

Background: Insulin resistance is a risk factor for Alzheimer disease.
Results: AMPKSer-485 is responsible for AMPK-mediated Tau phosphorylation.
Conclusion: Abnormal phosphorylation of AMPKSer-485 may be the link for the increased Alzheimer disease risk in metabolic syndrome
Significance: With the rapid increase in both metabolic syndrome and Alzheimer disease, it is crucial to understand the underling mechanism linking two diseases
Keywords: Alzheimer disease, insulin resistance, metabolic syndrome, neuron, phosphorylation
Abstract
Metabolic syndrome (MetS) is a cluster of cardiovascular risk factors including obesity, diabetes, and dyslipidemia, and insulin resistance (IR) is the central feature of MetS. Recent studies suggest that MetS is a risk factor for Alzheimer disease (AD). AMP-activated kinase (AMPK) is an evolutionarily conserved fuel-sensing enzyme and a key player in regulating energy metabolism. In this report, we examined the role of IR on the regulation of AMPK phosphorylation and AMPK-mediated Tau phosphorylation. We found that AMPKSer-485, but not AMPKThr-172, phosphorylation is increased in the cortex of db/db and high fat diet-fed obese mice, two mouse models of IR. In vitro, treatment of human cortical stem cell line (HK-5320) and primary mouse embryonic cortical neurons with the AMPK activator, 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR), induced AMPK phosphorylation at both Thr-172 and Ser-485. AMPK activation also triggered Tau dephosphorylation. When IR was mimicked in vitro by chronically treating the cells with insulin, AICAR specifically induced AMPKSer-485, but not AMPKThr-172, hyperphosphorylation whereas AICAR-induced Tau dephosphorylation was inhibited. IR also resulted in the overactivation of Akt by AICAR treatment; however, preventing Akt overactivation during IR prevented AMPKSer-485 hyperphosphorylation and restored AMPK-mediated Tau dephosphorylation. Transfection of AMPKS485A mutant caused similar results. Therefore, our results suggest the following mechanism for the adverse effect of IR on AD pathology: IR → chronic overactivation of Akt → AMPKSer-485 hyperphosphorylation → inhibition of AMPK-mediated Tau dephosphorylation. Together, our results show for the first time a possible contribution of IR-induced AMPKSer-485 phosphorylation to the increased risk of AD in obesity and diabetes.
Introduction
Alzheimer disease (AD)2 is a progressive neurodegenerative disease characterized by loss of memory and other cognitive functions necessary to perform complex daily activities (1). The most prominent neuropathological features of AD are the appearance of senile plaques composed of amyloid β peptides and neurofibrillary tangles derived from the aggregation of the microtubule-associated protein Tau (2, 3). AD is the most common form of dementia, accounting for over 70% of all cases, and it currently affects 5.4 million Americans. The incidence is expected to reach over 13.8 million by 2050, and it is estimated that the cost for caring for people with AD will dramatically increase from $203 billion for 2013 to $1.2 trillion by 2050.
Insulin resistance (IR) is defined as a state of reduced responsiveness of target tissue(s) to normal circulating levels of insulin. It is the central feature of metabolic syndrome (MetS), a constellation of disorders related to an increased risk of cardiovascular disease, and it is the major contributor for the development of diabetes (4). Multiple studies report that patients with MetS have an increased risk of developing AD compared with age- and gender-matched controls (5). In addition, accumulating evidence suggests that AD is closely related to dysfunction of both insulin signaling and glucose metabolism in the brain, prompting some investigators to refer to AD as type 3 diabetes or an insulin-resistant brain state (6, 7). Epidemiological studies also demonstrate an association between obesity caused by caused by dietary fat intake and an increased risk for AD (8), and multiple studies report that patients with diabetes have a 50–75% increased risk of developing AD compared with age- and gender-matched control groups (9–13). In parallel, a study of the Mayo Clinic AD Patient Registry revealed that 80% of AD patients have either type 2 diabetes or impaired fasting glucose (14).
There are many theories to explain the connection between AD and diabetes (and MetS in general), including impaired brain insulin signaling and the resulting IR (15, 16), mitochondrial dysfunction (17, 18), and disturbance in lipid and cholesterol metabolism (19). One theory suggests perturbed brain energy metabolism as the cause of pathogenic development of AD and cognitive impairment in general (20). Disruption in energy metabolism increases IR, affects glucose metabolism, and causes mitochondrial dysfunction, all of which are risk factors for AD (21). AMP-activated protein kinase (AMPK) is an evolutionarily conserved fuel-sensing enzyme and a key player in regulating energy metabolism (22). AMPK is activated during energy shortages and suppressed in energy surplus by sensing cellular AMP/ATP ratios. AMPK functions to restore cellular energy metabolism by suppressing energy consuming anabolic pathways and stimulating catabolic energy producing pathways (23).
In AD, the role of AMPK is controversial, with the reports suggesting both beneficial and detrimental effects on the progression of AD (20). It is reported that AMPK activation reduces amyloid β production in rat cortical neurons (24) and that leptin and resveratrol reduce amyloid β levels and Tau phosphorylation through AMPK activation (25–27). In contrast, AMPK can directly phosphorylate Tau, and amyloid β-induced increases in Tau phosphorylation require AMPK-activated Ca2+/calmodulin-dependent protein kinase kinase β (28). Furthermore, increased AMPK activation is observed in tangle- and pretangle-bearing neurons in AD brain (29). This activation of AMPK requires phosphorylation at threonine 172 (AMPKThr-172), a residue in its α-subunit, by upstream kinases such as liver kinase B1 (LKB1) or Ca2+/calmodulin-dependent protein kinase kinase β (21, 30). AMPK is also phosphorylated at serine 485 (AMPKSer-485) by Akt, which reduces Thr-172 phosphorylation and inhibits AMPK activation (31, 32); however, the direct contribution of Ser-485 on AMPK activation has not been extensively studied.
In this study, we report a novel role of AMPKSer-485 on Tau phosphorylation. We demonstrate that AMPKSer-485, but not AMPKThr-172, phosphorylation is increased in two animal models of diabetes and obesity. Our results also indicate that activation of AMPK by the AMPK activator AICAR decreases Tau phosphorylation and that this change is specifically correlated with changes in AMPKSer-485 phosphorylation. Finally, we demonstrate that Akt hyperactivation caused by IR may be responsible for the increased AMPKSer-485 phosphorylation and subsequent inhibition of AICAR-mediated Tau dephosphorylation. These results show for the first time how AMPKSer-485 phosphorylation may contribute to the increased risk of AD in obesity and diabetes.
Experimental Procedures
Antibodies and Chemicals
Polyclonal antibodies against phosphorylated Tau (Ser(P)-199/202, Ser(P)-262, Ser(P)-396, and Thr(P)-231) were purchased from Life Technologies Inc. Tau1 (recognizing dephosphorylated Tau), Tau5 (for total Tau), and anti-GAPDH antibodies were from Millipore (Billerica, MA). LY294002 and U0126 were also from Millipore. Ser(P)-485-AMPK and actin antibodies were from Abcam (Cambridge, MA). The antibodies for Thr(P)-172-AMPK, and AMPK were purchased from Cell Signaling (Beverly, MA). AICAR was purchased from Sigma-Aldrich. All other chemicals were purchased from either Sigma-Aldrich or Fisher Scientific (Fair Lawn, NJ).
Mouse Models and Brain Preparation
BKS-db/db and db+ (BKS.Cg-m+/+ Leprdb/J, JAX mice stock #000642) mice were purchased from Jackson Laboratory (Bar Harbor, ME). C57BL6 (B6) mice, a commonly used model to study diet-induced obesity (20, 21, 22, 23), were also from Jackson Laboratory. Obesity was induced in B6 mice by placing them on a high fat diet (HFD) consisting of 54% kcal from fat (D05090701; Research Diets Inc., New Brunswick, NJ;) at 4 weeks of age until 24 weeks of age, whereas control mice were fed a standard diet consisting of 10% kcal from fat (D12450B; Research Diets Inc.) (Table 1). Fasting blood glucose levels were measured every 4 weeks using a standard glucometer (OneTouch; LifeScan Inc., Milpitas, CA). All mice were housed in a pathogen-free environment and cared for following the University of Michigan Committee on the Care and Use of Animals guidelines.
TABLE 1.
Diet composition
| D12450B | D05090701 | |||
|---|---|---|---|---|
| gm | kcal | gm | kcal | |
| Product | ||||
| Protein | 19 | 20 | 25 | 20 |
| Carbohydrate | 67 | 70 | 32 | 26 |
| Fat | 4 | 10 | 30 | 54 |
| Total | 100 | 100 | ||
| kcal/gm | 3.85 | 5.04 | ||
| Ingredient | ||||
| Casein, 80 mesh | 200 | 800 | 200 | 800 |
| l-Cystein | 3 | 12 | 3 | 12 |
| Corn starch | 315 | 1260 | 56.3 | 225 |
| Maltodextrin 10 | 35 | 140 | 125 | 500 |
| Sucrose | 350 | 1400 | 68.8 | 275 |
| Cellulose, BW200 | 50 | 0 | 50 | 0 |
| Soybean oil | 25 | 225 | 25 | 225 |
| Lard | 20 | 180 | 220 | 1980 |
| Mineal mix S10026 | 10 | 0 | 10 | 0 |
| Dicalcium phosphate | 13 | 0 | 13 | 0 |
| Calcium carbonate | 5.5 | 0 | 5.5 | 0 |
| Potassium citrate, 1 H2O | 16.5 | 0 | 16.5 | 0 |
| Vitamin mix V10001 | 10 | 10 | 10 | 40 |
| Choline bitartrate | 2 | 0 | 2 | 0 |
| FD&C yellow dye #5 | 0.05 | 0 | 0.025 | 0 |
| FD&C red dye #40 | 0 | 0 | 0.025 | 0 |
| FD&C blue dye #1 | 0 | 0 | 0 | 0 |
| Total | 1055.05 | 4057 | 805.15 | 4057 |
Mice were euthanized per our published protocols with an overdose of sodium pentobarbital (33, 34). For Western immunoblotting analyses, brains were cut in half, the cortex and hippocampus were separated, and then tissues were snap frozen in liquid nitrogen and stored at −80 °C until use. At least six animals were analyzed for each treatment, and three or four representative Western blotting results are shown on each figure.
Cell Culture and Treatments
HK-532 human cortical stem cells were provided by Neuralstem, Inc. (Rockville, MD). Cells were incubated in differentiation medium (NSDM) for 7–10 days, and the medium was changed to NSDM without insulin 24 h before treatment.
Primary cortical neurons were prepared from embryonic day 15 embryos of Sprague-Dawley rats. Cells were maintained in feed medium (neurobasal media; Invitrogen) supplemented with B27 without antioxidant (Invitrogen) and other supplements as described previously (35) for 6 days before being used in experiments. Culture medium was changed to treatment medium (feed medium without B27 and antibiotics) 18 h before insulin and/or AICAR treatments.
Construction, Production, and Infection of Lentiviral Expression Vectors
Human AMPKa1 cDNA was amplified from SH-SY5Y human neuroblastoma cell cDNA and directionally subcloned into the XhoI and NotI sites of the lentiviral expression vector pLVX-IRES-mCherry (Clontech). The S485A and S485D mutants of AMPK were created by the QuikChange method (Stratagene, La Jolla, CA). All constructs were confirmed by sequencing. Lentiviral stocks were generated by the University of Michigan Vector Core. HK-532 cells were infected with 10× viral stock 3 days into differentiation for 6 h. The cells were harvested 5 days after infection following insulin and/or AMPK treatment as described in the results. For the generation of the stable clones, cells were selected by puromycin resistance.
Western Immunoblotting
Western immunoblotting was performed as described previously (35). Mouse cortex was homogenized using a plastic pestle in a microcentrifuge tube in T-PER tissue protein extraction reagent (Pierce) with a protease inhibitor tablet. Samples were briefly sonicated and centrifuged at 13,000 rpm for 20 min at 4 °C, and supernatants were collected. Cultured cells were lysed in radioimmune precipitation assay buffer (Pierce) with protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Lysates were collected, briefly sonicated, and centrifuged at 13,000 rpm for 15 min at 4 °C prior to the collection of supernatants. Protein concentration of the brain and cortical neuron lysates were measured with the Pierce 660-nm protein assay reagent (Thermo Scientific, Rockford, IL). The lysates were separated by SDS-PAGE and transferred to nitrocellulose membranes. After blocking with 3% BSA in TBS with 0.1% Tween 20, nitrocellulose membranes were incubated with the appropriate primary antibodies at 4 °C overnight followed by secondary antibodies conjugated with horseradish peroxidase (Santa Cruz) at room temperature for 2 h. Signals were visualized using enhanced chemiluminescence reagents (ECL; Amersham Bioscience) or SuperSignal West Femto maximum sensitivity substrate (Pierce), depending on the signal strength. Images were captured using the Chemidoc XRS system and analyzed by Quantity One software (Bio-Rad). In some experiments, the nitrocellulose membranes were incubated at 60 °C for 15 min in stripping solution (2% SDS, 100 mm dithiothreitol, and 100 mm Tris, pH 6.8) and then utilized for immunoblotting with an additional antibody. All experiments were repeated at least three times, and representative results are presented in the figures.
Statistical Analysis
At least six animals were used for statistical analysis for in vivo experiments. All in vitro experiments were repeated at least three times and presented as the means ± S.E. Statistical analysis was performed by either one-way analysis of variance with Tukey's postanalysis or Student's t test depending on the number of comparison groups, using GraphPad Prism software (GraphPad Software Inc., San Diego, CA). Statistical significance was defined as p < 0.05.
Results
db/db mice demonstrates an elevation of plasma insulin at ∼2 weeks and of blood sugar at 4–6 weeks. By 8 weeks of age, hyperinsulinemia and hyperglycemia were stable phenotypes of the db/db mice (36). At 24 weeks, db/db mice display the full blown diabetes phenotypes with the increased body weight and blood glucose and glycated hemoglobulin (Fig. 1A). However, as the diabetic phenotype continues to deteriorate the mice prone to a spontaneous death, so we harvested the mice at 24 weeks. Mice fed with high fat diet usually display signs of prediabetes starting around 16 weeks of age (37, 38). Their blood glucose level and body weight continue to increase up to a year (39). We observed that HFD mice display increased body weight (Fig. 1A) and impaired glucose tolerance test (Fig. 1B) but no significant changes in blood glucose and glycated hemoglobulin levels indicating that they are prediabetic.
FIGURE 1.

Metabolic phenotyping of db/db and HFD-fed mice. A, body weight, blood glucose, and glycated hemoglobulin (GHb) of 24-week-old db/db (left panels) mice and mice fed with HFD for 20 weeks (right panels). B, HFD mice display impaired glucose tolerance test (GTT). At least six mice were used for each group. *, p < 0.05 by t test
AMPK is an evolutionarily conserved serine/threonine kinase that plays a pivotal role in maintaining cellular homeostasis (22); therefore, we first examined the changes in AMPK phosphorylation in the brain of two IR mouse models; diabetic mice (db/db) or obese mice (HFD-fed). AMPK phosphorylation at Thr-172 in the cortex of db/db mice was not significantly different from that in control db+ mice (Fig. 2A). Similarly, mice fed a HFD for 24 weeks did not affect AMPKThr-172 phosphorylation in the cortex (Fig. 2B). In contrast, however, AMPKSer-485 was significantly increased in both db/db (2-fold) and obese (1.7-fold) mouse cortices compared with control animals (Fig. 2, A and B). In addition, Tau phosphorylation was increased in HFD mouse cortex compared with controls (Fig. 2C). Increased Ser/Thr phosphorylation of IRS proteins is one of the key features of IR (40). We observed consistently increased IRS phosphorylation (Fig. 2D). Basal phosphorylation of Akt was also increased in HFD mouse cortex; the increase in ERK phosphorylation was less significant compared with control animals. We previously reported similar changes in Tau phosphorylation as well as insulin signaling components in db/db mouse brains (36). These results imply that MetS factors, such as diabetes and obesity, may underlie the observed changes in AMPK and Tau phosphorylation.
FIGURE 2.

Increased AMPKSer-485 and Tau phosphorylation in db/db and HFD mouse brains. Cortex from 24-week-old db/db (A) or 20-week-old HFD (B–D) mouse were homogenized in TPER buffer. The lysates were immunoblotted with the indicated antibodies. The relative density of phosphorylated AMPK over total AMPK (A and B) and phosphorylated Tau (pTau) over total Tau (Tau5) (C) from the same mouse was measured after immunoblotting. Phosphorylation of IRS-1, Akt, and ERK, the important signaling components of insulin signaling, was also examined from control and HFD cortex (D). The results are means ± S.E. of at least six animals per group, and the representative blots are shown. *, p < 0.05 by t test.
To examine the role of AMPK on Tau phosphorylation in more detail, we used in vitro cell culture models utilizing HK-532 cortical stem cells and primary rat cortical neurons. Treatment of HK-532 cells with the AMPK activator, AICAR, resulted in time- and concentration-dependent increases in AMPK phosphorylation at both Thr-172 and Ser-485 (Fig. 3A). AMPK phosphorylation was maintained at least 6 h after AICAR treatment. Because of serine and threonine phosphorylation, total AMPK immunoblotting resulted in a diffuse pattern. Acetyl-CoA carboxylase phosphorylation by AMPK at Ser-79, a surrogate marker for AMPK activation (41), was also increased following AICAR treatment in a manner that corresponded to the observed AMPK phosphorylation (Fig. 3A). In parallel, AMPK activation following treatment of HK-532 cells with AICAR for 2 h reduced Tau phosphorylation at Ser-199/202, Thr-231, and Ser-396 (Fig. 3B). Tau1 immunoreactivity is also increased after AICAR treatment (Fig. 3B), consistent with the preferential reaction of the Tau1 antibody with Tau when it is dephosphorylated at serines 195, 198, 199, and 202 (42). Similar changes in AMPK and Tau phosphorylation were observed in primary cortical neurons by AICAR treatment (Fig. 3C).
FIGURE 3.

AICAR treatment increases AMPK phosphorylation and decreases Tau phosphorylation. A, HK-532 cells were differentiated for 1 week and treated with 1 mm AICAR for 0–6 h (left panel) or increasing concentrations of AICAR for 2 h (middle panel). The results of AMPK phosphorylation with 1 mm AICAR for 2 h treatment are shown in the graph on the right. B, HK-532 cells were treated with 1 mm AICAR for 2 h, and Tau phosphorylation was examined. C, primary rat embryonic cortical neurons were treated with 1 mm AICAR for 0–6 h. Cell lysates were prepared in radioimmune precipitation assay buffer and immunoblotted with the indicated antibodies, Anti-actin immunoblots demonstrate the equal protein loading. *, p < 0.05 by t test.
IR is at the core of MetS (4); therefore, we next tested the effect of IR on AMPK-mediated Tau dephosphorylation utilizing HK-532 cortical stem cells and primary rat cortical neurons. We previously reported that chronic insulin treatment of dorsal root ganglia and primary cortical neurons results in a blunted insulin response, suggesting that these models are an excellent means to study neuronal IR in vitro (43, 44). To examine the effect of IR on AMPK phosphorylation, we treated HK-532 cells without or with 50 nm insulin overnight and then with 1 mm AICAR for 0, 2, or 6 h. The increase in AMPKThr-172 phosphorylation by AICAR was not significantly different with or without insulin pretreatment (Fig. 4, A and B, upper panel); however, interestingly, AICAR-induced AMPKSer-485 phosphorylation was significantly higher after insulin pretreatment (Fig. 4, A and B, lower panel). As expected, AICAR treatment resulted in Tau dephosphorylation, and induction of IR by chronic insulin treatment prevented AICAR-induced Tau dephosphorylation (Fig. 4, C and D). We also observed a similar inhibitory effect of IR on AICAR-induced Tau dephosphorylation in embryonic cortical neurons (Fig. 4E). Together, these results suggest a connection between increased AMPKSer-485 phosphorylation and Tau phosphorylation, which is abolished in the setting of IR.
FIGURE 4.

Insulin pretreatment specifically increases AMPKSer-485 phosphorylation and prevents AICAR-induced Tau dephosphorylation. A, HK-532 cells were treated without (− Ins) or with (+ Ins) 50 nm insulin overnight and then treated with 1 mm AICAR for 0, 2, or 6 h. B, when the cells were pretreated with 50 nm insulin overnight, AICAR treatment specifically increased AMPKSer-485 phosphorylation. *, p < 0.05 compared with Ser-485 phosphorylation without insulin pretreatment at same time point. C, the cells were treated without or with 50 nm insulin overnight and then treated with 1 mm AICAR for 2 h. Insulin pretreatment prevented AICAR-induced Tau dephosphorylation. D, densitometric analysis of phosphorylated Tau (pTau). E, insulin pretreatment also prevented AICAR-induced Tau dephosphorylation in embryonic rat primary cortical neurons (eCN). *, p < 0.05 compared with control by t test.
To examine the possible mechanism behind IR-mediated changes in AMPK and Tau phosphorylation, we next looked at the contribution of insulin signaling pathway components. To first confirm that the cells are insulin-responsive, we treated HK-532 cells with insulin and observed an increase in Akt phosphorylation, as well as a slight increase in ERK phosphorylation (Fig. 5A). It is reported that Akt is the kinase responsible for AMPKSer-485 phosphorylation and the subsequent inhibition of AMPK activity (32), and it is also reported that AMPK can activate PI3-K/Akt pathway (45, 46). Here, we demonstrate that AICAR treatment of HK-532 cells also increases Akt phosphorylation, whereas insulin pretreatment significantly increased AICAR-mediated Akt phosphorylation (Fig. 5B). AICAR treatment did not increase ERK phosphorylation with or without insulin pretreatment (Fig. 5B). We previously reported that prevention of Akt hyperactivation restored insulin responsiveness and reversed IR in DRG and cortical neurons (43, 44). Similarly, treatment of HK-532 cells with insulin along with LY294002 to inhibit the hyperactivation of Akt prevented the overphosphorylation of AMPKSer-485 by AICAR (Fig. 5, C and D, panel b). As expected, AMPKThr-172 phosphorylation was not affected by insulin or LY294002 treatment (Fig. 5, C and D, panel a). LY294002 treatment also restored AICAR-mediated dephosphorylation of Tau (Fig. 5, C and D, panel c). Treatment with the MAPK inhibitor, U0126, had no effect on AMPKSer-485 or Tau phosphorylation (Fig. 5C). These results strongly suggest that hyperactivation of Akt increases AMPKSer-485 phosphorylation and ultimately leads to the inhibition of AMPK-mediated dephosphorylation of Tau.
FIGURE 5.

Hyperactivation of Akt by insulin pretreatment is responsible for the increased AMPKSer-485 phosphorylation in HK-532 cells. A, HK-532 cells were treated with 20 nm insulin and Akt, and ERK phosphorylation was examined. B, HK-532 cells were incubated overnight without or with 50 nm insulin (Ins) and then treated with 1 mm AICAR for 2 h. Insulin pretreatment significantly increased AICAR induced Akt phosphorylation. *, p < 0.05; #, p < 0.01 by t test. C, the cells were incubated with insulin along with 20 μm LY294002 (LY) or U0126 (U) overnight. The cells were washed and treated with 1 mm AICAR for 2 h. D, inhibiting Akt overactivation during insulin pretreatment by LY294002 prevented AMPKSer-485 hyperphosphorylation (b) and restored AICAR-induced Tau dephosphorylation. (c) but did not affect AMPKThr-172 phosphorylation (a). *, p < 0.05 compared with insulin + LY294002 + AICAR; #, p < 0.05 compared with insulin or insulin + AICAR; v, p < 0.05 compared with control.
Finally, to confirm the specific contribution of AMPKSer-485 phosphorylation, we transfected HK-532 cells with lentiviral vectors expressing nonphosphorylatable mutants of AMPKSer-485, AMPK S485A. Transfection of AMPK S485A decreased basal as well as AICAR-induced AMPKSer-485 phosphorylation and slightly increased AMPKThr-172 phosphorylation (Fig. 6A). AMPK S485A transfection suppressed AMPKSer-485 hyperphosphorylation after insulin pretreatment (Fig. 6B). Tau phosphorylation levels correlated well with the reduced AMPKSer-485 phosphorylation, and more importantly, AMPK S485A transfection restored the ability of AICAR to reduce Tau phosphorylation after insulin pretreatment (Fig. 6C). Stable transfection of S485A, but not S485D, mutant also reversed the inhibitor effect of IR on AICAR-induced Tau phosphorylation (Fig. 6, D and E). These results emphasize the direct and important role of Ser-485 in AMPK-mediated Tau phosphorylation.
FIGURE 6.

Preventing AMPKSer-485 hyperphosphorylation by mutating Ser-485 restored AICAR-induced Tau dephosphorylation by insulin pretreatment. A, HK-532 cell were infected with a lentiviral vector expressing S485A AMPK mutants or control for 5 days. S485A mutant transfection reduced both basal and AICAR-induced Ser-485 phosphorylation by AICAR. The cells were treated without or with 50 nm insulin overnight and then treated with 1 mm AICAR for 2 h. B and C, cell lysates were examined for AMPK (B) and Tau (C) phosphorylation. D, HK-532 cells were transfected with S485A or S485D AMPK mutant, and the stable clones were selected by puromycin. AICAR-induced Tau dephosphorylation was examined after without or with 50 nm overnight insulin pretreatment. E, densitometry of the result of stable AMPK mutant transfection. *, p < 0.05; #, p < 0.005.
Discussion
It is now well established that MetS, including obesity and diabetes, is a risk factor for AD (5, 6). IR is at the core of MetS, and studies suggest that IR has an important role for the development/acceleration of AD and further emphasize the need for precise regulation of energy expenditure (6, 7). There is significant gap in our knowledge, however, about the molecular link between IR and increased risk of AD. Considering the important role of AMPK in energy metabolism, we can naturally speculate that it is the crucial link between IR and AD. In the current study, we report that the activation of AMPK by AICAR resulted in Tau dephosphorylation and that chronic insulin treatment prevented AICAR-induced Tau dephosphorylation. Furthermore, we demonstrated for the first time that chronic insulin treatment specifically increased AICAR-induced AMPKSer-485 phosphorylation, consistent with what is observed in the brains of the diabetes and obesity mouse models, and preventing hyperactivation of Akt during chronic insulin treatment prevented the overphosphorylation of Ser-485 and subsequently restored the ability of AMPK to reduce Tau phosphorylation. Together, these results emphasize the important role of AMPKSer-485 in AMPK regulation of Tau phosphorylation, especially in IR.
Given that most of the studies of AMPK are focused on peripheral tissues, there is very limited knowledge about the regulation AMPK in brain regions other than the hypothalamus (47, 48). Our results from mouse brains demonstrate that MetS induces a specific increase in AMPKSer-485 phosphorylation in the cortices of both db/db and obese mice. We also observed increased Akt phosphorylation in these brains caused by IR (44), suggesting that increased Akt may contribute to the increased AMPKSer-485 phosphorylation (31, 32). In a very recent report, Arnold et al. (49) demonstrated for the first time that AMPK phosphorylation is increased in the brain of HFD-fed mice; however, their results only report AMPKThr-172 phosphorylation,3 which showed no changes in our study. This discrepancy may arise from apparent differences in the feeding scheme of the HFD between our study and theirs regarding the fat content (54% versus 60%), duration of feeding (24 weeks versus 17 days), and when the diet was initiated (4 weeks versus 8 weeks). Similarly, in another report, intracerebroventricular injection of streptozotocin decreased AMPK phosphorylation and increased Tau phosphorylation, both of which were reversed by AICAR administration (50). Because there are very few studies about AMPK in the brain, more research is required to obtain consistent results.
The exact contribution of AMPK to AD pathology is still controversial, with the results supporting both beneficial and detrimental effects (20, 24–29). Our results demonstrate a causal relationship between increased AMPKSer-485 and Tau phosphorylation in db/db and obese mouse brains. Our current study does not clearly describe whether Tau phosphorylation is regulated directly by AMPK, especially AMPKSer-485 phosphorylation, or through other kinases. However, the close correlation between AMPK and Tau phosphorylation observed in vivo along with our in vitro results strongly suggest the important contribution of AMPKSer-485 on Tau phosphorylation. Most of the studies about the connection between AMPK and AD pathology focus only on AMPKThr-172 phosphorylation. It is possible that the status of AMPKSer-485 phosphorylation (which was not examined in previous reports) might have resulted in the contradictory findings.
The important role of AMPKSer-485 phosphorylation on Tau phosphorylation is supported by our in vitro results. In agreement with published reports (26, 27), we show that activation of AMPK by AICAR reduced Tau phosphorylation, whereas induction of IR correlates with a specific increase in AMPKSer-485 phosphorylation and prevents AICAR-induced Tau dephosphorylation. However, our results demonstrating that increased AMPKSer-485 phosphorylation during IR did not affect AMPKThr-172 phosphorylation conflict with previous reports demonstrating that increases in AMPKSer-485 phosphorylation are generally accompanied by decreased AMPKThr-172 phosphorylation (31, 32, 51). This may possibly be due to the relatively small increase in AMPKSer-485 phosphorylation observed during IR, or it could be that AMPKSer-485-mediated regulation of Tau phosphorylation is independent of AMPKThr-172 phosphorylation. Thus, the precise role of AMPK on Tau phosphorylation is still controversial and likely depends on the context of the experiments.
AMPKThr-172 is phosphorylated by LKB1 or Ca2+/calmodulin-dependent protein kinase kinase β (21, 30), whereas AMPKSer-485 is phosphorylated by Akt (31, 32). Our in vitro data further demonstrate that AMPK reciprocally increases the phosphorylation and activation of Akt. Interestingly, AMPK activation during IR significantly increased Akt phosphorylation, correlating well with the specific increase of AMPKSer-485 phosphorylation. Although some studies suggest cooperative interactions between AMPK and Akt activation (45, 46), we believe, at least in our system, that AICAR-mediated Akt activation serves as a negative feedback regulator that limits AMPK activity. Indeed, we observe that AMPKThr-172 phosphorylation returns to basal levels after long term (>8 h) AICAR treatment (data not shown). Thus, IR induces overactivation of Akt and a subsequent increase in AMPKSer-485 phosphorylation, which results in the disruption of AMPK signaling.
In the current study, we further demonstrate that Akt hyperphosphorylation by AMPK in IR results in inhibition of AMPK-mediated Tau dephosphorylation and that AMPKSer-485 phosphorylation has a direct role in Tau phosphorylation using two Ser-485 phosphorylation-resistant AMPK mutant constructs. There are reports for both increased (28, 29) and decreased (25–27, 50) Tau phosphorylation by AMPK activation; however, none of the studies examined the role of AMPKSer-485 phosphorylation, and very few examined the connection between IR and AMPK in the brain (49, 50). Of note, insulin antagonizes AMPK activation by increased phosphorylation of AMPKSer-485 (31, 52); therefore, we believe that IR overactivates Akt, resulting in increased AMPKSer-485 phosphorylation and subsequent inhibition of Tau dephosphorylation.
Our results are summarized in the model (Fig. 7). In normal conditions, AMPK regulates Tau phosphorylation through balanced phosphorylation of Thr-172 and Ser-485. AMPK-mediated Akt phosphorylation may act as a negative feedback mechanism for a precise regulation of AMPK activity. In the MetS status, IR induces impaired insulin/Akt signaling, with supporting data for not only reduced Akt activation but also instances of chronic overactivation of Akt (43, 44). Overactivation of Akt in IR subsequently leads to the hyperphosphorylation of AMPKSer-485. These changes result in the elevated phosphorylation of Tau. It is not clear from our study whether AMPKSer-485 affects Tau phosphorylation through inhibitory action on AMPKThr-172 or direct independent action on Tau. Our results suggest for the first time a possible contribution of AMPKSer-485 phosphorylation toward the increased risk of AD in IR associated with diabetes and obesity. Furthermore, this study supports a new direction of investigation into the role of AMPK in AD and possibly other neurodegenerative diseases as well.
FIGURE 7.
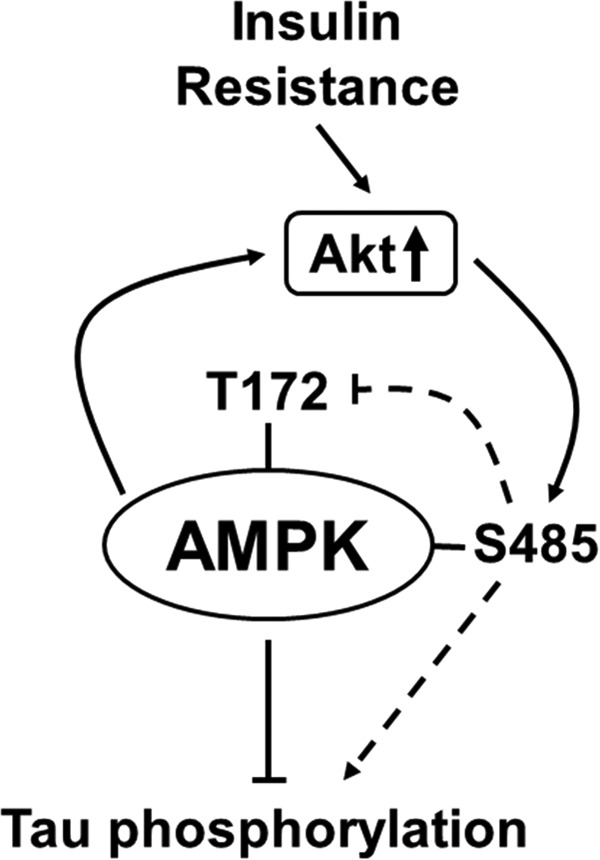
Model summarizing our study. IR induces chronic overactivation of Akt and subsequent hyperphosphorylation of AMPKSer-485. These changes result in the elevated phosphorylation of Tau and may contribute the progression of AD.
Author Contributions
B. K. designed and coordinated the study and wrote the paper. C. F.-G. designed and constructed vectors for expression of mutant AMPK and analyzed the mutant phenotypes on Fig 6. C. P. and C. B. provided technical assistance and preparation of the figures. E. L. F. revised the article and provided critical feedback.
This work was supported, in whole or in part, by National Institutes of Health Grants 1DP3DK094292 and 1R24082841 (to E. L. F.). This work was also supported by the Program for Neurology Research and Discovery. The authors declare that they have no conflicts of interest with the contents of this article.
S. E. Arnold, I. Lucki, B. R. Brookshire, G. C. Carlson, C. A. Browne, H. Kazi, S. Bang, B. R. Choi, Y. Chen, M. F. McMullen, and S. F. Kim, personal communication.
- AD
- Alzheimer disease
- IR
- insulin resistance
- MetS
- metabolic syndrome
- AMPK
- AMP-activated kinase
- HFD
- high fat diet
- NSDM
- neural stem differentiation media
- AICR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside.
References
- 1. Querfurth H. W., LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 2. Johnson G. V., Stoothoff W. H. (2004) Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117, 5721–5729 [DOI] [PubMed] [Google Scholar]
- 3. Martin L., Latypova X., Terro F. (2011) Post-translational modifications of Tau protein: implications for Alzheimer's disease. Neurochem. Int. 58, 458–471 [DOI] [PubMed] [Google Scholar]
- 4. Duvnjak L., Duvnjak M. (2009) The metabolic syndrome: an ongoing story. J. Physiol. Pharmacol. 60, 19–24 [PubMed] [Google Scholar]
- 5. Kim B., Feldman E. L. (2012) Insulin resistance in the nervous system. Trends Endocrinol. Metab. 23, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frisardi V., Solfrizzi V., Capurso C., Imbimbo B. P., Vendemiale G., Seripa D., Pilotto A., Panza F. (2010) Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J. Alzheimers Dis. 21, 57–63 [DOI] [PubMed] [Google Scholar]
- 7. Lester-Coll N., Rivera E. J., Soscia S. J., Doiron K., Wands J. R., de la Monte S. M. (2006) Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J. Alzheimers Dis. 9, 13–33 [DOI] [PubMed] [Google Scholar]
- 8. Luchsinger J. A., Tang M. X., Shea S., Mayeux R. (2002) Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 59, 1258–1263 [DOI] [PubMed] [Google Scholar]
- 9. Ott A., Stolk R. P., van Harskamp F., Pols H. A., Hofman A., Breteler M. M. (1999) Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53, 1937–1942 [DOI] [PubMed] [Google Scholar]
- 10. Peila R., Rodriguez B. L., Launer L. J. (2002) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes 51, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 11. Biessels G. J., Kappelle L. J. (2005) Increased risk of Alzheimer's disease in type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 33, 1041–1044 [DOI] [PubMed] [Google Scholar]
- 12. Cox D. J., Kovatchev B. P., Gonder-Frederick L. A., Summers K. H., McCall A., Grimm K. J., Clarke W. L. (2005) Relationships between hyperglycemia and cognitive performance among adults with type 1 and type 2 diabetes. Diabetes Care 28, 71–77 [DOI] [PubMed] [Google Scholar]
- 13. Brands A. M., Biessels G. J., de Haan E. H., Kappelle L. J., Kessels R. P. (2005) The effects of type 1 diabetes on cognitive performance: a meta-analysis. Diabetes Care 28, 726–735 [DOI] [PubMed] [Google Scholar]
- 14. Janson J., Laedtke T., Parisi J. E., O'Brien P., Petersen R. C., Butler P. C. (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481 [DOI] [PubMed] [Google Scholar]
- 15. de la Monte S. M. (2009) Insulin resistance and Alzheimer's disease. BMB Rep. 42, 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hölscher C. (2011) Diabetes as a risk factor for Alzheimer's disease: insulin signalling impairment in the brain as an alternative model of Alzheimer's disease. Biochem. Soc. Trans. 39, 891–897 [DOI] [PubMed] [Google Scholar]
- 17. Galindo M. F., Ikuta I., Zhu X., Casadesus G., Jordán J. (2010) Mitochondrial biology in Alzheimer's disease pathogenesis. J. Neurochem. 114, 933–945 [DOI] [PubMed] [Google Scholar]
- 18. Wang X., Su B., Lee H. G., Li X., Perry G., Smith M. A., Zhu X. (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 29, 9090–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlsson C. M. (2010) Type 2 diabetes mellitus, dyslipidemia, and Alzheimer's disease. J. Alzheimers Dis. 20, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salminen A., Kaarniranta K., Haapasalo A., Soininen H., Hiltunen M. (2011) AMP-activated protein kinase: a potential player in Alzheimer's disease. J. Neurochem. 118, 460–474 [DOI] [PubMed] [Google Scholar]
- 21. Cai Z., Yan L. J., Li K., Quazi S. H., Zhao B. (2012) Roles of AMP-activated protein kinase in Alzheimer's disease. Neuromolecular Med. 14, 1–14 [DOI] [PubMed] [Google Scholar]
- 22. Carling D., Mayer F. V., Sanders M. J., Gamblin S. J. (2011) AMP-activated protein kinase: nature's energy sensor. Nat. Chem. Biol. 7, 512–518 [DOI] [PubMed] [Google Scholar]
- 23. Mihaylova M. M., Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Won J. S., Im Y. B., Kim J., Singh A. K., Singh I. (2010) Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem. Biophys. Res. Commun. 399, 487–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vingtdeux V., Giliberto L., Zhao H., Chandakkar P., Wu Q., Simon J. E., Janle E. M., Lobo J., Ferruzzi M. G., Davies P., Marambaud P. (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 285, 9100–9113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greco S. J., Sarkar S., Johnston J. M., Tezapsidis N. (2009) Leptin regulates Tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 380, 98–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greco S. J., Hamzelou A., Johnston J. M., Smith M. A., Ashford J. W., Tezapsidis N. (2011) Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce Tau phosphorylation and beta-amyloid in neurons. Biochem. Biophys. Res. Commun. 414, 170–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thornton C., Bright N. J., Sastre M., Muckett P. J., Carling D. (2011) AMP-activated protein kinase (AMPK) is a Tau kinase, activated in response to amyloid beta-peptide exposure. Biochem. J. 434, 503–512 [DOI] [PubMed] [Google Scholar]
- 29. Vingtdeux V., Davies P., Dickson D. W., Marambaud P. (2011) AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies. Acta Neuropathol. 121, 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carling D., Sanders M. J., Woods A. (2008) The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. (Lond.) 32, S55–S59 [DOI] [PubMed] [Google Scholar]
- 31. Horman S., Vertommen D., Heath R., Neumann D., Mouton V., Woods A., Schlattner U., Wallimann T., Carling D., Hue L., Rider M. H. (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase α-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 281, 5335–5340 [DOI] [PubMed] [Google Scholar]
- 32. Ning J., Xi G., Clemmons D. R. (2011) Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 152, 3143–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leinninger G. M., Backus C., Sastry A. M., Yi Y. B., Wang C. W., Feldman E. L. (2006) Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol. Dis. 23, 11–22 [DOI] [PubMed] [Google Scholar]
- 34. Vincent A. M., McLean L. L., Backus C., Feldman E. L. (2005) Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 19, 638–640 [DOI] [PubMed] [Google Scholar]
- 35. Kim B., Backus C., Oh S., Feldman E. L. (2013) Hyperglycemia-induced Tau cleavage in vitro and in vivo: a possible link between diabetes and Alzheimer's disease. J. Alzheimers Dis. 34, 727–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim B., Backus C., Oh S., Hayes J. M., Feldman E. L. (2009) Increased Tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150, 5294–5301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watcho P., Stavniichuk R., Ribnicky D. M., Raskin I., Obrosova I. G. (2010) High-fat diet-induced neuropathy of prediabetes and obesity: effect of PMI-5011, an ethanolic extract of Artemisia dracunculus L. Mediators Inflamm. 2010, 268547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Obrosova I. G., Ilnytska O., Lyzogubov V. V., Pavlov I. A., Mashtalir N., Nadler J. L., Drel V. R. (2007) High-fat diet induced neuropathy of pre-diabetes and obesity: effects of “healthy” diet and aldose reductase inhibition. Diabetes 56, 2598–2608 [DOI] [PubMed] [Google Scholar]
- 39. Ahrén J., Ahrén B., Wierup N. (2010) Increased beta-cell volume in mice fed a high-fat diet: a dynamic study over 12 months. Islets 2, 353–356 [DOI] [PubMed] [Google Scholar]
- 40. Copps K. D., White M. F. (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ha J., Daniel S., Broyles S. S., Kim K. H. (1994) Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 269, 22162–22168 [PubMed] [Google Scholar]
- 42. Szendrei G. I., Lee V. M., Otvos L., Jr. (1993) Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J. Neurosci. Res. 34, 243–249 [DOI] [PubMed] [Google Scholar]
- 43. Kim B., McLean L. L., Philip S. S., Feldman E. L. (2011) Hyperinsulinemia induces insulin resistance in dorsal root ganglion neurons. Endocrinology 152, 3638–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim B., Sullivan K. A., Backus C., Feldman E. L. (2011) Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid. Redox. Signal. 14, 1829–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kimura T., Tomura H., Sato K., Ito M., Matsuoka I., Im D. S., Kuwabara A., Mogi C., Itoh H., Kurose H., Murakami M., Okajima F. (2010) Mechanism and role of high density lipoprotein-induced activation of AMP-activated protein kinase in endothelial cells. J. Biol. Chem. 285, 4387–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jessen N., Pold R., Buhl E. S., Jensen L. S., Schmitz O., Lund S. (2003) Effects of AICAR and exercise on insulin-stimulated glucose uptake, signaling, and GLUT-4 content in rat muscles. J. Appl. Physiol. 94, 1373–1379 [DOI] [PubMed] [Google Scholar]
- 47. Dagon Y., Hur E., Zheng B., Wellenstein K., Cantley L. C., Kahn B. B. (2012) p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab. 16, 104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xue B., Kahn B. B. (2006) AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J. Physiol. 574, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnold S. E., Lucki I., Brookshire B. R., Carlson G. C., Browne C. A., Kazi H., Bang S., Choi B. R., Chen Y., McMullen M. F., Kim S. F. (2014) High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 67, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Du L. L., Chai D. M., Zhao L. N., Li X. H., Zhang F. C., Zhang H. B., Liu L. B., Wu K., Liu R., Wang J. Z., Zhou X. W. (2015) AMPK activation ameliorates Alzheimer's disease-like pathology and spatial memory impairment in a streptozotocin-induced Alzheimer's disease model in rats. J. Alzheimers Dis. 43, 775–784 [DOI] [PubMed] [Google Scholar]
- 51. Hurley R. L., Barré L. K., Wood S. D., Anderson K. A., Kemp B. E., Means A. R., Witters L. A. (2006) Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 281, 36662–36672 [DOI] [PubMed] [Google Scholar]
- 52. Kovacic S., Soltys C. L., Barr A. J., Shiojima I., Walsh K., Dyck J. R. (2003) Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 278, 39422–39427 [DOI] [PubMed] [Google Scholar]