

Background: The intrinsically disordered domain 2 of NS5A is required for HCV replication.
Results: We characterized a short structural motif in the domain 2 of NS5A.
Conclusion: This structural motif in NS5A-D2 is essential for RNA replication.
Significance: This work provides a molecular basis for further understanding of the function of the intrinsically disordered domain 2 of HCV NS5A protein.
Keywords: cyclophilin, hepatitis C virus (HCV), intrinsically disordered protein, nuclear magnetic resonance (NMR), prolyl isomerase, protein motif, viral protein, RNA replication
Abstract
Hepatitis C virus (HCV) nonstructural protein 5A (NS5A) and its interaction with the human chaperone cyclophilin A are both targets for highly potent and promising antiviral drugs that are in the late stages of clinical development. Despite its high interest in regards to the development of drugs to counteract the worldwide HCV burden, NS5A is still an enigmatic multifunctional protein poorly characterized at the molecular level. NS5A is required for HCV RNA replication and is involved in viral particle formation and regulation of host pathways. Thus far, no enzymatic activity or precise molecular function has been ascribed to NS5A that is composed of a highly structured domain 1 (D1), as well as two intrinsically disordered domains 2 (D2) and 3 (D3), representing half of the protein. Here, we identify a short structural motif in the disordered NS5A-D2 and report its NMR structure. We show that this structural motif, a minimal Pro314–Trp316 turn, is essential for HCV RNA replication, and its disruption alters the subcellular distribution of NS5A. We demonstrate that this Pro-Trp turn is required for proper interaction with the host cyclophilin A and influences its peptidyl-prolyl cis/trans isomerase activity on residue Pro314 of NS5A-D2. This work provides a molecular basis for further understanding of the function of the intrinsically disordered domain 2 of HCV NS5A protein. In addition, our work highlights how very small structural motifs present in intrinsically disordered proteins can exert a specific function.
Introduction
Hepatitis C virus (HCV)5 is a small single-stranded RNA virus that chronically infects 130–170 million people worldwide. It thereby constitutes a serious global health challenge, because infection can lead to severe liver diseases such as chronic hepatitis, cirrhosis, and hepatocellular carcinoma (1). The HCV genome encodes for 10 mature proteins: the structural proteins Core, E1, and E2; the viroporin p7; and the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B. NS proteins are involved in polyprotein processing and viral genome replication (2). Because of increased molecular knowledge about the HCV life cycle, the treatment of HCV infection has been largely improved in recent years, with the approval of efficient direct acting antivirals targeting the viral protease NS3/4A, the RNA polymerase NS5B, and the NS5A protein (3–5). Future HCV regimens will most probably include a combination of direct acting antivirals and/or host-targeted antivirals to treat all HCV genotypes and to increase the genetic barrier to resistance mutations (6). In this respect, NS5A is of particular interest because it is the target of efficient direct acting antivirals (e.g. daclatasvir) (7) and its interaction with the human cyclophilin A (CypA), an essential peptidyl-prolyl cis-trans isomerase (PPIase) (8, 9), is also targeted by the most advanced host-targeted antiviral (alisporivir) (10, 11).
NS5A is a multifunctional but still enigmatic protein. Despite a lack of known enzymatic activity, the protein is essential for HCV genome replication (12), is involved in the production of new virions (13), and has been shown to modulate numerous viral and host processes (14). NS5A is a multidomain phosphoprotein (15) anchored via an N-terminal helix in the endoplasmic reticulum (16). The cytoplasmic part of NS5A is composed of a well structured domain 1 (D1) and two intrinsically disordered domains (D2 and D3) that exist as highly dynamic interconverting conformers. NS5A-D1, which is the target of daclatasvir (7, 17), contains a zinc finger motif and possesses RNA binding properties (18). Crystallographic studies revealed that this domain could adopt at least three different homodimeric conformations (19–21), underscoring the multifunctionality of the protein. By using several biophysical techniques such as NMR spectroscopy, circular dichroism, and gel filtration chromatography, NS5A-D2, which is essential for viral RNA replication (12), and NS5A-D3, which is involved in production and assembly of new virions (13, 22), were shown to be mainly disordered (23–25). In this respect, NS5A is the HCV protein with the highest percentage of its primary sequence predicted to be disordered (26). In vitro, both NS5A-D2 and -D3 have been shown to directly interact with the host CypA and to be substrates of its PPIase enzymatic activity (27, 28).
Intrinsically disordered proteins or regions (IDPs/IDRs) have been involved in numerous human diseases such as cancer, neurodegenerative diseases and diabetes (29). The functions attributed to IDPs/IDRs are most often related to interactions with proteins or nucleic acids in regulatory and signaling networks (30, 31). IDRs are abundant in RNA viruses (26, 32, 33), allowing them to minimize their genome while keeping multiple biological functions. IDRs indeed can establish numerous interactions (thereby acting as hub proteins), can cope with high mutation rates (caused by error-prone RNA-polymerases), can evade host immune system, and can adapt to different environments (inside and outside the cell). Whereas it is now well recognized that IDPs/IDRs are functional despite the lack of three-dimensional fold, the identification of the features that carry the function(s) remains challenging (34). Whether short linear motifs (35) or small residual structures (e.g. MoRFs, molecular recognition features (36); PreMos, pre-structured motifs (37)) that fold upon binding would carry the functions in IDRs is still under debate (34). To date, these features cannot be identified or even predicted in all of these proteins/domains. Current knowledge of IDPs/IDRs features is still limited and must be improved to decipher their multiple functionalities.
Here we report the identification as well as the molecular and functional description of a short stable structural motif in the intrinsically disordered NS5A-D2 and show its essential role in HCV RNA replication. This motif, a Pro314–Trp316 turn denoted PW turn, pre-exists in the isolated protein domain prior to binding to any potential target. Using NMR spectroscopy, we determined the structure of this PW turn, which explains the sequence conservation in this region, as well as the absolute requirement for these peculiar residues in the RNA replication process. We show that this NS5A-D2 PW turn is directly involved in the binding of host CypA (27) and that it influences the CypA PPIase activity regarding residue Pro314.
Experimental Procedures
Production and Purification of HCV NS5A-D2
The domain 2 of the HCV NS5A WT protein from Con1 strain (euHCVdb; GenBankTM accession number AJ238799, genotype 1b) was expressed in Escherichia coli BL21(DE3) cells using a pT7.7 expression vector containing the synthetic coding sequence. The resulting recombinant domain 2 of HCV NS5A (NS5A-D2 WT; residues 245–341) has extra M- and -LQHHHHHH extensions at the N and C termini, respectively. The NS5A-D2 I315G mutation was introduced in the WT plasmid by site-directed mutagenesis using the following forward and reverse primers: 5′-CGT GCA ATG CCG GGC TGG GCC CGT CCG GAT TAC AAC C-3′ and 5′-GGT TGT AAT CCG GAC GGG CCC AGC CCG GCA TTG CAC G-3′. To produce 15N- or 15N,13C-labeled NS5A-D2 (WT o I315G) the cells were grown at 37 °C in a M9-based semi-rich medium (M9 medium supplemented with [15N]-NH4Cl (1 g/liter), d-[13C6]-glucose (2 g/liter) (for 13C labeling only), Isogro [15N], or [13C,15N] powder growth medium (1 g/liter, 10%; Sigma-Aldrich). At an A600 nm of 0.8, the protein production was induced with 0.4 mm isopropyl-1-thio-β-d-galactopyranoside, and cells were harvested by centrifugation at 4 h postinduction. Following cell lysis and subsequent removal of cell debris by centrifugation, the resulting supernatant was heated at 75 °C for 15 min and then cooled down on ice. Precipitated material was removed by centrifugation. The clarified supernatant, which contains soluble NS5A-D2, was submitted to Ni2+ affinity chromatography (HisTrap column, 1 ml; GE Healthcare Europe). Following SDS-PAGE analysis, selected fractions containing NS5A-D2 were pooled and dialyzed against 30 mm Na2HPO4/NaH2PO4, pH 6.8, 50 mm NaCl, 1 mm THP (Tris(hydroxypropyl)phosphine), 2 mm EDTA. The protein was concentrated up to 200–400 μm using a Vivaspin 15 concentrator (cutoff, 5 kDa) (Satorius Stedim Biotech, Aubagne, France), filtered at 0.2 μ, flash frozen in liquid nitrogen, and then stored at −80 °C. Protein concentration was estimated based on UV absorbance at A280 nm.
Production and Purification of CypA
The production and purification of both unlabeled and 15N-labeled CypA were performed as previously described in Hanoulle et al. (27).
Unlabeled and Doubly 15N,13C-labeled Peptides
Unlabeled synthetic peptides, PepD2-WT (KFPRAMPIWARPDYNPPLLE) and PepD2-I315G (KFPRAMPGWARPDYNPPLLE), corresponding to residues 308–327 of NS5A-D2 Con1 were obtained from GenCust (Luxembourg). The purity was checked by reverse phase HPLC and mass spectrometry as >95%. 15N,13C-labeled PepD2-WT and PepD2-I315G peptides were produced in E. coli BL21(DE3) using a modified pET32a expression vector (Novagen). Briefly, the peptides were each produced at the C terminus of a fusion protein harboring a N-terminal histidine tag, the thioredoxin protein, and a linker containing both a thrombin (LVPRGS) and a chemical (DP) cleavage sites. Recombinant bacteria were grown in [15N,13C]-M9-based semi-rich medium (see above), and protein production was induced by isopropyl-1-thio-β-d-galactopyranoside. Following cells lysis, the fusion protein was first purified by Ni2+ affinity chromatography (HisTrap column, 1 ml; GE Healthcare Europe) and dialyzed against buffer 50 mm Tris-Cl, pH 6.8, 200 mm NaCl. The acid-sensitive Asp-Pro cleavage site was cleaved by the addition of 0.1% TFA and heating at 75 °C for 4.5 h, thereby releasing the peptide of interest from the fusion protein. The cleavage was checked by SDS-PAGE analysis. After pH neutralization, the His-TrxA moiety was then removed from the peptide sample by incubation with Ni2+-loaded chelating Sepharose beads (GE Healthcare) and filtration (0.2 μ). The clarified sample was acidified with 0.1% TFA and loaded on a preparative C18 reverse phase column (Zorbax 300SB C18 9.4/250; Agilent). The purified 15N,13C-labeled peptide was eluted using an acetonitrile gradient and then analyzed by mass spectrometry. Following lyophilization, the peptide was dissolved in 30 mm NaH2PO4/Na2HPO4, pH 6.8, 50 mm NaCl, 1 mm THP, 2 mm EDTA. The resulting 15N,13C-labeled PepD2 and PepD2-I315G peptides thus contain an extra N-terminal proline residue resulting from the DP chemical cleavage site.
Conservation and Variability of NS5A-D2 Sequence from Various HCV Genotypes
The NS5A-D2 sequence (residues 248–341) from HCV Con1 strain (AJ238799; genotype 1b) is numbered as in the full-length NS5A protein. The amino acid repertoire was deduced from the ClustalW multiple alignments of 28 representative NS5A sequences from all confirmed HCV genotypes and subtypes (see the European HCV Database (38)) using the Network Protein Sequence Analysis (39) webserver tools. Amino acids observed at a given position in less than two distinct sequences are not included. The degree of amino acid conservation at each position can be inferred from the extent of variability (with the observed amino acid listed in decreasing order of frequency from top to bottom) together with the similarity index according to ClustalW convention (asterisk, invariant; colon, highly similar; dot, similar).
NMR Spectroscopy
Spectra were acquired at 298 K on either a Bruker Avance 600 MHz or a Bruker 900 MHz NMR spectrometer, both equipped with a cryogenic triple resonance probe (Bruker, Karlsruhe, Germany). The proton chemical shifts were referenced using the methyl signal of sodium 3-trimethylsillyl-[2,2,3,3-d4]propionate at 0 ppm. Spectra were processed and analyzed with the Bruker TopSpin software package 2.1. Assignments of NS5A-D2 Con1 (WT) backbone resonances were achieved using the product plane method (40) with two-dimensional 1H,15N HSQC and three-dimensional 1H,15N,13C HNCO, HNCACO, HNCACB, HNCOCACB, and HNCANNH spectra acquired at 600 MHz on a 180 μm 15N,13C-labeled NS5A-D2 Con1 sample at 298 K. NMR data (assignments and structural constraints) corresponding to the peptide PepD2-WT (residues 308–327) were acquired as three data sets. The two first sets, which contain 1H-1H TOCSY, 1H-1H NOESY, 1H-15N HSQC, and 1H-13C HMQC spectra at 600 MHz and 1H-13C HMQC, 1H-13C HMQC-TOCSY, and 1H-13C HMQC-NOESY spectra at 900 MHz, were acquired on the unlabeled peptide at natural abundance. The last data set was acquired on a uniformly 15N,13C-labeled peptide and comprises 1H-15N HSQC and three-dimensional HNCACB, HN(CO)CACB, HNCO, and HNHA sequences. On the mutant peptide PepD2-I315G (residues 308–327), two data sets were recorded at 600 MHz. The first one, acquired on unlabeled peptide, contains 1H-1H TOCSY, 1H-1H NOESY, and 1H-13C-HMQC spectra, whereas the second one was acquired on a uniformly 15N,13C-labeled peptide and comprises 1H-15N HSQC and three-dimensional HNCACB, HNCO, and HNHA spectra.
NMR-derived Restraints and Structure Calculation
From the different NMR data sets acquired both on unlabeled and 15N,13C-labeled peptide PepD2-WT (residues 308–327, Con1), distance-based (NOEs) and backbone dihedral angle-based experimental restraints were derived. NOE intensities used as input for structure calculations were obtained from the NOESY spectrum recorded with a 400-ms mixing time. NOEs were partitioned into three categories of intensity that were converted into distances ranging from a common lower limit of 1.8 Å to upper limits of 2.8, 3.9, and 5.0 Å, respectively. Protons without stereospecific assignments were treated as pseudoatoms, and correction factors were added to the upper distance constraints (41). Additionally dihedral angle constraints calculated with Talos (42) from 1H, 15N, and 13C chemical shifts were introduced. Three-dimensional structures were generated from NOE distances and dihedral angles with the standard torsion angle molecular dynamics protocol in the XPLOR-NIH 2.30 program (43) using the standard force field and default parameter set. A set of 50 structures was initially calculated to widely sample the conformational space, and the structures with no distance restraint violations were retained. The 28 final selected structures were compared by pairwise root mean square deviation over the backbone atom coordinates (N, Cα, and C′). Statistical analyses, superimposition of structures, and structural analyses were performed with MOLMOL (44) and the PDB Validation Server. Ramachandran analysis performed on the 28 final structures (560 residues with 364 non-glycine and non-proline residues) showed that 51.9, 44.2, 1.4, and 2.5% of the residues were in most favored, allowed, generously allowed, and disallowed regions, respectively. The PyMOL software (PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger) was used for molecular graphics.
CαZ·NZ Exchange NMR Spectroscopy
To detect the CypA-catalyzed PPIase activity directly on proline resonances, we used a proline-directed zz-exchange experiment. Briefly, the pulse sequence corresponds to a two-dimensional 1H,15N-Hα(Cα)N experiment with an additional CαZ·NZ magnetization transfer period (150 ms). During this period, the magnetization (along the z-axis) can transfer between the 1Hα-15N resonances corresponding to the different conformers (trans, cis) of a given proline residue. This method has the advantage of detecting a PPIase activity directly on proline resonances and not on resonances from neighboring residues, thus avoiding the possibility of misinterpreting exchange signals in proline-rich regions.
Accession Codes
NMR assignment of the backbone atoms (HN, NH, Cα, Cβ, C′) of the domain 2 of NS5A (Con1 strain) have been deposited in the Biological Magnetic Resonance Data Bank under accession code 19055. NMR assignment of the peptide PepD2-WT (residues 308–327 of the domain 2 of NS5A, Con1 strain) has been deposited in the Biological Magnetic Resonance Data Bank under accession code 19059, and the coordinates of the NMR structure have been deposited in the Protein Data Bank under accession code 2M5L.
Circular Dichroism Spectroscopy
Circular dichroic spectra were recorded at 293 K with a model CD6 spectropolarimeter (Jobin Yvon-SPEX-Horiba). In the far ultraviolet region (200–250 nm) measurements were made in a 10-μm-path length quartz cell. In the near ultraviolet region (250–320 nm) measurements were made in a 1-mm-path length quartz cell. Spectra were acquired with a step of 0.5 nm. To compare the signal of both samples, the concentration of the samples (1.6 mm) were determined and adjusted precisely by two methods: their absorbance at 280 nm and the integration of their 1H-NMR signal relatively to a standard. Baseline runs were made prior to each sample run, and the baseline was subtracted to obtain the final spectrum. Near and far UV intensities were expressed in terms of specific ellipticity (per decimole of amino acid residue), [θ]MRW = [θ]/N, where N is the total number of amino acids.
Fluorescence Spectroscopy
Steady state fluorescence of the Trp residue was measured at 293 K on a PTI fluorescence spectrometer (PTI Monmouth Junction, NJ). To excite specifically the Trp residue, the excitation was set at 295 nm, and the emission scanned from 305 to 500 nm (0.5-nm stepwise). The excitation and emission slit widths were set to 2 and 4 nm, respectively. Excitation and emission polarizers were used to measure the steady state fluorescence anisotropy (excitation and emission set at 295 and 350 nm, respectively) on 125 μm peptide samples in 1-cm-path length cells. We determined for these wavelengths a G factor of 1.19.
Plasmid Constructs
All nucleotide and amino acid numbers refer to the HCV Con1 strain. Plasmids encoding the subgenomic luciferase reporter replicon (pFK_i389LucNS3–3′_Con1_ET_δg) and the nonreplicative mutant (pFK_i389LucNS3–3′_NS5B_GND_Con1_ET_δg) have been described previously (45). Single amino acid substitutions in NS5A-D2 were generated by PCR-based site-directed mutagenesis and, after restriction with BclI and XhoI, the fragment was inserted into the pFK_i389LucNS3–3′_Con1_ET_δ vector. All constructs were verified by nucleotide sequence analysis.
In Vitro Transcription, Electroporation of HCV Replicons, and Replication Assay
The protocol used for generation and electroporation of HCV RNAs has been described elsewhere (46). For transient replication assays, 400 μl of single cell suspensions of (107cells/ml) Huh-7 cells were mixed with 5 μg in vitro transcribed subgenomic replicon RNA and transfected by electroporation. After transfection, cells were resuspended in 41 ml of complete DMEM, and 1.5 ml of cell suspension was seeded in duplicate in 12-well plates. To measure luciferase activity, the cells were washed with PBS and lysed 4, 24, 48, 72, and 96 h after electroporation by addition of 350 μl of lysis buffer (0.1% Triton X-100, 25 mm glycylglycine, pH 7.8, 15 mm MgSO4, 4 mm EGTA, and 1 mm DTT). Cells were immediately frozen at −70 °C, and after thawing, 100 μl of lysate was mixed with 360 μl of assay buffer (25 mm glycylglycine, 15 mm MgSO4, 4 mm EGTA, 1 mm DTT, 2 mm adenosine triphosphate, and 15 mm K2PO4, pH 7.8). Luciferase activity was measured for 20 s in a luminometer (Lumat LB9507; Berthold, Freiburg, Germany) after addition of 200 μl of luciferin solution (200 mm luciferin, 25 mm glycylglycine, pH 8.0). Replication efficiency was determined by normalizing the relative light units of the different time points to the respective value obtained at 4 h.
Immunofluorescence Analysis
All nucleotide and amino acid residue numbers refer to the Con1 genome (GenBankTM accession number AJ238799). The pTM NS3–5B ET Con1 vector allowing the expression of the HCV nonstructural proteins NS3 to 5B containing the replication-enhancing mutations ET (E1202G, T1280I in NS3 and K1846T in NS4B) was used to insert the I315G and P314A mutations into the NS5A domain 2 coding region. To generate pTM NS3–5B ET/I315G or ET/P314A, the XhoI/SpeI fragments from constructs pFKI389Luc/NS3–3′/Con1/ET/dg/I315G or P314A were inserted into the XhoI/SpeI-digested pTM NS3–5B ET Con1 plasmid. Nucleotide sequences of all constructs were verified by sequence analysis (GATC, Konstanz, Germany). Details about cloning strategies are available upon request.
Huh7-Lunet/T7 cells (1 × 105) were seeded onto glass coverslips in 24-well plates 1 day before prior to transfection. Cells were transfected with pTM-NS3–5B-based expression vectors by using the TransIT LT1 transfection reagent (Mirus Bio LLC, Madison, WI) according to the providers' instructions. Immunofluorescence staining for detection of NS5A was performed as described elsewhere (47). Briefly, cells were fixed with 4% paraformaldehyde for 20 min, washed three times with PBS, and permeabilized with 50 μg/ml digitonin for 5–10 min. Cells were incubated in blocking solution (3% BSA in PBS) for 30 min and subsequently in 1% BSA/PBS containing primary antibodies for 1 h at room temperature. NS5A was detected by using a NS5A-specific monoclonal antibody (9E10, generous gift from Charles M. Rice) at a final concentration of 1:10,000. The nuclei were stained with DAPI for 1 min. The cells were washed and mounted with Fluoromount G (Southern Biotechnology Associates, Birmingham, AL), and pictures were acquired with a Leica SP2 confocal laser scanning microscope using a 63× objective.
Results
Identification of Nondisordered Residues in NS5A-D2
The 1H,15N HSQC spectrum of NS5A-D2 (Con1 strain) displays a narrow 1HN chemical shift dispersion limited to 1 ppm, as expected for a mainly disordered domain (Fig. 1A). All the backbone amide resonances, except for the 12 proline residues, were assigned (Biological Magnetic Resonance Data Bank accession code 19055) with the product plane method (40). Two resonances corresponding to Trp316 and Ala317 in the main CypA binding site (27) are unusually upfield shifted (Fig. 1B). Indeed, comparison of experimental 1H and 15N chemical shifts with their corresponding neighbor-corrected IDP values (ncIDP) (48) singles out the Trp316 residue (Fig. 1D). Amino acid sequence analysis with a MoRFs predictor does, however, not reveal any peculiarity for these residues (data not shown). Secondary structure propensity analysis (Fig. 1C), based on experimental 13Cα and 13Cβ chemical shifts, shows the presence of two residual α-helices in NS5A-D2 (residues 250–267 and 299–305), in agreement with the results of Feuerstein et al. (49), but does not highlight residual structure for Trp316 or Ala317. Nonetheless, residues Trp316 and Ala317 are strictly conserved over all HCV genotypes (Fig. 1B), underscoring their importance.
FIGURE 1.

NMR analyses of HCV NS5A-D2. A, 1H,15N HSQC spectrum of NS5A-D2. B, NS5A-D2 amino acid sequence (residues 248–345) from HCV Con1 strain (genotype 1b). Underneath is the amino acid repertoire deduced from ClustalW multiple alignments of reference sequences from all confirmed genotypes and subtypes. The black box corresponds to the PepD2-WT peptide encompassing the CypA binding site. C, secondary structure propensity analysis of the 13Cα and 13Cβ NMR chemical shift of NS5A-D2. Values close to 0 correspond to fully disordered residues, whereas positive and negative scores represent helical propensities and extended regions, respectively. D, deviations of combined 1H,15N chemical shifts of NS5A-D2 compared with neighbor-corrected IDP chemical shift library.
NMR Structural Model of the Proline-Tryptophane Turn in NS5A-D2
To further characterize this particular region, centered on WTrp16 and Ala317, a peptide encompassing NS5A residues 308–327 was chemically synthesized and designated PepD2-WT. The 1H,15N HSQC spectrum of this peptide overlaps with that of the full-length NS5A-D2, demonstrating that it behaves similarly as the corresponding region in the full-length NS5A-D2 (Fig. 2). Homo- and heteronuclear NMR experiments recorded on both unlabeled and doubly 15N,13C-labeled pepD2-WT allowed us to measure several parameters such as 1H,15N,13C chemical shifts (Biological Magnetic Resonance Data Bank accession code 19059) and NOE contacts (Figs. 3 and 4) that have been used as experimental constraints for three-dimensional structure calculation. The final set of 28 low energy structures that fulfill the experimental restraints displays a well defined small structural motif (from Met313 to Ala317) in PepD2-WT (Fig. 5 and Table 1; PDB code 2M5L), whereas the N and C termini remain highly flexible. The most characteristic feature of this motif corresponds to the interaction of Trp316 side chain with the Pro314 residue, as shown by the following selected NOE cross-peaks: Trp316-Hδ1/Pro314-Hβ-γ and Trp316-Hϵ1/Pro314-Hβ-γ (Fig. 4B). The same NOE contacts made by Trp316-Hϵ1 were also identified in spectra of full-length NS5A-D2, further validating the PW turn as a structural feature of NS5A-D2 (Fig. 4B). The aromatic side chain of Trp316 adopts a near perpendicular orientation relative to the cyclic Pro314 residue. This hydrophobic interaction between Pro314 and Trp316 results in a turn that is also constrained by contacts between the side chain of Ala317 with these of Pro314 and Met313, as shown by the NOE contacts Met313-Hγ/Ala317-Hβ and Pro314-Hδ/Ala317-Hβ (Figs. 4B and 5). Residues Pro314, Trp316, and Ala317 are strictly conserved among all HCV genotypes (Fig. 1B) and have been shown to be crucial for viral replication (12). In contrast, position 315 is quite variable because residues Ile, Val, Pro, and Ala are observed in different genotypes. This is consistent with the side chain of Ile315 pointing outward the PW turn and being not directly involved in its stabilization. In the PDB database, we found that the sequence P[IVPA]WA (taking into account all existing residues at position 315 for the different HCV genotypes) is present in 100 entries corresponding to 89 unique proteins. In 36 of them, this sequence adopts a similar PW turn structure as identified here in NS5A-D2. For example, similar PW turn structures were found in: triosephosphate isomerase from Staphylococcus aureus MRSA252 (PDB code 3M9Y, sequence PIWA); triosephosphate isomerase of Tenebrio molitor (PDB code 2I9E, sequence PVWA); urocanase from Geobacillus stearothermophilus (PDB code 1X87, sequence PAWA); and triosephosphate isomerase from Trypanosoma brucei brucei (PDB code 1ML1, sequence PPWA). Moreover, this structural motif is involved in the function of the triosephosphate isomerases) (see “Discussion”) (50). These data strongly suggest a functional role for the turn in NS5A-D2.
FIGURE 2.

NMR comparison of HCV NS5A-D2 and PepD2-WT. Superimposition of 1H,15N HSQC NMR spectra of 15N-labeled NS5A-D2 (blue) and PepD2-WT (308KFPRAMPIWARPDYNPPLLE327, residues 308–327 of NS5A-D2) (red). The spectra were acquired on a 600MHz NMR spectrometer. Assignments of NS5A-D2 and PepD2-WT resonances are indicated as black and red labels, respectively (according to the full-length NS5A protein numbering). Assignments of NS5A-D2 and PepD2-WT have been deposited in the Biological Magnetic Resonance Data Bank with the accession codes 19055 and 19059, respectively. Beyond the two N- and C-terminal residues, PepD2-WT resonances match with the corresponding resonances in NS5A-D2.
FIGURE 3.

NMR chemical shifts and coupling constant analyses of PepD2-WT and PepD2-I315G. A and B, the deviations between experimental and calculated random coil chemical shift values were plotted for PepD2-WT (A) and PepD2-I315G (B). The theoretical random coil values were calculated using the neighbor-corrected intrinsically disordered protein chemical shift library (48) from the amino acids sequence of both peptides. The neighbor-corrected IDP analyses were performed for combined NH, Hα, and Cα chemical shifts and showed that a small region around residue Trp316 exhibits unexpected chemical shifts for a disordered protein. C, The 3JHN-Hα coupling constants (Hz) of PepD2-WT (in black) and PepD2-I315G (in gray), which are related to the backbone dihedral angle φ are plotted along the peptide sequences.
FIGURE 4.

Definition of the structural element identified in NS5A-D2. A, isomeric state of proline residues in PepD2-WT. The figure corresponds to the superimposition of 1H,1H TOCSY (in blue) and 1H,1H NOESY (400-ms mixing time) (in red) spectra recorded on PepD2-WT (1 mm) and centered on the proline Hα/Hδ region. The NOE cross-peaks between the Hδ of all proline residues (i.e. Pro310, Pro314, Pro319, Pro323, and Pro324) and the Hα of their respective i − 1 residue indicate that all the proline residues are mainly in trans conformation. B, typical NOE contacts that define the structural element identified in NS5A-D2. Each panel corresponds to the superimposition of 1H,1H TOCSY (blue) and 1H,1H NOESY (400-ms mixing time) (red) spectra recorded on PepD2-WT at 600 MHz. These NOE contacts have been used as experimental distance constraints to calculate the structure of PepD2-WT. The green box highlights the NOE contacts recorded on the full-length 15N-labeled NS5A-D2 domain via a 15N NOESY HSQC experiment. The NOE pattern in the full-length domain is similar to that of the PepD2-WT peptide.
FIGURE 5.
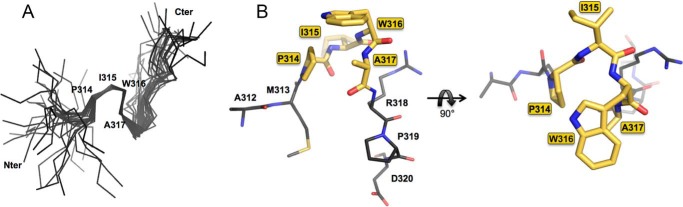
A PW turn in the intrinsically disordered HCV NS5A-D2. A, superimposition of Cα traces of the 28 final structures of PepD2-WT (308–327) showing the well defined small structural motif from Pro314 to Ala317. B, structure of the PW turn motif (in yellow) identified in NS5A-D2 (in gray) in stick representation.
TABLE 1.
Statistics of the NMR structure model calculations of PepD2-WT (308–327) Con1
| NS5A-D2 (308–327) | |
|---|---|
| NMR experimental constraints | |
| Distance restraints NOE | |
| Intraresidue | 0 |
| Sequential | 87 |
| Medium range | 28 |
| Long range | 6 |
| Total distance restraints | 121 |
| Dihedral angle constraints | |
| φ angles | 10 |
| Ψ angles | 10 |
| Statistics for the final X-PLOR structures | |
| Number of structures in the final set | 28 |
| X-PLOR energy (kcal·mol−1) | −5.5 ± 9.9 |
| NOE violations | |
| Number > 0.5 Å | None |
| Root mean square deviation (Å) | 0.084 ± 0.007 |
| Dihedral angle violations | |
| Number > 5° | ≤1 |
| Root mean square deviation (°) | 1.95 ± 0.61 |
| Deviation from idealized geometry | |
| Angles (°) | 0.66 ± 0.02 |
| Impropers (°) | 0.382 ± 0.014 |
| Bonds (Å) | 0.0036 ± 0.0002 |
| Root mean square deviation (Å) | |
| Backbone (C′, Cα, N) | |
| Residues 314–317 | 0.25 ± 0.10 |
| All residues | 4.20 ± 1.10 |
| All heavy atoms | |
| Residues 314–317 | 0.51 ± 0.25 |
| All residues | 5.49 ± 1.07 |
| Ramachandran statisticsa | |
| Residues in most favored regions (%) | 51.9 |
| Residues in allowed regions (%) | 44.2 |
| Residues in generously allowed regions (%) | 1.4 |
| Residues in disallowed regions (%) | 2.5 |
a Non-proline, non-glycine, and non-end residues (364 residues).
Mutation I315G Is Incompatible with the PW Turn
When extending our search in the PDB database to the sequence PXWA, where X is any amino acid, we noted that the structural PW turn is largely disfavored when the (i + 1) position relative to the proline is either an aromatic or a glycine residue. Indeed the PW turn fold was found in 0 of 14, 0 of 12, 1 of 5, and 2 of 21 entries (unique protein) for residue X being Tyr, Phe, Trp, or Gly, respectively. To investigate whether a Gly at this position would be incompatible with the turn formation, a peptide with the I315G mutation was produced. Differences between the PepD2-WT and PepD2-I315G were pronounced, with significant changes for the amide proton of Trp316 and Ala317 and for all ring protons of Pro314 (Fig. 6). The NMR chemical shift (1H, 15N, and 13C) values for the peptide PepD2-I315G were indeed close to random coil values (Figs. 3 and 6). The characteristic NOE contact between Hγ-M313 and Hβ-A317 is absent in the 1H,1H NOESY spectrum of PepD2-I315G, confirming that the PW turn is not present despite the presence of the crucial Pro314 and Trp316 residues. Moreover, far and near UV circular dichroism and fluorescence spectroscopy analyses revealed a marked difference between the two peptides, notably with a different relative orientation of the aromatic residues and a reduced anisotropy (i.e. more compact shape) for PepD2-WT, arguing again for a loss of the PW turn structure in the I315G mutant (Fig. 7). The I315G mutation therefore provides an efficient means to evaluate the functional role of the turn while intervening minimally with the primary sequence.
FIGURE 6.

NMR comparison of PepD2 WT and I315G mutant. A, overlay of 1H,15N HSQC NMR spectra of 15N-labeled PepD2-WT (blue) and PepD2-I315G (red) showing the consecutive perturbations for Trp316 and Ala317. B, overlay of 1H,1H TOCSY spectra of PepD2-WT (blue) and PepD2-I315G (red) centered on the Hδ of proline residues. C, overlay of 1H,13C HMQC spectra of PepD2-WT (blue) and PepD2-I315G (red) centered on the Cβ and Cγ of proline residues. D, overlay of 1H,13C-HMQC spectra of PepD2-WT (blue) and PepD2-I315G (red) centered on the Hβ of Trp316. E, overlay of 1H,1H TOCSY (blue or red) and NOESY (violet or orange) spectra corresponding to PepD2-WT (left) and PepD2-I315G (right). F, I315G-induced chemical shift perturbations on proline resonances of PepD2 (residues 308–327). The figure corresponds to the superimposition of two-dimensional 1H,15N-Hα(Cα)N NMR spectra of PepD2-WT (in blue) and PepD2-I315G (in red).
FIGURE 7.

CD and fluorescence spectroscopy analyses of PepD2-WT and I315G mutant. A and B correspond to far and near UV CD analyses of PepD2-WT (plain black lines) and PepD2-I315G (dashed gray lines). A, the far CD spectra of both peptides are significantly different in intensity even though the concentrations were adjusted precisely (see “Experimental Procedures”). A small difference in shape is observable. B, the near UV CD spectra of both peptides are also very different in intensity and shape. The tryptophan (Trp316), tyrosine (Tyr321), and phenylalanine (Phe309) may all contribute to the CD signal in the 260–280-nm region (71). The spatial arrangement of these groups in the protein determines the sign and intensity of the CD bands in the near UV (71). Therefore, the near UV CD spectrum of a protein or a peptide reflects its conformation. From this CD analysis, we concluded that the aromatic residues Phe309, Trp316, and Tyr321 have different relative orientations in PepD2-WT and PepD2-I315G. This result is consistent with the presence or absence of a structural motif. C, tryptophan fluorescence spectroscopy. When excited at 295 nm, both peptides display a maximum of emission at 350 nm, in agreement with the expected exposition of their single Trp residue to the water solvent. The anisotropy observed for the PepD2-WT (0.033) was significantly inferior to that observed for the PepD2-I315G (0.044). Because both peptides are small (20 residues) and have the same size, the fluorescence anisotropy of their single Trp reflects their global motions in solution. The PepD2-WT peptide is thus more compact in solution than the PepD2-I315G peptide. When measured in the presence of 3 m guanidine HCl, the anisotropy of both peptides became higher and equivalent (0.053). These results are consistent with the presence of a local structuration in PepD2-WT in the absence of guanidine.
The PW Turn Is Essential for HCV RNA Replication
Because NS5A-D2 has been shown to be essential for viral replication, we compared RNA replication efficiency of NS5A WT and the I315G mutant by using a subgenomic (NS3-NS5B, Con1) replicon and Huh-7 cells. As illustrated in Fig. 8, the sole mutation I315G in NS5A almost completely abolished viral replication that was close to the background as determined with the replicon encoding an inactive NS5B RNA-polymerase (GND mutant). The replication levels corresponding to the I315G and P314A mutations, respectively, were similar. Note that in the I315G mutant, residues Pro314 and Trp316, which have been previously identified as crucial for replication (12, 51), are unaffected. Hence, the short PW turn structural motif in the mainly disordered NS5A-D2 domain plays an essential role for HCV RNA replication.
FIGURE 8.
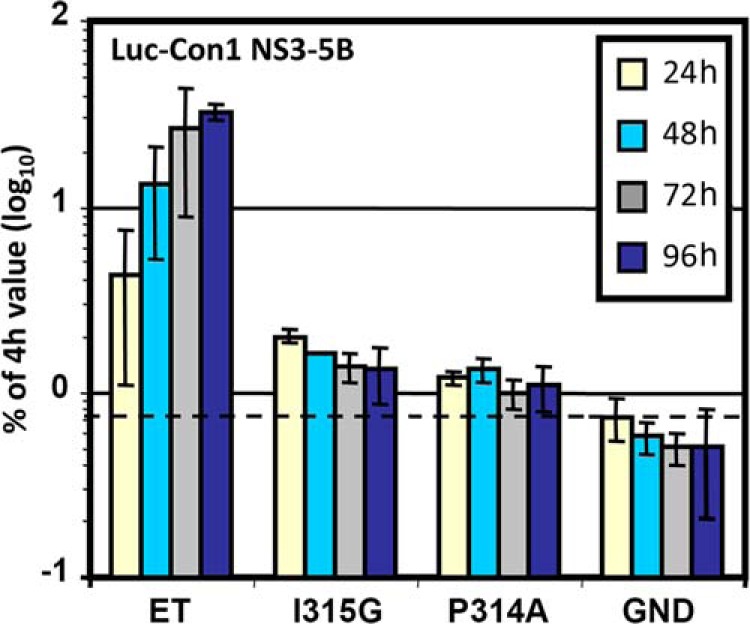
Effect of the I315G substitution on HCV RNA replication. NS3–5B subgenomic Con1 replicons containing cell culture adaptive mutations (E1202G, T1280I, and K1846T) designated ET (45) and additional mutations I315G or P314A in NS5A or a defective polymerase mutant (GND) were electroporated into Huh-7 cells. The cells were lysed at the given time points, and the luciferase activities were determined. The data were normalized to the respective 4-h value reflecting transfection efficiency. Mean values of quadruplicate measurements of two independent experiments are shown. The error bars indicate standard deviations. The horizontal dashed line indicates the background.
Impact of Mutations I315G and P314A in NS5A Domain 2 on Subcellular Distribution of NS5A
To evaluate the impact of I315G and P314A mutations on the subcellular localization of NS5A, we expressed mutant NS3–5B polyproteins in Huh7/Lunet T7 cells and performed immunofluorescence assays using specific NS5A antibodies. First, expression of wild type and NS5A mutants was analyzed by Western blot. Detection of comparable amounts of NS5A indicated no initial effect of mutations on polyprotein cleavage (Fig. 9A). NS5A localization pattern in cells expressing NS3–5B WT is characterized by a dispersed distribution of dot-like structures, similar to that described in replicon cells (47) (Fig. 9B, left column). Interestingly, expression of mutations disrupting the structural motif in NS5A domain 2 resulted in an increased number of cells exhibiting altered distribution of the viral protein (Fig. 9B, right column). This altered phenotype correlated with a strong accumulation of large structures where NS5A is present, reminiscent of the cluster distribution in cells treated with NS5A and Cyp inhibitors (46, 52, 53). Notably, the proportion of cells with the cluster phenotype varied from 24 to 67% for I315G and P314A mutants, respectively, compared with 5% for WT (Fig. 9C). Therefore, disruption of the structural motif by mutation I315G or P314A results in a higher abundance of a clustered NS5A distribution.
FIGURE 9.
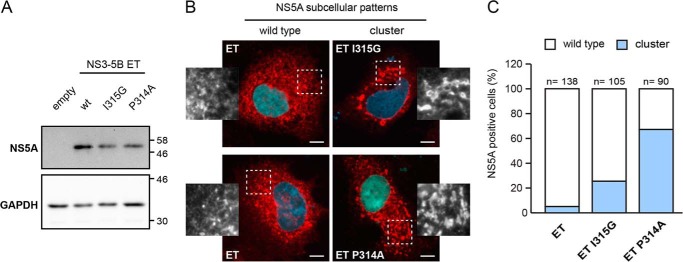
Impact of mutations within the structural motif in disordered domain 2 on subcellular localization of HCV NS5A. Huh-7 Lunet/T7 cells were transfected with empty vector (empty) or plasmids encoding the NS3 to NS5B polyprotein of HCV genotype 1b (Con1) containing mutations ET (E1202G, T1280I, and K1816T; designated ET wt) or mutations I315G (ET I315G) or P314A (ET P314A) in the NS5A sequence. 23 h after transfection, the cells were lysed or alternatively fixed for immunofluorescence analysis. A, NS5A expression was confirmed by immunoblot using antibodies directed against NS5A. GAPDH served as loading control. Apparent molecular masses (kDa) are indicated on the right. B, NS5A subcellular distribution (red) in cells expressing wild type (wt) NS5A (left column) or mutants (right column) I315G (upper right) and P314A (lower right) was analyzed by immunofluorescence; nuclei were stained with DAPI (blue). White boxed areas are shown as enlargements in the corresponding side panels. Scale bar, 10 μm. Note the wild type dotted-like staining pattern in ET wild type-transfected cells compared with the formation of large NS5A “clusters” in cells expressing NS5A-D2 mutant polyproteins. C, quantitative analysis of NS5A staining patterns in cells expressing ET wild type or ET I315G or P314A NS5A mutant polyproteins. For each construct, NS5A-positive cells (cell numbers indicated on top of the bars) were counted and classified into WT (white) and clustered (blue) phenotypes.
The PW Turn Is Required for Proper Interaction with Host CypA
Because the structural motif is located in the CypA binding site (8, 27, 51) and CypA is a mandatory host factor for HCV replication, we investigated the possibility that the structural PW turn element was involved in the binding of CypA. NMR titration experiments were recorded on 15N-labeled CypA with increasing amounts of either unlabeled PepD2-WT or PepD2-I315G. The chemical shift perturbations of CypA residues near the binding site, from the free position toward the ligand-saturated frequency, allowed the determination of the dissociation constant (KD) (Fig. 10). The affinity between CypA and the PW turn containing PepD2-WT peptide (KD = 0.5 mm) is three times better than the one involving the random coil PepD2-I315G peptide (KD = 1.4 mm). The structural motif hence is required for proper interaction with the host chaperone CypA. Its absence in the NS5A I315G mutant does, however, not completely abolish the interaction. Indeed, our experimental setup measures the sole contribution of the structural motif, because in each peptide there are the same five proline residues that participate in the CypA binding.
FIGURE 10.
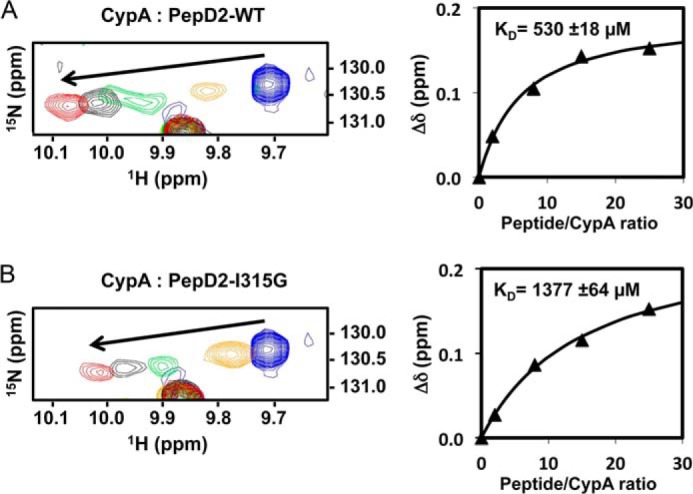
Interaction of CypA with PepD2-WT (A) and PepD2-I315G (B). Left panels correspond to the overlay of 1H,15N HSQC spectra of 15N-CypA (0.1 mm) (centered on the W121sc resonance) acquired in presence of increasing amounts of unlabeled PepD2 (0, 0.2, 0.8, 1.5, and 2.5 mm). Right panels show the titration curves where the 1H,15N combined chemical shift perturbations (Δδ, ppm) were plotted as a function of the peptide/CypA ratios. The KD values correspond to the average (± S.D.) calculated over four CypA resonances (Trp121sc, Q63, Met100, and Glu120).
The PW Turn Modulates the CypA PPIase Activity on Pro314
Because the binding and PPIase enzymatic activity of CypA are tightly linked (54), we assessed the cis/trans isomerization activity of CypA on the proline residues in the 308–327 region of NS5A-D2. Because the 20-mer peptide contains 5 prolines, assessing their conformers on the basis of the 1H,15N HSQC peaks of neighboring residues proved too ambiguous. We therefore adapted the 1Hα-(13Cα)-15N experiment to include a 13Cβ transfer step to assess the proline conformation (cis/trans) or an exchange period to monitor the PPIase activity on the 1Hα-15N correlations directly on proline resonances. In the 15N,13C-labeled PepD2-WT spectra, we found several populations for the different proline residues (Fig. 11A). The major population always corresponds to the trans (T) conformer, as in the PW turn structural model (Figs. 4A and 5). The minor populations correspond to a cis (c) or an additional trans (t) conformer for every prolines (Fig. 12A). These latter trans (t) conformers reflect the long range effect of the cis/trans equilibrium of neighboring prolines on the proline chemical shift. CypA-catalyzed exchange was detected between T and c (T ↔ c) for Pro310, Pro314, Pro319, Pro323, but not for Pro324 and between T and t (T ↔ t) for both Pro314 and Pro324 (Fig. 12, A and B). PepD2-WT and PepD2-I315G showed marked differences for 1H and 15N chemical shift values of Pro314 and to a lesser extent of Pro319 (Fig. 6F). We identified a major (T) and a minor (c) population for each proline residue in this 15N,13C-PepD2-I315G peptide, but the second minor (t) population was only observed for Pro324, which belongs to the Pro323-Pro324 sequence motif (Figs. 11B and 12C). The absence of the minor (t) population for Pro314 in PepD2-I315G suggests that the loss of the PW turn structural motif uncouples Pro314 from the neighboring prolines. The addition of catalytic amounts of CypA to PepD2-I315G led to T ↔ c exchange peaks for all prolines except Pro324 (as for WT) and to a T ↔ t exchange peak for Pro324 (Fig. 12, C and D). The CypA-catalyzed T ↔ c exchange on Pro314 is significantly faster when the structural motif is absent in the mutant peptide (Fig. 12, B and D).
FIGURE 11.

Assignments of the proline resonances in PepD2-WT (A) and PepD2-I315G (B). The panels correspond to two-dimensional 1H,15N-Hα(Cα)N NMR spectra centered on the Hα-δ/N region of proline residues, which have been recorded on 15N,13C-doubly labeled peptide. For a given proline, we observe at its 15N frequency two resonances corresponding to its own (i) Hα and the Hα of the preceding (i − 1) residue (in the 4.0–5.0-ppm range). This experiment also connects the Hδ protons (in the 3.0–3.9-ppm range) to the same nitrogen frequency because the coupling constant between Cα-N and Cδ-N are similar. A, in PepD2-WT, we find several signals, corresponding to different populations, for each proline residue: one major and two minor signals. The major population always corresponds to the trans conformer (T) (labeled in red), whereas the minor populations correspond to either cis (c) (labeled in green) or trans (t) (labeled in gray) conformers. B, in PepD2-I315G we assigned a major (T, labeled in red) and a minor (c, labeled in green) population for each proline residues and a second minor (t, labeled in gray) population only for Pro324, which is in a PP motif. Signals of ∼132 ppm (15N) correspond to the extra proline at the extreme N terminus from the DP cleavage site. Resonance assignments and determination of the proline conformations were based on a three-dimensional 1H,13C,15N-HαCαCβN NMR experiment.
FIGURE 12.

I315G mutation enhances the CypA-catalyzed cis/trans isomerization on Pro314. A and C, tables summarizing all the CypA-catalyzed exchange processes that we have detected between the different proline populations in PepD2-WT (A) and in PepD2-I315G (C), respectively. B and D, superimposition of two-dimensional 1H,15N-Hα(Cα)N NMR spectra with a CαZ.NZ magnetization transfer period (150 ms) in absence (in red) or in presence of catalytic amount of unlabeled CypA (in blue), recorded on 15N,13C-PepD2-WT (B) and 15N,13C-PepD2-I315G (D). B and D correspond to zoomed regions centered on Pro314 and Pro323 resonances. NMR exchange cross-peaks connecting the trans (T) and cis (c) conformers of Pro314 (orange dashed frames and open circles) are more intense in the case of PepD2-I315G (D) than with PepD2-WT (B), whereas the normalized cross-peaks corresponding to the Pro323 isomerization (black dashed frames) are of the same intensity.
Discussion
NS5A is an essential protein that is involved in several steps of the HCV life cycle, such as RNA replication and new viral particle production (12, 13), but for which no enzymatic activity and no precise molecular function(s) have been identified so far. NS5A possesses, in addition to a well structured domain (D1), two additional domains (D2 and D3) that are mainly intrinsically disordered (23–25) and that we previously characterized by NMR spectroscopy (27, 28). IDPs/IDRs exist as highly dynamic and interconverting conformers and thus escape to the rule “one three-dimensional structure, one function.” The features that are responsible for the biological functions of this peculiar class of proteins remain to be better characterized (34). Here we present the identification, structure, and functional role of a short PW turn structural element within the mainly disordered domain 2 of the HCV NS5A protein.
NS5A-D2 from the HCV Con1 strain (genotype 1b) is mainly disordered (Fig. 1), as has previously been shown for the HCV strains H77 (genotype 1a) (23), JFH-1 (genotype 2a) (24), and HC-J4 (genotype 1b) (49). Similar to another genotype 1b HCV strain (49), secondary structural propensity analysis (55) indicates that NS5A-D2 (Con1) does contain two residual α-helices corresponding to residues 250–267 and 299–305 (Fig. 1C). Whereas no special features are detected with the latter method or with MoRF predictors for residues Pro314, Trp316, and Ala317, we demonstrate here that these residues are part of a minimal structural element that we name a PW turn. Among these crucial residues, the proline corresponds to the major disorder-promoting amino acid, whereas the tryptophan is the main order-promoting residue (34, 56, 57), which might explain why the PW turn escapes to the MoRF predictors. Residues Met313 to Ala317 of NS5A-D2 fold as a turn, which is mainly characterized by the hydrophobic interaction between the cyclic Pro314 residue and the aromatic side chain of Trp316 (Fig. 5). This PW turn in NS5A-D2 shares features of the Trp cage fold, which corresponds to the encapsulation of a Trp side chain by several proline residues (i.e. the cage) (58, 59). This motif has been described to be sufficient to efficiently promote the folding of miniproteins (59). The PW turn identified in NS5A-D2 can be seen as a minimal Trp cage, with the interaction of a single proline residue with a tryptophan side chain. Contrary to MoRFs and PreMos in IDPs/IDRs, which are thought to fold upon binding to a partner, the PW turn of NS5A-D2 already exists as a stable structural element in solution before any particular binding event.
In viruses that have a high mutation rate in their genome because of error-prone polymerases, the IDPs/IDRs have been proposed to have evolved to minimize the possible deleterious effects of these frequent mutations (30, 33). The PW turn nevertheless contains three residues, Pro314, Trp316, and Ala317, that are strictly conserved across all HCV genotypes (Fig. 1B). In contrast, position 315 is quite variable because Ile, Val, Pro, and Ala can be observed. In our NMR PW turn structure model, the side chain of Ile315 points outward of the turn and does not participate in the motif formation, thereby explaining the sequence variability observed at this position. In the PDB database, we identified 36 unique folded proteins in which the primary sequence P[IVPA]WA adopts a PW turn fold similar to that observed in NS5A-D2 (e.g. in PDB codes 3M9Y, 2I9E, 1X87 and 1ML1, with the primary sequences being PIWA, PVWA, PAWA, and PPWA, respectively). The motif is notably conserved in the family of the triosephosphate isomerases from bacteria and eukaryotes and corresponds to the N-terminal hinge at the base of catalytic loop 6, which can exist in open or closed conformations (50, 60), thereby regulating the catalytic loop 6 motions (61). While the manuscript was in preparation, Chung et al. (62) reported the structure of a complex between the cellular MOBKL1B protein and two copies of a HCV NS5A-D2 derived peptide (residues 308–327, strain Jc1 genotype 2a). Although no functional role for this MOBKL1B-NS5A interaction was found, in one of the bound peptides the residues 310PAWA313 (PDB code 4J1V chain G) adopt the PW turn motif we here describe for the free peptide in solution, further confirming that the PW turn is an essential structural element over all HCV genotypes. Altogether, these results strongly suggest a functional role for the turn in NS5A-D2. On the basis of our search in the PDB database, we found that an aromatic or Gly residue in position equivalent to 315 disfavors the PW turn. We produced a NS5A-D2 I315G mutant and showed that this mutation efficiently disrupts the PW turn structure while maintaining all strictly conserved residues (Figs. 3 and 6). This mutant, without the PW turn motif, does not support viral RNA replication in a subgenomic replicon assay (Fig. 8). Tellinghuisen et al. (12) have previously observed that the NS5A-D2 P314A and W316A mutants impaired RNA replication, whereas the I315A mutant did not. Their results are consistent with the disruption of the PW turn in the two first mutants and with the fact that an alanine at position 315 is still compatible with this PW turn structure. It hence unambiguously indicates that the structural motif rather than the primary sequence is important for the biological activity of NS5A.
NS5A mutations (I315G or P314A) disrupting the PW turn structure lead to an altered subcellular distribution of NS5A, forming predominantly large clusters as compared with the small speckles formed by WT NS5A (Fig. 9). Electron microscopy analyses suggest that NS5A-positive structures correspond, in part, to double membrane vesicles, which are a hallmark of the membranous HCV replication factory, designated membranous web (52). A NS5A “cluster” phenotype similar to that of the PW turn mutants was previously found in cells treated with NS5A or Cyp inhibitors (46, 52, 53), as well as in PI4KIIIα knockdown cells. This phenotype was shown to correspond to lipid droplets or aberrant double membrane vesicles, respectively (47). Both CypA and PI4KIIIα are essential host cell factors for HCV replication. In case of the latter, kinase activity is enhanced upon HCV infection by interaction with NS5A and NS5B (63, 64), resulting in increased PI4P levels. Although NS5A-PI4KIIIα interaction occurs through NS5A domain I, it is also required for proper membranous web morphology, and therefore, we assessed the effect of I315G and P314A mutations on PI4KIIIα activity by quantifying the intracellular PI4P levels. As expected, expression of WT NS3–5B enhanced PI4P levels (2.6-fold on average) and induced its dispersion throughout the cytoplasm with partial colocalization with NS5A (47). Mutants I315G and P314A also stimulated PI4P production (3.2- and 1.8-fold, respectively) compared with control cells, albeit to different extents (data not shown). Collectively, these results argue against a direct effect of mutations in the structural motif of NS5A-D2 on PI4KIIIα activation. Therefore, inhibition of HCV replication, at least of the genotype 1b isolate Con1, by I315G and P314A mutations (Fig. 8) is probably due to disruption of the structural motif in NS5A-D2 and consequently to the abolishment of the functional interaction with CypA that is required for the establishment of HCV replication sites (46).
Regions in viral proteins associated with a higher sequence conservation have been proposed to be required for interaction with host proteins, which do not evolve as rapidly (32). We previously identified the 308–327 region of NS5A-D2 as the main binding site for the human CypA, which is a mandatory host factor for viral RNA replication (27). Several Pro to Ala mutations in this NS5A region have been evaluated in terms of CypA binding (51). The reduced CypA binding observed for P310A in the JFH-1 strain (equivalent to Pro314 in Con1) might be due to the loss of the Pro as anchoring residue but also to the disruption of the structural PW turn. Here, on the basis of the comparative titration experiment results with NS5A-D2 WT and the mutant NS5A-D2 I315G (which still contains all the proline residues), we show that the proper binding to CypA requires the presence of the PW turn structure (Fig. 10). Grisé et al. (51) have shown that two peptides derived from the cellular Supervillin and Nsun5 proteins containing the linear sequence ALPAW were able to bind to CypA in vitro. Interestingly, we found that these Pro and Trp residues in the Supervillin structure (PDB code 2K6N) (65), but not in the Nsun5 structure (PDB code 2B9E), fold into a PW turn similar to that we here identified in NS5A-D2. The sole Pro to Ala mutation in this sequence motif efficiently abolished the interaction with CypA in the case of the Supervillin peptide, whereas all of the five prolines have to be mutated in the case of the Nsun5 peptide (51). These observations further support our conclusion that the structural PW turn motif beyond the linear sequence is required for proper binding of CypA.
The binding and enzymatic activity of PPIases are inherently tightly coupled, raising questions about their respective role in functional processes in which they are involved (54). For HIV-1, the binding of CypA to the viral Capsid protein seems required (66), whereas the infection of E. coli by the filamentous phage fd is regulated by the Cyclophilin18-catalyzed cis/trans-isomerization of its Pro213 (67, 68). Regarding HCV and its mandatory host factor CypA, this important question remains to be answered (69). However, because mutations in the active site of CypA abolishing its PPIase activity also interfere with its binding property (69, 70), it is challenging to distinguish between these two possible mechanisms, which are moreover not mutually exclusive. We show that CypA-catalyzed cis/trans (c ↔ T) isomerization of Pro314 is significantly faster when the structural motif is absent (Fig. 12), suggesting that it is the interaction of the PW turn structure with CypA that is required for HCV RNA replication rather than the CypA-catalyzed cis/trans isomerization of Pro314. Although we showed a correlation between the CypA binding to the PW turn, the subcellular distribution of NS5A, which is related to the membranous web formation, and the HCV RNA replication, we cannot exclude the possibility that the PW turn structure might also be involved in the binding of other proteins, as, for example, the NS5B polymerase.
In conclusion, we have identified a small well defined PW turn structure in the intrinsically disordered domain 2 of HCV NS5A protein. This PW turn is conserved across all HCV genotypes and plays a crucial functional role, because it is required for viral RNA replication. This minimal structural element is directly involved in the binding of the host CypA, which is mandatory for HCV replication, and plays a role in NS5A subcellular distribution. Our work thereby provides a molecular basis for the understanding of the role of NS5A in the HCV replication and also highlights how very small structural motifs can carry specific function(s) in IDPs.
Author Contributions
X. H., G. L., R. B., and F. P. designed the study and wrote the paper. M. D. and X. H. purified proteins and peptides and performed the NMR analyses shown in Figs. 1–4, 6, 11, and 12. V. M. and R. B. designed, performed, and analyzed the cellular experiments shown in Figs. 8 and 9. R. M. and F. P. calculated and analyzed the NMR structure models of PepD2-WT shown in Fig. 5 and Table 1. P. A. and G. L. designed, performed, and analyzed the exchange NMR spectroscopy experiments shown in Figs. 11 and 12. I. H. designed and constructed vectors for expression of mutant proteins and analyzed the mutant phenotypes in bacteria. H. L. provided assistance for NMR experiments and contributed to the preparation of the figures. A. L. designed, performed, and analyzed the CD and fluorescence spectroscopy experiments shown in Fig. 7. All authors analyzed the results and approved the final version of the manuscript.
The work was supported by French National Agency for Research Grant ANR-11-JSV8-005 and French National Agency for Research on AIDS and Viral Hepatitis Grant A02007-2 and A02011-2. The NMR facility was supported by the European Community, the CNRS (TGIR RMN THC, FR-3050), the Universities of Lille1 and Lille2, the Région Nord-Pas-de-Calais (France), and the Institut Pasteur de Lille. This work was also supported by Mapping 128 Project ANR-11-BINF-003 (to F. P. and R. M.) and Deutsche Forschungsgemeinschaft Grants TRR83 and TP13 (to R. B.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 2M5L) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NMR assignment of the backbone atoms (HN, NH, Cα, Cβ, C′) of the domain 2 of NS5A (Con1 strain) and NMR assignment of the peptide PepD2-WT (residues 308–327 of the domain 2 of NS5A, Con1 strain) have been deposited in the Biological Magnetic Resonance Data Bank under accession codes 19055 and 19059, respectively.
- HCV
- hepatitis C virus
- Cyp
- cyclophilin
- HSQC
- heteronuclear single quantum correlation
- HMQC
- heteronuclear multiple-quantum correlation
- IDP
- intrinsically disordered protein
- IDR
- intrinsically disordered region
- MoRF
- molecular recognition feature
- PPIase
- peptidyl-prolyl cis/trans isomerase
- PW turn
- proline-tryptophane turn
- TOCSY
- total correlation spectroscopy
- PDB
- Protein Data Bank.
References
- 1. Gravitz L. (2011) A smouldering public-health crisis. Nature 474, S2–S4 [DOI] [PubMed] [Google Scholar]
- 2. Moradpour D., Penin F., Rice C. M. (2007) Replication of hepatitis C virus. Nat. Rev. Microbiol. 5, 453–463 [DOI] [PubMed] [Google Scholar]
- 3. Bartenschlager R., Lohmann V., Penin F. (2013) The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 11, 482–496 [DOI] [PubMed] [Google Scholar]
- 4. Scheel T. K., Rice C. M. (2013) Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pawlotsky J.-M. (2014) New hepatitis C virus (HCV) drugs and the hope for a cure: concepts in anti-HCV drug development. Semin. Liver Dis. 34, 22–29 [DOI] [PubMed] [Google Scholar]
- 6. Lange C. M., Jacobson I. M., Rice C. M., Zeuzem S. (2014) Emerging therapies for the treatment of hepatitis C. EMBO Mol. Med. 6, 4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao M., Nettles R. E., Belema M., Snyder L. B., Nguyen V. N., Fridell R. A., Serrano-Wu M. H., Langley D. R., Sun J. H., O'Boyle D. R., 2nd, Lemm J. A., Wang C., Knipe J. O., Chien C., Colonno R. J., Grasela D. M., Meanwell N. A., Hamann L. G. (2010) Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang F., Robotham J. M., Grise H., Frausto S., Madan V., Zayas M., Bartenschlager R., Robinson M., Greenstein A. E., Nag A., Logan T. M., Bienkiewicz E., Tang H. (2010) A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 6, e1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaul A., Stauffer S., Berger C., Pertel T., Schmitt J., Kallis S., Zayas M., Lopez M. Z., Lohmann V., Luban J., Bartenschlager R. (2009) Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5, e1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paeshuyse J., Kaul A., De Clercq E., Rosenwirth B., Dumont J. M., Scalfaro P., Bartenschlager R., Neyts J. (2006) The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 43, 761–770 [DOI] [PubMed] [Google Scholar]
- 11. Gallay P. A., Lin K. (2013) Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des. Devel. Ther. 7, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tellinghuisen T. L., Foss K. L., Treadaway J. C., Rice C. M. (2008) Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 82, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Appel N., Zayas M., Miller S., Krijnse-Locker J., Schaller T., Friebe P., Kallis S., Engel U., Bartenschlager R. (2008) Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4, e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moradpour D., Penin F. (2013) Hepatitis C virus proteins: from structure to function. Curr. Top. Microbiol. Immunol. 369, 113–142 [DOI] [PubMed] [Google Scholar]
- 15. Ross-Thriepland D., Harris M. (2014) Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J. Virol. 88, 1421–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Penin F., Brass V., Appel N., Ramboarina S., Montserret R., Ficheux D., Blum H. E., Bartenschlager R., Moradpour D. (2004) Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 279, 40835–40843 [DOI] [PubMed] [Google Scholar]
- 17. Ascher D. B., Wielens J., Nero T. L., Doughty L., Morton C. J., Parker M. W. (2014) Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 4, 4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L., Hwang J., Sharma S. D., Hargittai M. R., Chen Y., Arnold J. J., Raney K. D., Cameron C. E. (2005) Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280, 36417–36428 [DOI] [PubMed] [Google Scholar]
- 19. Tellinghuisen T. L., Marcotrigiano J., Rice C. M. (2005) Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Love R. A., Brodsky O., Hickey M. J., Wells P. A., Cronin C. N. (2009) Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambert S. M., Langley D. R., Garnett J. A., Angell R., Hedgethorne K., Meanwell N. A., Matthews S. J. (2014) The crystal structure of NS5A domain 1 from genotype 1a reveals new clues to the mechanism of action for dimeric HCV inhibitors. Protein Sci. 23, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tellinghuisen T. L., Foss K. L., Treadaway J. (2008) Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4, e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang Y., Ye H., Kang C. B., Yoon H. S. (2007) Domain 2 of nonstructural protein 5A (NS5A) of hepatitis C virus is natively unfolded. Biochemistry. 46, 11550–11558 [DOI] [PubMed] [Google Scholar]
- 24. Hanoulle X., Badillo A., Verdegem D., Penin F., Lippens G. (2010) The domain 2 of the HCV NS5A protein is intrinsically unstructured. Protein Pept. Lett. 17, 1012–1018 [DOI] [PubMed] [Google Scholar]
- 25. Hanoulle X., Verdegem D., Badillo A., Wieruszeski J. M., Penin F., Lippens G. (2009) Domain 3 of non-structural protein 5A from hepatitis C virus is natively unfolded. Biochem. Biophys. Res. Commun. 381, 634–638 [DOI] [PubMed] [Google Scholar]
- 26. Fan X., Xue B., Dolan P. T., LaCount D. J., Kurgan L., Uversky V. N. (2014) The intrinsic disorder status of the human hepatitis C virus proteome. Mol. Biosyst. 10, 1345–1363 [DOI] [PubMed] [Google Scholar]
- 27. Hanoulle X., Badillo A., Wieruszeski J. M., Verdegem D., Landrieu I., Bartenschlager R., Penin F., Lippens G. (2009) Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284, 13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verdegem D., Badillo A., Wieruszeski J. M., Landrieu I., Leroy A., Bartenschlager R., Penin F., Lippens G., Hanoulle X. (2011) Domain 3 of NS5A protein from the hepatitis C virus has intrinsic α-helical propensity and is a substrate of cyclophilin A. J. Biol. Chem. 286, 20441–20454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uversky V. N., Oldfield C. J., Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 30. Xue B., Williams R. W., Oldfield C. J., Goh G. K., Dunker A. K., Uversky V. N. (2010) Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept. Lett. 17, 932–951 [DOI] [PubMed] [Google Scholar]
- 31. Gunasekaran K., Tsai C. J., Kumar S., Zanuy D., Nussinov R. (2003) Extended disordered proteins: targeting function with less scaffold. Trends Biochem. Sci. 28, 81–85 [DOI] [PubMed] [Google Scholar]
- 32. Tokuriki N., Oldfield C. J., Uversky V. N., Berezovsky I. N., Tawfik D. S. (2009) Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 34, 53–59 [DOI] [PubMed] [Google Scholar]
- 33. Alves C., Cunha C. (2012) Order and disorder in viral proteins: new insights into an old paradigm. Fut. Virol. 7, 1183–1191 [Google Scholar]
- 34. Uversky V. N. (2013) A decade and a half of protein intrinsic disorder: biology still waits for physics: protein intrinsic disorder. Protein Sci. 22, 693–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T. J. (2012) Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 36. Vacic V., Oldfield C. J., Mohan A., Radivojac P., Cortese M. S., Uversky V. N., Dunker A. K. (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee S.-H., Kim D.-H., Han J. J., Cha E.-J., Lim J.-E., Cho Y.-J., Lee C., Han K.-H. (2012) Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 13, 34–54 [DOI] [PubMed] [Google Scholar]
- 38. Combet C., Garnier N., Charavay C., Grando D., Crisan D., Lopez J., Dehne-Garcia A., Geourjon C., Bettler E., Hulo C., Le Mercier P., Bartenschlager R., Diepolder H., Moradpour D., Pawlotsky J. M., Rice C. M., Trépo C., Penin F., Deléage G. (2007) euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 35, D363–D366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Combet C., Blanchet C., Geourjon C., Deléage G. (2000) NPS@: network protein sequence analysis. Trends Biochem. Sci. 25, 147–150 [DOI] [PubMed] [Google Scholar]
- 40. Verdegem D., Dijkstra K., Hanoulle X., Lippens G. (2008) Graphical interpretation of Boolean operators for protein NMR assignments. J. Biomol. NMR. 42, 11–21 [DOI] [PubMed] [Google Scholar]
- 41. Wüthrich K. (1986) NMR of proteins and nucleic acids, The George Fisher Baker Non-resident Lectureship in Chemistry at Cornell University, Wiley, New York [Google Scholar]
- 42. Cornilescu G., Delaglio F., Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 43. Schwieters C. D., Kuszewski J. J., Marius Clore G. (2006) Using Xplor: NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 [Google Scholar]
- 44. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 45. Lohmann V., Hoffmann S., Herian U., Penin F., Bartenschlager R. (2003) Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77, 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Madan V., Paul D., Lohmann V., Bartenschlager R. (2014) Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 146, 1361–1372 [DOI] [PubMed] [Google Scholar]
- 47. Reiss S., Harak C., Romero-Brey I., Radujkovic D., Klein R., Ruggieri A., Rebhan I., Bartenschlager R., Lohmann V. (2013) The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 9, e1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tamiola K., Acar B., Mulder F. A. (2010) Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 132, 18000–18003 [DOI] [PubMed] [Google Scholar]
- 49. Feuerstein S., Solyom Z., Aladag A., Favier A., Schwarten M., Hoffmann S., Willbold D., Brutscher B. (2012) Transient structure and SH3 interaction sites in an intrinsically disordered fragment of the hepatitis C virus protein NS5A. J. Mol. Biol. 420, 310–323 [DOI] [PubMed] [Google Scholar]
- 50. Casteleijn M. G., Alahuhta M., Groebel K., El-Sayed I., Augustyns K., Lambeir A.-M., Neubauer P., Wierenga R. K. (2006) Functional role of the conserved active site proline of triosephosphate isomerase. Biochemistry. 45, 15483–15494 [DOI] [PubMed] [Google Scholar]
- 51. Grisé H., Frausto S., Logan T., Tang H. (2012) A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates alisporivir susceptibility. J. Virol. 86, 4811–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berger C., Romero-Brey I., Radujkovic D., Terreux R., Zayas M., Paul D., Harak C., Hoppe S., Gao M., Penin F., Lohmann V., Bartenschlager R. (2014) Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 147, 1094–1105.e25 [DOI] [PubMed] [Google Scholar]
- 53. Anderson L. J., Lin K., Compton T., Wiedmann B. (2011) Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 8, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fischer G., Tradler T., Zarnt T. (1998) The mode of action of peptidyl prolyl cis/trans isomerases in vivo: binding vs. catalysis. FEBS Lett. 426, 17–20 [DOI] [PubMed] [Google Scholar]
- 55. Marsh J. A., Singh V. K., Jia Z., Forman-Kay J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Campen A., Williams R. M., Brown C. J., Meng J., Uversky V. N., Dunker A. K. (2008) TOP-IDP-Scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 15, 956–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Theillet F.-X., Kalmar L., Tompa P., Han K.-H., Selenko P., Dunker A. K., Daughdrill G. W., Uversky V. N. (2013) The alphabet of intrinsic disorder I: act like a pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins. 1, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barua B., Lin J. C., Williams V. D., Kummler P., Neidigh J. W., Andersen N. H. (2008) The Trp-cage: optimizing the stability of a globular miniprotein. Protein Eng. Des. Sel. PEDS. 21, 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neidigh J. W., Fesinmeyer R. M., Andersen N. H. (2002) Designing a 20-residue protein. Nat. Struct. Mol. Biol. 9, 425–430 [DOI] [PubMed] [Google Scholar]
- 60. Kursula I., Salin M., Sun J., Norledge B. V., Haapalainen A. M., Sampson N. S., Wierenga R. K. (2004) Understanding protein lids: structural analysis of active hinge mutants in triosephosphate isomerase. Protein Eng. Des. Sel. 17, 375–382 [DOI] [PubMed] [Google Scholar]
- 61. Rozovsky S., Jogl G., Tong L., McDermott A. E. (2001) Solution-state NMR investigations of triosephosphate isomerase active site loop motion: ligand release in relation to active site loop dynamics. J. Mol. Biol. 310, 271–280 [DOI] [PubMed] [Google Scholar]
- 62. Chung H.-Y., Gu M., Buehler E., MacDonald M. R., Rice C. M. (2014) Seed sequence-matched controls reveal limitations of small interfering RNA knockdown in functional and structural studies of hepatitis C virus NS5A-MOBKL1B interaction. J. Virol. 88, 11022–11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.-S., Longerich T., Diehl S., Ramirez F., Balla T., Rohr K., Kaul A., Bühler S., Pepperkok R., Lengauer T., Albrecht M., Eils R., Schirmacher P., Lohmann V., Bartenschlager R. (2011) Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 9, 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berger K. L., Kelly S. M., Jordan T. X., Tartell M. A., Randall G. (2011) Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85, 8870–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brown J. W., Vardar-Ulu D., McKnight C. J. (2009) How to arm a supervillin: designing F-actin binding activity into supervillin headpiece. J. Mol. Biol. 393, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Luban J., Bossolt K. L., Franke E. K., Kalpana G. V., Goff S. P. (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 73, 1067–1078 [DOI] [PubMed] [Google Scholar]
- 67. Eckert B., Martin A., Balbach J., Schmid F. X. (2005) Prolyl isomerization as a molecular timer in phage infection. Nat. Struct. Mol. Biol. 12, 619–623 [DOI] [PubMed] [Google Scholar]
- 68. Weininger U., Jakob R. P., Eckert B., Schweimer K., Schmid F. X., Balbach J. (2009) A remote prolyl isomerization controls domain assembly via a hydrogen bonding network. Proc. Natl. Acad. Sci. 106, 12335–12340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chatterji U., Bobardt M., Selvarajah S., Yang F., Tang H., Sakamoto N., Vuagniaux G., Parkinson T., Gallay P. (2009) The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284, 16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Braaten D., Ansari H., Luban J. (1997) The hydrophobic pocket of cyclophilin is the binding site for the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 71, 2107–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Strickland E. H. (1974) Aromatic contributions to circular dichroism spectra of proteins. CRC Crit. Rev. Biochem. 2, 113–175 [DOI] [PubMed] [Google Scholar]