and rabbit antibodies against GPS2 have been described previously (8, 24). siRNAs against human TBL1 and PRMT6 were purchased from Ambion, and Siah1 and Siah2 were from Qiagen. Nonspecific scrambled siRNA and siLUC were included as negative controls in each experiment.
Cloning and Site-directed Mutagenesis
Deletant expression vectors for GPS2 were generated by PCR amplification of the murine full-length GPS2 cDNA and subcloning into the pCMX-HA-FLAG mammalian expression vector using standard molecular cloning techniques (49). All vectors were validated by sequencing prior to use. Transient transfection in mammalian cells was performed for 24 h using Lipofectamine 2000 reagent according to the protocol of the manufacturer (Promega).
Protein Extracts, Immunoprecipitation, and Western Blotting
For fractionated nuclear and cytosolic protein extraction, cells were rinsed in PBS, harvested, and lysed by syringe homogenization in hypotonic buffer (10 mm Hepes (pH 7.9), 1 mm EDTA, 210 mm mannitol, 70 mm sucrose, 50 mm NaF, 0.5 mm PMSF, and protease inhibitor mixture (Roche)). After precipitation of the nuclei by low-speed centrifugation, the supernatant containing cytosolic proteins was recovered, and the nuclear pellet was lysed for 20 min in high-salt buffer (20 mm Tris-HCl (pH 8), 25% glycerol, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm DTT, 2 mm Na3VO4, 50 mm NaF, 1 mm PMSF, and protease inhibitor mixture), followed by removal of all membrane debris by high-speed centrifugation. The concentration of fractionated protein extracts was measured using a colorimetric Bio-Rad assay. Extracts were boiled in SDS sample buffer and loaded directly onto precast Bio-Rad gels. For immunoprecipitation, protein extracts were incubated with the specific antibody overnight at 4 °C after adjusting the buffer to a final concentration of 150 mm NaCl and 0.5% Nonidet P-40 and then incubated for 2 h with protein A/G-agarose beads (Santa Cruz Biotechnology), washed extensively, separated by electrophoresis, transferred onto PVDF membranes (Millipore), and subjected to Western blotting following standard protocols. For in vivo methylation assays, HEK293T cells were lysed 48 h after transfection in 20 mm Tris (pH 7.4), 120 mm NaCl, 1% Triton X-100, and EDTA-free protease inhibitors (Complete, Roche) and sonicated three times for 10 s each. Cell lysates were immunoprecipitated at 4 °C for 2 h with anti-FLAG M2-conjugated agarose beads (catalog no. A2220, Sigma). For protein extraction from the adipose tissue of wild-type and Siah2 KO mice, the epididymal adipose tissue depot was homogenized in a denaturing buffer (50 mm Tris-Cl (pH 7.4) with 150 mm NaCl, 1 mm EDTA, 1% Igepal CA 630, 0.5% sodium deoxycholate, and 0.1% SDS) with 10 mm NEM (N-ethylmaleimide), 1 mm PMSF, 10 mg/ml aprotinin, 1 mg/ml pepstatin, 5 mg/ml leupeptin, and 2 mm sodium orthovanadate. The lysates were centrifuged at 14,000 × g for 10 min at 4 °C, and the supernatant protein concentrations were determined by BCA assay (Thermo Scientific) prior to Western blotting.
Recombinant Protein Expression, in Vitro Binding Assay, and in Vitro Arginine Methylation Assay
Fusion proteins containing GST bound to GPS2 or to TBL1 (either full-length, Nt,5 or Ct) (24) were expressed in Escherichia coli BL21 and purified from homogenized lysates with glutathione-agarose beads (Sigma) as described previously (24). For interaction studies, the immobilized GST-fusion proteins were incubated for 2 h with HA-GPS2 full-length or mutants that were generated by in vitro translation-transcription (Promega TnT quick-coupled transcription/translation system). After extensive washes with GST binding buffer, the interacting proteins were separated by SDS-PAGE, and Western blotting was performed with HA-HRP or FLAG-HRP antibodies. Methylation of GST-GPS2 and other substrates was performed as described previously (27). Briefly, 1 μg of substrate was incubated with 1 μl of S-adenosyl-l-[methyl-3H]methionine (PerkinElmer Life Sciences) and 0.5 μg of purified recombinant enzyme for 1 h at 30 °C. The reaction was stopped by addition of sample loading buffer (80 mm Tris-HCl (pH 6.8), 30% glycerol, 10% SDS, 0.6 m DTT, and 0.012% bromphenol blue). Samples were separated on a 10% SDS-polyacrylamide gel and transferred to a PVDF membrane. EN3HANCETM (PerkinElmer Life Sciences) was added to the membrane according to the instructions of the manufacturer. The PVDF membrane was then exposed to an x-ray film.
RNA Isolation and Quantitative PCR Analysis
Cells were lysed directly on the plate in the presence of highly denaturing buffer, and the lysate was homogenized through a QIAshredder spin column (RNeasy kit, Qiagen). Ethanol was added to adjust the binding conditions, and the sample was applied to an RNeasy spin column for binding of total RNA according to the protocol of the manufacturer (Qiagen). First-strand cDNA synthesis from the total RNA template was performed using iScript Reverse Transcriptase Supermix (Bio-Rad), followed by SYBR Green quantitative PCR amplification on a Viia7 thermocycler (Life Technologies). Normalization was performed using specific amplification of the Cyclophilin A gene. PCRs were performed in triplicate for each biological replicate, and the results are presented as mean ± S.D. The p value was calculated using Student's t test.
Results
Identification of the GPS2 Minimal Nuclear Localization Domain (NLD)
With the final goal of identifying the molecular determinants of GPS2 nuclear localization, we first overexpressed either HA-tagged GPS2 full-length or deletants corresponding to the Nt and Ct domains in 293T cells. Western blot analysis of fractionated extracts indicated that full-length protein and the Nt domain were present in both nuclear extracts (NEs) and cytosolic extracts (CEs), whereas the Ct domain was observed only in the cytoplasm (Fig. 1A), suggesting that the N-terminal domain is required for GPS2 nuclear targeting.
FIGURE 1.

TBL1 interaction with the GPS2 amino terminus is required for GPS2 stabilization. A, Western blot analysis of NEs and CEs from 293T cells transfected with HA-tagged GPS2 WT, Nt, or Ct. Blotting for tubulin and HDAC2 provided controls for CEs and Nes, respectively. B, Western blot analysis of 293T cells transfected with either HA-GPS2 or serial 20-aa deletions (aa 1–20, 1–40, 1–60, and 1–80) reveals that the first 40 aa are dispensable for GPS2 nuclear localization, whereas deletions beyond aa 60 impair protein expression. C and D, GPS2 protein level in nuclear extracts is decreased when TBL1 is down-regulated via specific siRNA (C) or upon genetic ablation in whole cell extracts from mouse ES cells (D) (the asterisk indicates a nonspecific band). E, Coomassie Blue staining of a 10% protein gel shows purification of recombinant GST-GPS2, alone, or coexpressed with GST-TBL1 from BL21 E. coli.
To further investigate the mechanism of GPS2 nuclear localization, serial deletions of the Nt domain were generated by progressively removing 20 amino acid sequences. As shown in Fig. 1B, both GPS2-Δ1–20 and GPS2-Δ1–40 are correctly targeted to the nucleus, indicating that the first 40 aa are dispensable for nuclear compartmentalization. However, the relevance of the following residues in terms of nuclear localization could not be evaluated because no protein expression was detected upon transfection of the GPS2-Δ1–60 and GPS2-Δ1–80 constructs in 293T or HeLa cells (Fig. 1B and data not shown). Because the very N-terminal region of GPS2 is critical for interacting with TBL1 (aa 1–53) and for promoting the correct assembly of the NCoR-SMRT corepressor complex (aa 53–90) (28), we hypothesized that the interaction with TBL1 might be required for GPS2 stabilization. In support of this hypothesis, we observed that the expression of GPS2-Δ1–40 is also reduced compared with wild-type GPS2 (Fig. 1B), that down-regulation of TBL1 via small interfering RNA leads to a decrease in the expression of GPS2 (Fig. 1C), and GPS2 protein level is strongly reduced upon TBL1 genetic ablation in mouse ES cells (Fig. 1D) (24). Conversely, coexpression of recombinant GPS2 together with TBL1 in E. coli results in GPS2 stabilization (Fig. 1E) in a similar fashion as what has been reported previously (28). Because these observations supported the hypothesis that the interaction with TBL1 via the very N terminus of GPS2 might be required for GPS2 stabilization, we generated a new construct by reintroducing amino acids 1–60 in the vector containing the Ct sequence of GPS2 (Fig. 2A). As expected, introducing this small domain was sufficient to rescue the binding of GPS2 to the Nt domain of TBL1 in a GST pulldown assay (Fig. 2B). More importantly, expression of the fusion protein could be detected in both the nucleus and cytosol (Fig. 2C). Therefore, our results confirm that the interaction with TBL1 is required for protein stabilization and identify aa 40–60 as the minimal NLD required for GPS2 targeting to the nucleus (as summarized in Fig. 2A).
FIGURE 2.

Characterization of the GPS2 nuclear localization domain. A, schematic of GPS2 structure, including the coiled coil (CC) domain, and constructs utilized in Fig. 1 with a summary of the results in terms of nuclear localization (NL) of the different mutants that led to the identification of aa 40–60 as the minimal NLD. B, GST pulldown assays confirming that GPS2 Δ60–154 interacts with TBL1 Nt as well as GPS2 wild-type. The interaction with GST-TBL1 Ct was measured as a negative control. C, Western blot for HA-GPS2 in HeLa NEs and CEs showing that adding aa 1–60 to the GPS2 Ct domain (GPS2 Δ60–154) rescues GPS2 expression and targeting to the nucleus. D, site-directed mutagenesis of a putative NLS (K52A/K53A) within the NLD does not impair GPS2 nuclear localization. E, coimmunoprecipitation (IP) of TBL1 and GPS2 from HeLa nuclear extracts upon transfection with control siRNA (sictl) or siUbc9 and HA-GPS2 showing that inhibiting sumoylation does not impair GPS2 nuclear expression or disrupt the interaction between GPS2 and TBL1. F, GST pulldown assay testing the direct interaction between GST-TBL1 Nt and recombinant GPS2 (wild-type, K45R, K71R, or double K45R/K71R mutants).
Inspection of the short amino acid sequence of the GPS2 minimal NLD revealed a classic nuclear localization signal (NLS) located between aa 49–56. To test whether this motif is responsible for GPS2 targeting to the nucleus, we generated a specific mutation of the NLS (ERRKKKE to ERRAAKE) by site-directed mutagenesis and tested it in 293T cells. Surprisingly, both the wild-type GPS2 (WT) and the NLS mutant (NLS-mut) were equally able to localize to the nucleus (Fig. 2D). Because similar results were obtained upon additional mutation of a close, but less conserved, NLS located between aa 98–104 (data not shown), we conclude that nuclear targeting of GPS2 is not prevented by the removal of specific nuclear localization signals. This suggests that GPS2 nuclear localization could be unregulated or could depend on the interaction with other nuclear factors, i.e. other components of the NCoR-SMRT complex like TBL1, rather than being mediated by a classic NLS.
GPS2 Protein Stability Is Regulated by Interaction with TBL1 and Proteasomal Degradation via Ubiquitination of the C-terminal Domain
To further investigate how the interaction with TBL1 affects GPS2 stability and cellular localization, we focused on a recent paper reporting that sumoylation of GPS2 on Lys-45 and Lys-71 participates in regulating GPS2 interaction with TBL1 and repressive activity in the nucleus (19). Interestingly, substitution of Lys-45/71 with arginine residues (2KR mutant) appeared to affect GPS2 shuttling between the nucleus and cytoplasm (19). Because Lys-45 is located within the newly identified NLD, we considered the possibility that sumoylation contributes to regulating GPS2 stabilization and/or nuclear localization via interaction with TBL1. However, in our hands, down-regulation of the SUMO-conjugating enzyme Ubc9 by siRNA has no effect on GPS2 nuclear expression or on the interaction with TBL1 (Fig. 2F), indicating that sumoylation is not required for GPS2 nuclear localization or for the interaction with TBL1. To better understand these discrepancies, we tested sumoylation mutants equivalent to those described previously (K45R, K71R, and 2KR or K45/71R) (19) for direct interaction with TBL1. As shown in Fig. 2F, mutating K71R significantly decreases the direct interaction between GPS2 and TBL1 in a GST pulldown assay, whereas no changes were noticed upon mutation of K45R alone. Together, these data indicate that the integrity of Lys-71 is required for the direct interaction between GPS2 and TBL1 and confirm that the reported decreased half-life of the double mutant (19) is likely caused by decreased interaction between GPS2 and TBL1 but in a sumoylation-independent manner.
Stabilization of GPS2 upon coexpression with TBL1 in bacteria (Fig. 1E), together with the report of a tight association between TBL1 and GPS2 in the assembly of the NCoR-SMRT core complex (28), suggests that the interaction among these cofactors may be required for proper protein folding and that GPS2-unregulated destabilization in the absence of TBL1 may result from reduced complex formation. However, the alternative hypothesis of GPS2 being actively degraded by the 26S proteasome via an ubiquitination-dependent mechanism also exists (3, 8, 19, 22). To directly assess whether GPS2 instability in the absence of TBL1 was mediated by proteasomal degradation, we first looked for GPS2 polyubiquitination. Highly stringent immunoprecipitation of GPS2 from cells overexpressing HA-tagged ubiquitin or from untransfected cells confirmed that GPS2 is polyubiquitinated in both cases (Fig. 3, A and B).
FIGURE 3.

GPS2 proteasomal degradation depends on polyubiquitination at the carboxyl terminus domain. A and B, Western blot (WB) analysis for HA-tagged ubiquitin (Ub) or endogenous ubiquitin showing polyubiquitination of GPS2 as immunoprecipitated (IP) from 293T cells either transfected with HA-Ub (A) or not transfected (B). C, Western blot analysis of full-length GPS2 (aa 1–327) and different deletants expressed in reticulocyte lysate by in vitro transcription and translation (TnT). D, the effect of site-directed mutagenesis of lysines 254, 300, and 327 on GPS2 expression levels in NEs and CEs was measured by Western blotting for HA. Blotting for tubulin and HDAC2 provides controls for CEs and Nes, respectively. E, gene expression analysis in 293T cells comparing the efficiency of GPS2 WT and the more stable triple lysine mutant in rescuing the effect of down-regulating endogenous GPS2 by siRNA on the activation of the proinflammatory target CCL2. Data are presented as mean ± S.D. *, p < 0.08.
Next we screened for polyubiquitination sites by comparing the predicted molecular weight of full-length GPS2 and different deletants of the Nt and Ct domains with the size of their products when expressed by in vitro TnT and assessed by Western blot analysis. Constructs containing only the Nt domain (aa 1–99 and 1–155) generate protein fragments of the expected size in addition to a single higher molecular weight band, which could correspond to the sumoylation described previously (19) or to monoubiquitination. Constructs encompassing the Ct domain (aa 155–327 and 212–327), instead, are translated into proteins with a smear of high molecular weight modifications, which could correspond to ladders of polyubiquitination (Fig. 3C). On the basis of these results, we reasoned that the C-terminal domain of GPS2 is likely encompassing one or more sites of polyubiquitination. In support of this hypothesis, the Ct fragment of GPS2 was significantly less stable than the full-length protein when expressed alone (Fig. 1A), further supporting a role for the C terminus of the protein in regulating GPS2 stability. Indeed, specific mutation of the three lysines present within the C terminus of GPS2 (Lys-254, Lys-300, and Lys-327) is sufficient to increase protein stabilization, suggesting that they serve a degradative role (Fig. 3D). Accordingly, the triple mutant (GPS2 K254A/K300A/K327A) is more efficient than the wild type when used to rescue the hyperinflammatory response that is activated, as reported previously, by specific down-regulation of GPS2 (8) (Fig. 3E). Therefore, our results indicate that GPS2 instability is promoted by an active degradation strategy that is mediated by polyubiquitination occurring within the C-terminal domain.
GPS2 Ubiquitination and Degradation by Siah2
Previous work from others and by us indicate that Drosophila seven-in-absentia homolog 2 (Siah2)-dependent ubiquitination promotes the dismissal of the NCoR-SMRT corepressor complex from target genes and is required for NCoR proteasomal degradation (24, 29). Because GPS2 and TBL1 are core components of the NCoR-SMRT corepressor complex, we hypothesized that Siah2, or the highly conserved homolog Siah1, could be responsible for catalyzing GPS2 polyubiquitination and degradation. To test this hypothesis, we looked at the stability of GPS2 in 293T cells either overexpressing or lacking these E3 ligases. As predicted by our hypothesis, the expression of GPS2 is significantly down-regulated upon overexpression of HA-tagged Siah ligases (HA-Siah1 and HA-Siah2), whereas TBL1 overexpression has a stabilizing effect (Fig. 4A). Conversely, transient transfection of a mixture of specific siRNA against Siah1 and Siah2 is sufficient to observe increased expression of GPS2 (Fig. 4B), confirming that Siah proteins endogenously regulate GPS2 protein levels. More importantly, the effect of Siah proteins is mediated by the ubiquitination of the lysines identified within the C terminus of GPS2, as indicated by the fact that overexpression of Siah1/2 does not induce destabilization of the triple mutant, whereas it is sufficient to prevent expression of the wild-type construct (Fig. 4C). Finally, we took advantage of the Siah2 KO mouse model (30) to assess whether Siah2 was the specific E3 ligase regulating GPS2 stability, as proposed previously for NCoR (29). As shown in Fig. 4D, the GPS2 protein level is increased significantly in the adipose tissue of Siah2 KO mice compared with their wild-type littermates. Therefore, these data demonstrate that GPS2 proteasomal degradation is mediated by Siah2-dependent polyubiquitination of the GPS2 C terminus.
FIGURE 4.
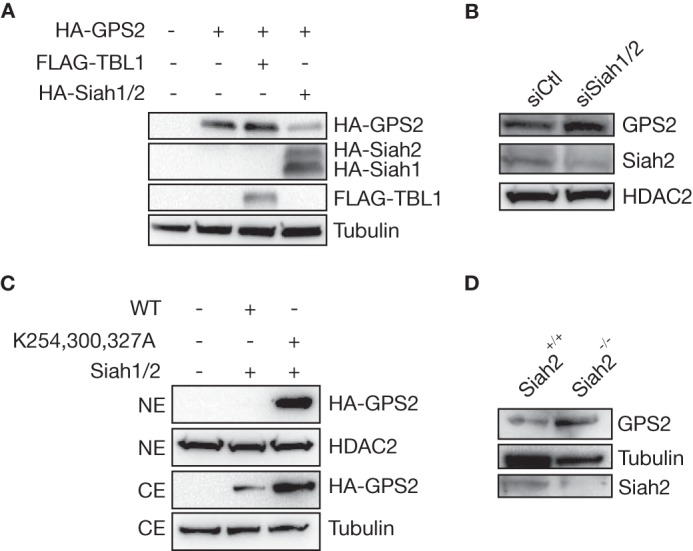
Siah2 is required for GPS2 degradation. A, transient transfection of equal amounts of HA-Siah1 and HA-Siah2 in 293T cells prevents the expression of HA-GPS2, whereas coexpressing GPS2 together with TBL1 increases its stabilization. Blotting for tubulin provided a loading control. B, the endogenous level of nuclear GPS2 increases upon Siah1 and Siah2 down-regulation via specific siRNA. Blotting for HDAC2 provided a loading control. C, Western blot analysis of CEs and NEs showing that overexpression of Siah1/2 in 293T cells fails to decrease the expression of a mutant GPS2 lacking the three C-terminal lysines (K254,300,327A). Blotting for tubulin and HDAC2 provided controls for CEs and Nes, respectively. D, Siah2 genetic ablation, as verified by blotting for Siah2, promotes an increased GPS2 protein level in extracts from mouse adipose tissue. Blotting for tubulin provided a loading control.
GPS2 Is Methylated in Vivo on Arg-312 and Arg-323
Interestingly, the three lysines required for GPS2 dependent polyubiquitination are flanking an arginine residue (Arg-323) that has been described recently as a site of differential dimethylation in a study on the human leukocyte antigen peptidome of melanoma cells (20). Methylation of GPS2 on Arg-312 has also emerged in large-scale studies of protein methylation (21, 31), suggesting that arginine methylation could be an important aspect of GPS2 posttranslational regulation. Because peptidomes are often the result of proteasomal degradation, we hypothesized that GPS2 methylation could contribute to regulating its stability. To address this question, we first confirmed that GPS2 is methylated in vivo by performing immunoprecipitation of HA-GPS2-FLAG followed by Western blotting for arginine methylation in 293T cells. Interestingly, even though several pan-arginine methyl-specific antibodies were tested, only an antibody raised against H3R17me2a worked well in recognizing methylated GPS2 (Fig. 5A). Alignment of the motifs around Arg-312 and Arg-323 with known arginine methylation sites on histone H3 and H4 confirms that the sequence around H3R17 most closely resembles the putative methylation sites on GPS2, with the conserved P in position −1 potentially being an important determinant of antibody specificity (Fig. 5B). Next we used the same strategy to validate the sites of methylation by comparing the GPS2 wild type with the R312A/R323A mutant. Loss of methylation signal upon mutagenesis of both arginines confirmed that Arg-312 and Arg-323 are the main methylation sites in vivo (Fig. 5C). In addition, the consistently lower expression level of the mutant protein compared with wild-type GPS2 suggests that loss of methylation promotes protein destabilization. To further explore this hypothesis and test whether methylation of both residues was required for protein stabilization, we generated single Arg-to-Ala mutants and tested their expression in 293T cells. Interestingly, mutation of Arg-312 did not affect GPS2 stability, whereas ablation of the Arg-323 methylation site alone was sufficient to destabilize GPS2 expression. As expected, expression of the R323A mutant was fully rescued by the specific proteasome inhibitor MG132 (Fig. 5D).
FIGURE 5.
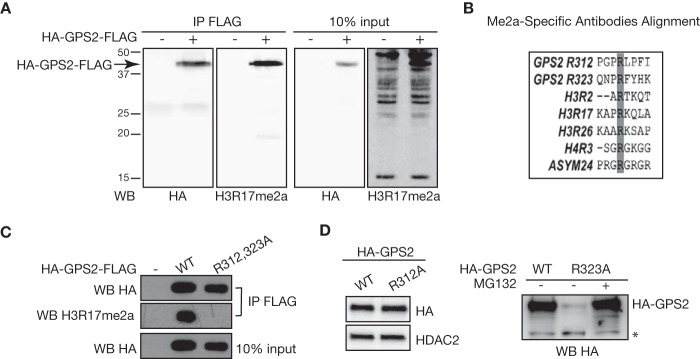
Methylation of GPS2 in vivo onArg-R312 and Arg-323. A, immunoprecipitation (IP) for FLAG followed by Western blotting with the pan-arginine methylation antibody H3R17me2a confirms in vivo methylation of HA-GPS2-FLAG. Western blotting (WB) for HA provided a positive control for the efficiency of immunoprecipitation. B, alignment of the motifs around the GPS2 putative methylation sites Arg-312 and Arg-323 with the arginine methylation sites recognized by the antibodies tested in A. C, confirmation of in vivo methylation of GPS2 on Arg-312 and Arg-323 in 293T cells transfected with FLAG-eYFP, HA-GPS2WT-FLAG, or the HA-GPS2R312/323K-FLAG double mutant. Immunoprecipitation was followed by Western blotting with the H3R17me2a antibody as in A. D, Western blotting for HA-GPS2 in 293T nuclear extracts showed that mutation of arginine 323 alone (GPS2-R323A) is sufficient to promote GPS2 degradation in a proteasome-dependent manner (right panel), whereas mutating R312A does not affect GPS2 protein expression (left panel). Blotting for HDAC2 provided a loading control. The asterisk indicates a nonspecific band.
PRMT6-mediated Methylation of Arg-323 Prevents Proteasome-dependent Degradation of GPS2
To further probe into this regulatory mechanism, we looked for the specific enzyme responsible for Arg-323 methylation of GPS2. Protein arginine N-methyltransferases (PRMTs) that can catalyze monomethylation and asymmetric dimethylation reactions belong to the type I PRMT family. PRMT1 is the major source of type I methyltransferase activity in mammalian cells. However, previous work has already eliminated PRMT1 as a candidate enzyme for Arg-323 methylation (20). Among the remaining type I PRMTs, PRMT6 stands out as an exclusively nuclear enzyme, whereas the others are mainly cytosolic or membrane-associated (33–36). Upon testing it in an in vitro methylation assay with recombinant proteins, we found that PRMT6 can directly methylate GPS2, whereas no effect was seen using PRMT1, as expected on the basis of published data (Fig. 6A) (20). Methylation is mainly observed on GPS2 fragments containing the C-terminal part of the protein (as indicated by Western blotting with an antibody directed against the last 20 aa) rather than on the full-length protein. This may depend on an allosteric effect with dimerization, as mediated by the GPS2 N terminus (37) or by the GST moiety, inhibiting full accessibility of the methylation site. Importantly, it also confirms that methylation occurs over the C terminus of the protein.
FIGURE 6.
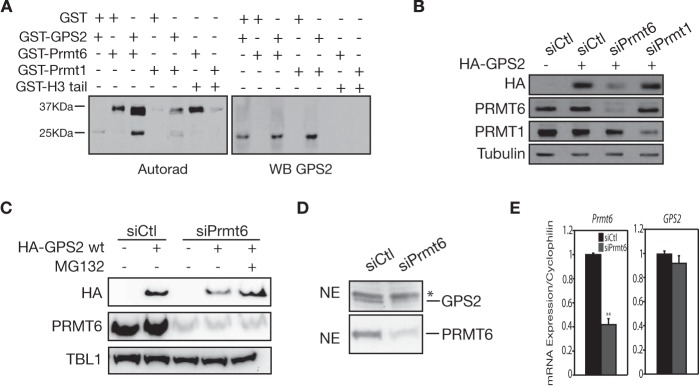
Methylation of GPS2 by PRMT6 prevents its proteasomal degradation. A, in vitro methylation assay showing GPS2 direct methylation by recombinant PRMT6. WB, Western blot. B, Western blot showing that expression of HA-GPS2 in transiently transfected 293T cells is significantly impaired upon down-regulation of PRMT6 but not of PRMT1. Blotting for PRMT1 and PRMT6 validated the efficiency of siRNAs, and blotting for tubulin provided a loading control. C, the same experimental approach was used to confirm that HA-GPS2 expression in the absence of PRMT6 can be rescued by treatment with the proteasome inhibitor MG132. Blotting for TBL1 was used as a loading control in this case. D, a significant decrease in GPS2 protein level was observed in 293T nuclear extracts upon PRMT6 down-regulation via specific siRNA, as validated by blotting for PRMT6. E, quantitative RT-PCR showing that down-regulation of PRMT6 in 293T cells by siRNA does not change the mRNA expression of GPS2. Data are mean ± S.D. **, p < 0.05.
Next, on the basis of the instability observed when methylation of Arg-323 is impaired by mutagenesis (Fig. 5D), we tested whether down-regulation of PRMT6 in HeLa cells would also impair GPS2 stability. As shown in Fig. 6, reduced expression of both endogenous (Fig. 6D) and HA-tagged GPS2 (Fig. 6, B and C) was observed upon specific down-regulation of PRMT6 by siRNA but not upon down-regulation of PRMT1 (Fig. 6B). Importantly, the reduction in protein level is due to proteasomal degradation and not to changes in gene expression, as proven by the following observations. Expression of the HA-GPS2 vector is rescued upon proteasome inhibition by MG132 (Fig. 6C), and mRNA expression of endogenous GPS2 is not affected by PRMT6 ablation, as measured by quantitative RT-PCR (Fig. 6E). Therefore, or results show that GPS2 is methylated in vivo on Arg-312 and Arg-323 and that methylation of Arg-323 by PRMT6 is required to prevent proteasomal degradation of GPS2.
TBL1 Protective Role toward GPS2 Proteasomal Degradation
Finally, because both the interaction with TBL1 and arginine methylation appear to regulate GPS2 stability in the nucleus, we asked whether the two mechanisms are linked, with methylation regulating the interaction with TBL1. As shown in Fig. 7, A and B, both mutation of R323A and down-regulation of PRMT6 by siRNA are sufficient to significantly impair the interaction with TBL1 when GPS2 expression is stabilized by MG132. Therefore, our results reveal a highly specific strategy, with the regulation of GPS2 stability by TBL1 being modulated via posttranslational modifications.
FIGURE 7.
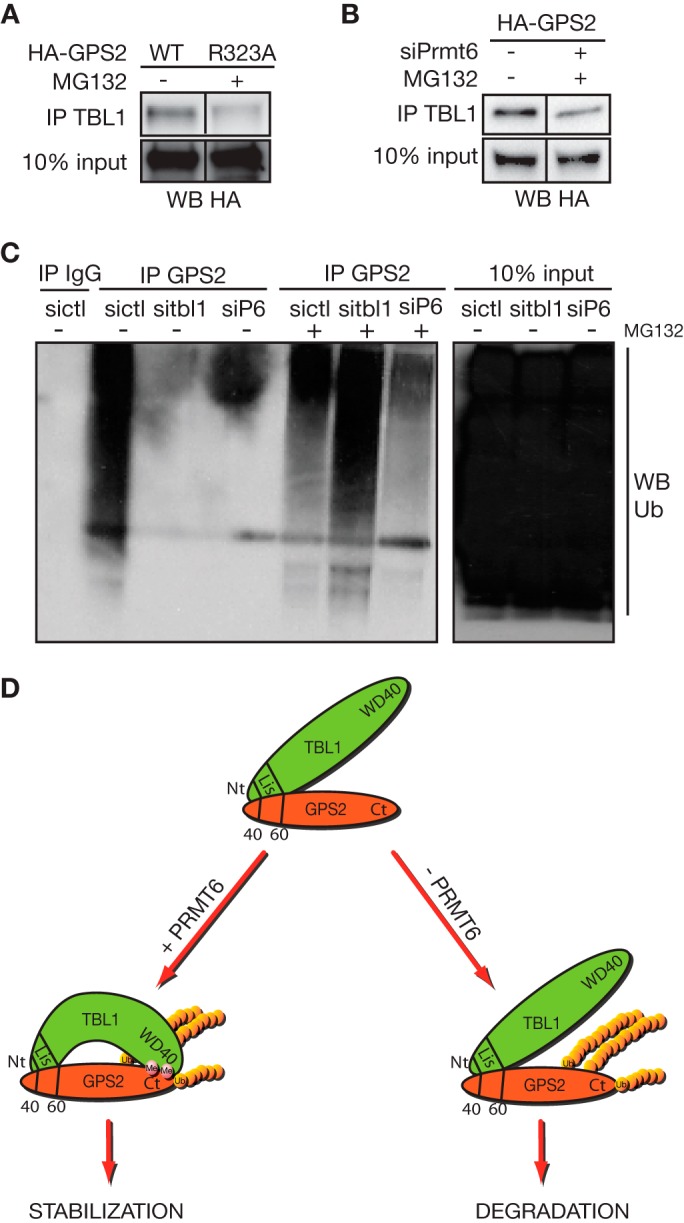
TBL1 protects polyubiquitinated GPS2 from proteasomal degradation. A, to compare equal amounts of GPS2 wild-type and R323A mutant in their ability to interact with TBL1, expression of the R323A mutant was rescued by MG132 (as shown in Fig. 5D), and the interaction was measured by coimmunoprecipitation (IP) in nuclear extracts from transiently transfected 293T cells. WB, Western blot. B, the same experimental approach is used to measure the effect of down-regulating PRMT6 via specific siRNA on the interaction between wild-type GPS2 and TBL1. In this case, expression of HA-GPS2 is rescued by MG132 when cells are depleted of PRMT6 (as shown in Fig. 5G). C, the polyubiquitination of GPS2 in absence of TBL1 or PRMT6 is assayed in 293T cells transfected with the specific siRNAs by GPS2 immunoprecipitation and Western blot against ubiquitin (Ub). D, proposed model of GPS2 alternative regulation by methylation and ubiquitination. GPS2 stability in the nucleus is strictly dependent on its interaction with the exchange cofactor TBL1. TBL1 direct interaction with GPS2 through the N-terminal domains of both proteins is further strengthened by regulated interaction between the respective C-terminal domains. This secondary interaction is regulated by PRMT6-mediated methylation of Arg-312/323 and is required for preventing GPS2 degradation. Ubiquitination of GPS2, as mediated by the E3 ligase Siah2, is not abrogated when the TBL1 protective role is removed, but degradation of polyubiquitinated GPS2 by the 26S proteasome is impaired under these conditions.
These findings of a TBL1 protective role toward GPS2 degradation upon Siah2-dependent ubiquitination were initially surprising since we had previously reported TBL1 being required for the ubiquitination and degradation of other components of the NCoR-SMRT complex (24). However, similar opposing roles have been reported for TBL1 in the regulation of β-catenin protein levels (16, 38–40). In particular, Dimitrova et al. (38) propose that TBL1 prevents polyubiquitination and degradation of β-catenin via competition between Siah-1 and TBL1 for binding to the region of β-catenin. To directly test whether the TBL1 protective role depends on preventing the polyubiquitination of GPS2 or the degradation of polyubiquitinated GPS2, we inhibited proteasome-dependent degradation with MG132 and then immunoprecipitated GPS2 from 293T cells in which we had selectively down-regulated TBL1 or PRMT6. As shown in Fig. 7C, the dramatic decrease in GPS2 polyubiquitination observed in the absence of TBL1 is fully rescued upon proteasome inhibition. This indicates that polyubiquitination itself is not impaired by lack of TBL1 but, rather, that TBL1 prevents the degradation of the polyubiquitinated protein. Furthermore, a similar decrease in the accumulation of polyubiquitinated GPS2 is observed upon PRMT6 down-regulation (Fig. 7C). Under these conditions, the polyubiquitination of GPS2 is also rescued by proteasome inhibition, although to a lesser extent, confirming that impairing the interaction with TBL1 by inhibiting GPS2 methylation provokes the same effect as down-regulating TBL1 itself.
Combined, our results indicate that methylation of GPS2 by PRMT6 is required to stabilize the interaction between TBL1 and GPS2 and to protect GPS2 from proteasomal degradation. Interestingly, specific motifs have been reported as having a special affinity for methylated residues, including WD40 domains like the one present in the C-terminal of TBL1 (32), suggesting that the TBL1 WD40 domain might act as a reader for methylated arginines in the C terminus. This would further stabilize the direct interaction between the N-terminal regions of GPS2 and TBL1 and provide a regulatory switch for modulating GPS2 stability upon cellular needs. Importantly, our results indicate that the regulated interaction with TBL1 does not prevent the ubiquitination of the GPS2 C terminus by Siah2 but, rather, that it appears to play a protective role against protein degradation by the proteasome (Fig. 7C).
Discussion
GPS2 is involved in a variety of important cellular processes, including gene expression, cell division, programmed cell death, and transduction of a number of signaling pathways. In particular, it has recently emerged as an important regulator of metabolic and inflammatory pathways in key organs, including the liver, adipose tissue, and the immune system (4, 6, 8, 11, 41). Previous work from our laboratory indicates that GPS2 relevance in many of these processes stems from its ability to cooperatively regulate ubiquitin signaling in different cellular compartments (8, 9). This suggests that strategies regulating GPS2 intracellular localization could have a profound effect on cellular homeostasis. In this study, we set out to dissect the molecular mechanisms controlling GPS2 intracellular localization and discovered that GPS2 nuclear targeting depends on a minimal NLD located at the very N terminus. Moreover, our results indicate that GPS2 nuclear localization does not require a classic NLS and suggest that it could be regulated via interaction with other nuclear proteins, possibly including components of the NCoR-SMRT corepressor complex, such as the exchange factor TBL1. However, because of the small size of GPS2, it is also possible that nuclear localization is achieved via passive diffusion through the nuclear membrane (Figs. 1 and 2).
Characterization of the interaction between GPS2 and TBL1 confirms that the interaction is mediated via a small N-terminal domain that is critically important for both GPS2 stability as well as the correct assembly of the NCoR-SMRT corepressor complex (28). In addition, our results indicate that residues outside of the core binding motif identified previously are important to mediate the direct interaction between GPS2 and TBL1. They also suggest that sumoylation does not play a critical role in regulating GPS2-TBL1 interaction in the nucleus, which is not surprising considering that only a small fraction of GPS2 is sumoylated at any given time, whereas GPS2 association with TBL1 is constantly required to prevent its destabilization.
In an effort to understand whether GPS2 instability in the absence of TBL1 was caused by an active degradative mechanism or by problems with correct protein folding, we observed that GPS2 was polyubiquitinated and subjected to proteasome-dependent degradation (Fig. 3). To our surprise, we found that Siah2 was responsible for GPS2 ubiquitination (Fig. 4). Siah2 is an E3 ubiquitin ligase mediating the ubiquitination of a number of key targets in cells, including the nuclear receptor corepressors NCoR and HDAC3 (29, 42). In particular, our previous work indicated that TBL1, together with its close homolog TBLR1, is required for the ubiquitination and the dismissal of the NCoR-SMRT corepressor complex from target genes upon induced gene activation. On the basis of these observations, we proposed previously that the F box domain in the N terminus of TBL1 was required to recruit the Siah2-UbcH5 complex to the NCoR complex in vivo in a similar fashion as the role reported for Ebi, the Drosophila homolog of TBL1, during the regulation of Tramtrack88 in Drosophila and β-catenin in mammalian cells (17, 24, 39, 40). Interestingly, the regulation of β-catenin activity by the SCF (Skp, Cullin, F-box containing)/TBL1 complex turned out more complex than initially envisioned because a later study suggests an unexpected role for TBL1 as a “protector” of β-catenin during Wnt signaling and indicates that Siah1 alone is able to ubiquitinate β-catenin, at least in vitro (38). This clearly raised the question of whether TBL1 function is to promote or antagonize polyubiquitination and degradation by the Siah complex. In the case of GPS2, the results presented here indicate that GPS2 is degraded rapidly in the absence of TBL1, supporting the idea that the regulated binding of TBL1 to GPS2 exerts a protective role toward degradation. However, the mechanism underlying the protective function of TBL1 seems to differ among different targets. In both cases, the recruitment of the E3 ligase complex seem to occur independently of TBL1, but our data indicate that GPS2 polyubiquitination is not affected by down-regulation of TBL1 (Fig. 7), implying that binding between TBL1 and GPS2 is not mutually exclusive with Siah2-dependent polyubiquitination, as reported previously in the case of β-catenin (38). Although further studies are required to address the molecular mechanism of the TBL1 protective role on different substrates, we speculate that, in this case, TBL1 interaction with GPS2 might allosterically prevent the recognition and binding of the polyubiquitinated protein to the proteasome.
Another outstanding question to be investigated further is whether the phenotypic effects observed upon down-regulation of TBL1 might be partially explained by reduced GPS2 levels (as shown in Fig. 1 in TBL1Δ/Y embryonic stem cells). For example, it is striking that TBLR1 adipose-specific knockout mice are defective in fasting-induced lipid mobilization because of inhibited lipolysis, which is reminiscent of the GPS2 requirement for the expression of the lipolysis rate-limiting enzymes ATGL (adipose triglyceride lipase) and HSL (hormone-sensitive lipase) in adipocytes (9, 43). Similarly, TBL1 has been proposed to promote the recruitment of NF-κB to target promoters in a similar fashion as GPS2 pioneering activity toward promoter-specific recruitment of PPARγ to a selected subset of target genes (9, 44). So far, GPS2 has been mainly associated with a repressive role, as part of the NCoR-SMRT complex, in the regulation of proinflammatory genes. However, it is possible that GPS2 also contributes to the activation of a subset of NF-κB targets, as described in the case of nuclear receptors (8, 9, 11).
Finally, our work identifies the arginine methyltransferase PRMT6 as a critical regulator of GPS2 protein stability and indicates that the TBL1-mediated protective effect is modulated by posttranslational modification of GPS2, making this regulatory network tightly controlled by the local nuclear environment. Asymmetric dimethylation of GPS2 on Arg-323 was originally discovered while analyzing the human leukocyte antigen peptidome of melanoma cells with the ultimate goal of identifying modifications that are specific to tumor cells and can be specifically recognized by the immune system (20). However, the enzyme responsible for modifying GPS2 was unknown. Here we show that GPS2 is methylated in vivo on Arg-312 and Arg-323, that recombinant PRMT6 can methylate GPS2 in vitro, and that the methylation of Arg-323 by PRMT6 is required to promote protein stabilization by TBL1 (Figs. 5 and 6). Although we cannot exclude that other type I methyltransferases might contribute to GPS2 methylation under certain conditions, i.e. in other cellular compartments or in a tissue-specific manner (33), our data suggest that PRMT6 is the main enzyme regulating GPS2 methylation and stability in the nucleus. Intriguingly, PRMT6 has been implicated in many processes related to GPS2 functions, including chromatin remodeling, cell cycle regulation, p53 activity, and transcriptional regulation by the NF-κB pathway (45–48), suggesting, as in the case of TBL1, that some of the effects observed when modulating PRMT6 levels may be attributed to changes in GPS2-mediated functions.
Overall, although our results confirm that the direct interaction of GPS2 with TBL1 occurs via the very N terminus region of GPS2, as reported previously (28), our data also indicate that the interaction between TBL1 and GPS2 is stabilized by the presence of a methylated arginine on the opposite end of GPS2, possibly because of the TBL1 WD40 domain acting as a “reader” of methylated arginine residues (32). Accordingly with this interpretation, PRMT6 down-regulation by siRNA leads to the rapid degradation of polyubiquitinated GPS2 in a manner closely mimicking what is observed upon transfection with siRNA against TBL1. In conclusion, our results reveal that a tightly regulated degradation process, promoted by the E3 ubiquitin ligase Siah2 and antagonized by the cofactor TBL1 in a PRMT6-dependent manner, defines GPS2 protein levels in the nucleus.
Author Contributions
M. D. C. and V. P. conceived and coordinated the study and wrote the paper. J. H. performed and analyzed most of the experiments characterizing GPS2 ubiquitination by Siah2, the GPS2 interaction with TBL1, and stabilization via methylation. H. E. J. performed most of the studies to identify and characterize the GPS2 minimal nuclear localization domain. M. N. and F. A. M. designed and conducted the studies characterizing GPS2 methylation by PRMT6. M. C. contributed to perform the initial analysis of GPS2 localization and sumoylation. Z. E. F. provided tissues from Siah2 KO mice. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank all past and present members of the Perissi and Varelas laboratories for insightful discussions and valuable inputs. We particularly thank Sherry Prasad and John Chamberland for assistance with cloning. The HA-Siah1 and HA-Siah2 constructs were provided by Dr. Ross A. Dickins.
This work was supported, in whole or in part, by National Institutes of Health Grants DK078756 and DK100422 (to V. P.). This work was also supported by Canadian Institutes of Health Research Operating Grant MOP-133442 (to F. A. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- Nt
- N terminus
- Ct
- C terminus
- NLD
- nuclear localization domain
- NE
- nuclear extract
- CE
- cytosolic extract
- aa
- amino acid(s)
- NLS
- nuclear localization signal
- SUMO
- small ubiquitin-like modifier
- PRMT
- protein arginine methyltransferase
- TBL1
- transducin β-like 1
- NCoR
- nuclear receptor corepressor
- SMRT
- silencing mediator of retinoic and thyroid hormone receptor
- WT
- wild type.
References
- 1. Spain B. H., Bowdish K. S., Pacal A. R., Staub S. F., Koo D., Chang C. Y., Xie W., Colicelli J. (1996) Two human cDNAs, including a homolog of Arabidopsis FUS6 (COP11), suppress G-protein- and mitogen-activated protein kinase-mediated signal transduction in yeast and mammalian cells. Mol. Cell. Biol. 16, 6698–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peng Y. C., Kuo F., Breiding D. E., Wang Y. F., Mansur C. P., Androphy E. J. (2001) AMF1 (GPS2) modulates p53 transactivation. Mol. Cell. Biol. 21, 5913–5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peng Y. C., Breiding D. E., Sverdrup F., Richard J., Androphy E. J. (2000) AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J. Virol. 74, 5872–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanyal S., Båvner A., Haroniti A., Nilsson L. M., Lundåsen T., Rehnmark S., Witt M. R., Einarsson C., Talianidis I., Gustafsson J. A., Treuter E. (2007) Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 15665–15670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang D., Yang Q., Ding Y., Cao X., Xue Y., Cheng Z. (2008) Cytological characterization of the tandem repetitive sequences and their methylation status in the Antirrhinum majus genome. Genomics 92, 107–114 [DOI] [PubMed] [Google Scholar]
- 6. Jakobsson T., Venteclef N., Toresson G., Damdimopoulos A. E., Ehrlund A., Lou X., Sanyal S., Steffensen K. R., Gustafsson J. A., Treuter E. (2009) GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Mol. Cell 34, 510–518 [DOI] [PubMed] [Google Scholar]
- 7. Lee T. H., Yi W., Griswold M. D., Zhu F., Her C. (2006) Formation of hMSH4-hMSH5 heterocomplex is a prerequisite for subsequent GPS2 recruitment. DNA Repair 5, 32–42 [DOI] [PubMed] [Google Scholar]
- 8. Cardamone M. D., Krones A., Tanasa B., Taylor H., Ricci L., Ohgi K. A., Glass C. K., Rosenfeld M. G., Perissi V. (2012) A protective strategy against hyperinflammatory responses requiring the nontranscriptional actions of GPS2. Mol. Cell 46, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cardamone M. D., Tanasa B., Chan M., Cederquist C. T., Andricovich J., Rosenfeld M. G., Perissi V. (2014) GPS2/KDM4A pioneering activity regulates promoter-specific recruitment of PPARγ. Cell Rep. 8, 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J., Kalkum M., Chait B. T., Roeder R. G. (2002) The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 9, 611–623 [DOI] [PubMed] [Google Scholar]
- 11. Toubal A., Clément K., Fan R., Ancel P., Pelloux V., Rouault C., Veyrie N., Hartemann A., Treuter E., Venteclef N. (2013) SMRT-GPS2 corepressor pathway dysregulation coincides with obesity-linked adipocyte inflammation. J. Clin. Invest. 123, 362–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang D., Harry G. J., Blackshear P. J., Zeldin D. C. (2008) G-protein pathway suppressor 2 (GPS2) interacts with the regulatory factor X4 variant 3 (RFX4_v3) and functions as a transcriptional co-activator. J. Biol. Chem. 283, 8580–8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo C., Li Y., Gow C. H., Wong M., Zha J., Yan C., Liu H., Wang Y., Burris T. P., Zhang J. (2015) The optimal corepressor function of nuclear receptor corepressor (NCoR) for peroxisome proliferator-activated receptor γ requires G-protein pathway suppressor 2. J. Biol. Chem. 290, 3666–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., Ozawa Y., Lee H., Wen Y. D., Tan T. H., Wadzinski B. E., Seto E. (2005) Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 19, 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi H. K., Yoo J. Y., Jeong M. H., Park S. Y., Shin D. M., Jang S. W., Yoon H. G., Choi K. C. (2013) Protein kinase A phosphorylates NCoR to enhance its nuclear translocation and repressive function in human prostate cancer cells. J. Cell. Physiol. 228, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 16. Choi H. K., Choi K. C., Yoo J. Y., Song M., Ko S. J., Kim C. H., Ahn J. H., Chun K. H., Yook J. I., Yoon H. G. (2011) Reversible SUMOylation of TBL1-TBLR1 regulates β-catenin-mediated Wnt signaling. Mol. Cell 43, 203–216 [DOI] [PubMed] [Google Scholar]
- 17. Perissi V., Scafoglio C., Zhang J., Ohgi K. A., Rose D. W., Glass C. K., Rosenfeld M. G. (2008) TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol. Cell 29, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang W., Ghisletti S., Saijo K., Gandhi M., Aouadi M., Tesz G. J., Zhang D. X., Yao J., Czech M. P., Goode B. L., Rosenfeld M. G., Glass C. K. (2011) Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature 470, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bi H., Li S., Wang M., Jia Z., Chang A. K., Pang P., Wu H. (2014) SUMOylation of GPS2 protein regulates its transcription-suppressing function. Mol. Biol. Cell 25, 2499–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jarmalavicius S., Trefzer U., Walden P. (2010) Differential arginine methylation of the G-protein pathway suppressor GPS-2 recognized by tumor-specific T cells in melanoma. FASEB J. 24, 937–946 [DOI] [PubMed] [Google Scholar]
- 21. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin D. Y., Teramoto H., Giam C. Z., Chun R. F., Gutkind J. S., Jeang K. T. (1997) A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor α. J. Biol. Chem. 272, 25816–25823 [DOI] [PubMed] [Google Scholar]
- 23. Chun A. C., Zhou Y., Wong C. M., Kung H. F., Jeang K. T., Jin D. Y. (2000) Coiled-coil motif as a structural basis for the interaction of HTLV type 1 Tax with cellular cofactors. AIDS Res. Hum. Retroviruses 16, 1689–1694 [DOI] [PubMed] [Google Scholar]
- 24. Perissi V., Aggarwal A., Glass C. K., Rose D. W., Rosenfeld M. G. (2004) A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116, 511–526 [DOI] [PubMed] [Google Scholar]
- 25. Huang W., Ghisletti S., Perissi V., Rosenfeld M. G., Glass C. K. (2009) Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol. Cell 35, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pascual G., Fong A. L., Ogawa S., Gamliel A., Li A. C., Perissi V., Rose D. W., Willson T. M., Rosenfeld M. G., Glass C. K. (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng D., Vemulapalli V., Bedford M. T. (2012) Methods applied to the study of protein arginine methylation. Methods Enzymol. 512, 71–92 [DOI] [PubMed] [Google Scholar]
- 28. Oberoi J., Fairall L., Watson P. J., Yang J. C., Czimmerer Z., Kampmann T., Goult B. T., Greenwood J. A., Gooch J. T., Kallenberger B. C., Nagy L., Neuhaus D., Schwabe J. W. (2011) Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J., Guenther M. G., Carthew R. W., Lazar M. A. (1998) Proteasomal regulation of nuclear receptor corepressor-mediated repression. Genes Dev. 12, 1775–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frew I. J., Hammond V. E., Dickins R. A., Quinn J. M., Walkley C. R., Sims N. A., Schnall R., Della N. G., Holloway A. J., Digby M. R., Janes P. W., Tarlinton D. M., Purton L. E., Gillespie M. T., Bowtell D. D. (2003) Generation and analysis of Siah2 mutant mice. Mol. Cell. Biol. 23, 9150–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uhlmann T., Geoghegan V. L., Thomas B., Ridlova G., Trudgian D. C., Acuto O. (2012) A method for large-scale identification of protein arginine methylation. Mol. Cell. Proteomics 11, 1489–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Migliori V., Mapelli M., Guccione E. (2012) On WD40 proteins: propelling our knowledge of transcriptional control? Epigenetics 7, 815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frankel A., Yadav N., Lee J., Branscombe T. L., Clarke S., Bedford M. T. (2002) The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem. 277, 3537–3543 [DOI] [PubMed] [Google Scholar]
- 35. Harrison M. J., Tang Y. H., Dowhan D. H. (2010) Protein arginine methyltransferase 6 regulates multiple aspects of gene expression. Nucleic Acids Res. 38, 2201–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee Y. H., Stallcup M. R. (2009) Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol. Endocrinol. 23, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breiding D. E., Sverdrup F., Grossel M. J., Moscufo N., Boonchai W., Androphy E. J. (1997) Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol. Cell. Biol. 17, 7208–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dimitrova Y. N., Li J., Lee Y. T., Rios-Esteves J., Friedman D. B., Choi H. J., Weis W. I., Wang C. Y., Chazin W. J. (2010) Direct ubiquitination of β-catenin by Siah-1 and regulation by the exchange factor TBL1. J. Biol. Chem. 285, 13507–13516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong X., Tsuda L., Zavitz K. H., Lin M., Li S., Carthew R. W., Zipursky S. L. (1999) ebi regulates epidermal growth factor receptor signaling pathways in Drosophila. Genes Dev. 13, 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsuzawa S. I., Reed J. C. (2001) Siah-1, SIP, and Ebi collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol. Cell 7, 915–926 [DOI] [PubMed] [Google Scholar]
- 41. Venteclef N., Jakobsson T., Ehrlund A., Damdimopoulos A., Mikkonen L., Ellis E., Nilsson L. M., Parini P., Jänne O. A., Gustafsson J. A., Steffensen K. R., Treuter E. (2010) GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRβ in the hepatic acute phase response. Genes Dev. 24, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao H. L., Ueki N., Hayman M. J. (2010) The Ski protein negatively regulates Siah2-mediated HDAC3 degradation. Biochem. Biophys. Res. Commun. 399, 623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rohm M., Sommerfeld A., Strzoda D., Jones A., Sijmonsma T. P., Rudofsky G., Wolfrum C., Sticht C., Gretz N., Zeyda M., Leitner L., Nawroth P. P., Stulnig T. M., Diaz M. B., Vegiopoulos A., Herzig S. (2013) Transcriptional cofactor TBLR1 controls lipid mobilization in white adipose tissue. Cell Metab. 17, 575–585 [DOI] [PubMed] [Google Scholar]
- 44. Ramadoss S., Li J., Ding X., Al Hezaimi K., Wang C. Y. (2011) Transducin β-like protein 1 recruits nuclear factor κB to the target gene promoter for transcriptional activation. Mol. Cell. Biol. 31, 924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Di Lorenzo A., Yang Y., Macaluso M., Bedford M. T. (2014) A gain-of-function mouse model identifies PRMT6 as a NF-κB coactivator. Nucleic Acids Res. 42, 8297–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kleinschmidt M. A., de Graaf P., van Teeffelen H. A., Timmers H. T. (2012) Cell cycle regulation by the PRMT6 arginine methyltransferase through repression of cyclin-dependent kinase inhibitors. PloS ONE 7, e41446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stein C., Riedl S., Rüthnick D., Nötzold R. R., Bauer U. M. (2012) The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res. 40, 9522–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Neault M., Mallette F. A., Vogel G., Michaud-Levesque J., Richard S. (2012) Ablation of PRMT6 reveals a role as a negative transcriptional regulator of the p53 tumor suppressor. Nucleic Acids Res. 40, 9513–9521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]